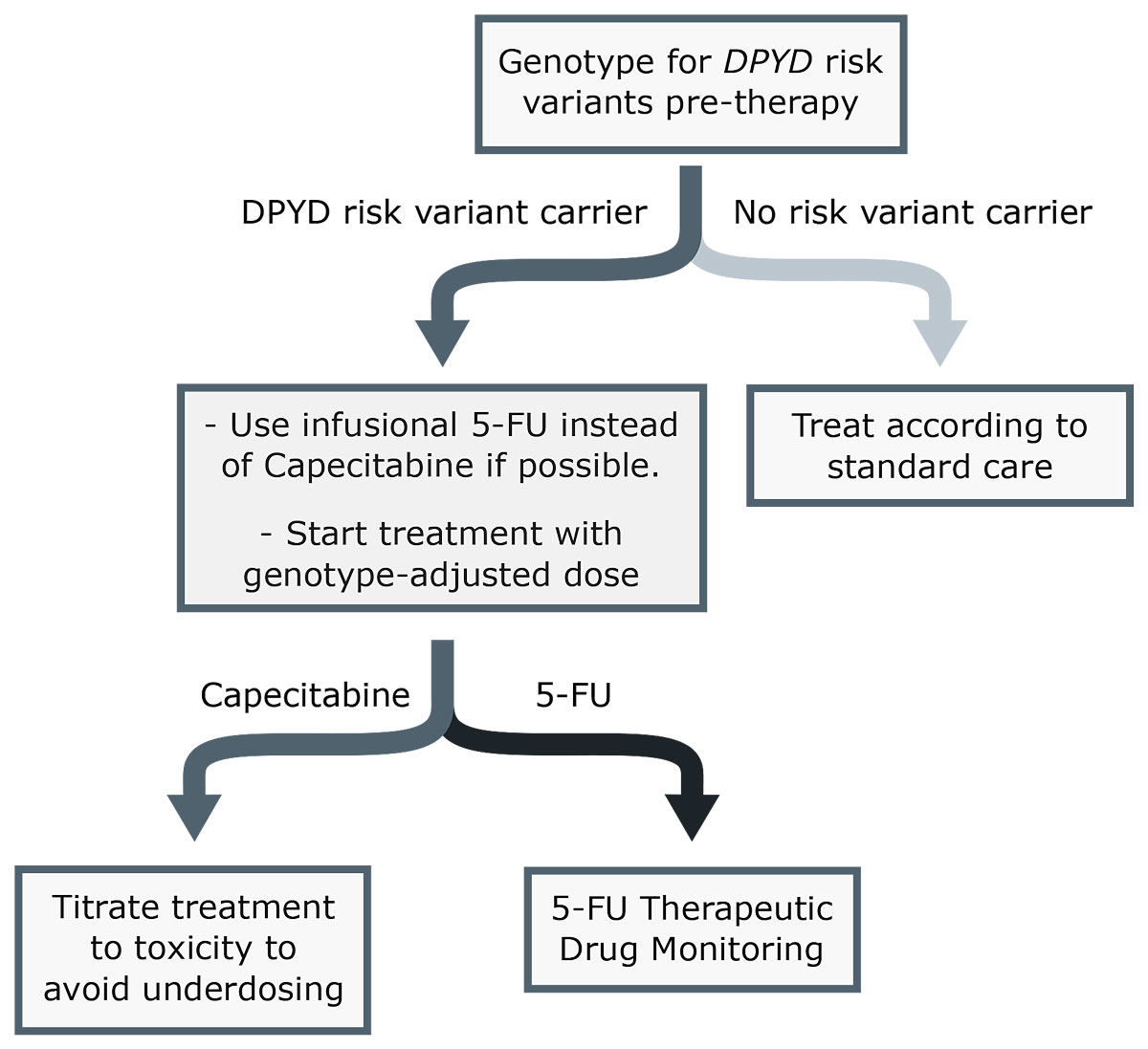
Figure 1 Simplified scheme for fluoropyrimidine dosing. 5-FU = 5-fluorouracil.
DOI: https://doi.org/10.4414/smw.2020.20375
The two fluoropyrimidines (FPs) 5-fluorouracil (5-FU) and capecitabine (Cap) are among the most frequently used chemotherapies in the treatment of colorectal, breast, and head and neck cancer [1]. Despite their good performance in diverse treatment settings, the occurrence of severe FP-related toxicities causing severe morbidity or treatment cessation is an important drawback of these drugs. Depending on the treatment regimen, and in particular on the drugs used in combination with the FPs, 10–40% of patients experience severe, and in rare cases (0.2–0.5%) even lethal, FP-related toxicity in early chemotherapy cycles [2–4]. Major toxicity from FP treatment primarily reflects excessive cell death in healthy tissue with rapidly dividing cells, such as the bone marrow and mucous membranes [5]. The most common side effects thus include dose-dependent hematological and gastrointestinal toxicities and skin reactions like hand-foot syndrome (HFS) [6].
In this context, it is important to note that the toxicity profiles of Cap and 5-FU differ considerably. For example, patients receiving Cap-based therapies have a three-fold higher risk of developing severe HFS [7]. A recently published phase three clinical trial of biliary tract cancer patients treated with Cap reported severe HFS as the most common toxicity (grade ≥3, 20% of patients). This study also reported that around 5% of patients discontinued treatment exclusively due to HFS, even if this toxicity was not life-threatening [8].
A recent study investigated the incidence of FP-related toxicities in France [9]. This study reported that one fifth, 14,700 out of 76,200 patients treated annually with 5-FU or Cap developed serious FP-associated adverse events (SAEs) within the first two cycles of chemotherapy. Gastrointestinal and haematological toxicities were reported as the most common SAEs (66.3%). SAEs were defined in this particular study as “any adverse event entailing hospitalisation or prolongation of hospital stay, permanent disability or invalidity, life-threatening prognosis, and death” [9]. The number of patients with a life-threatening prognosis, disability or death was estimated to be 1200 per year. Adjusting these findings to the Swiss population (France ∼65 million vs Switzerland ∼8.6 million), around ∼2000 FP-related SAEs are expected each year in Switzerland, with life-threatening toxicity, disability or death in approximately 160 patients.
The dose-dependence of FP-related toxicities is demonstrated by clinical studies showing a clear relationship between 5-FU exposure (i.e., area under the curve, AUC) and the severity of FP-related toxicity [10–12], with up to two-fold higher mean 5-FU AUCs observed in patients with life-threatening FP toxicity compared to patients with no or minimal toxicity from 5-FU-based chemotherapy [10].
In current clinical practice, FP dosing is based on a patient’s body surface area (BSA), with doses adjusted to meet a prespecified amount of mg/m2. However, pharmacokinetic studies have demonstrated that BSA-based dosing is very limited in its ability to account for inter-individual differences in FP plasma concentrations [13, 14]. In fact, Gamelin et al. found that systemic 5-FU concentrations vary up to 10-fold between patients receiving BSA-based dosing [14], strongly suggesting an extensive amount of FP pharmacokinetic variability which is not accounted for by BSA. As a result, 5-FU concentrations within the desired therapeutic target range are met only in approximately 15–20% of patients, based on current BSA-guided dosing practices [15, 16]. Several studies have shown the benefit of dose adjustments based on 5-FU plasma levels (therapeutic drug monitoring, TDM) over BSA-dosing, reporting a reduced incidence of FP toxicity, and even evidence for increased efficacy [16–20]. A recently published review of 5-FU TDM, therefore, concluded that TDM is strongly recommended for various 5-FU treatment regimens [21]. However, 5-FU TDM is currently not applied in clinical routine.
Patients with reduced activity of the rate-limiting enzyme for 5-FU catabolism – dihydropyrimidine dehydrogenase (DPD) – are at high risk of supratherapeutic drug concentrations under BSA-based standard dosing, and consequently are at risk of developing severe or sometimes even lethal FP-related toxicities. DPD activity is highly variable in the population [3]. This can be at least partly attributed to genetic variability in its encoding gene, DPYD. Specifically, numerous studies have demonstrated the clinical relevance of four genetic variants in DPYD, which are predictive for severe FP-related toxicities: c.1905+1G>A (rs3918290), c.1679T>G (rs55886062), c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, c.1236G>A/HapB3) [22]. It has also been shown that prospective genotyping of DPYD is feasible and even cost-effective [3, 23].
In Switzerland, pharmacogenetic testing of these four DPYD risk variants, as well as the measurement of 5-FU plasma levels (TDM), is covered by the compulsory health insurance [24], and analytical procedures have been implemented in clinical diagnostic laboratories [25, 26]. However, in spite of clinical practice recommendations for DPYD genotype-based FP dosing and TDM, and their potential for added therapeutic benefit, clinical uptake of these tests has been minimal so far in Switzerland. In the following, we evaluate different guidelines in the context of the current situation in Switzerland, discuss potential reasons for the lack of utilisation in clinical practice, and provide general recommendations for a patient-tailored FP therapy based on pharmacogenetic DPYD testing and 5-FU TDM.
Recently, the Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA) published a recommendation to screen for DPD deficiency in patients before the use of fluoropyrimidines [27]. These recommendations are likely to foster the uptake of DPYD genotyping/phenotyping and 5-FU TDM in Europe. Indeed, a consensus position paper in support of the EMA recommendations was published in June 2020 by the German Society for Haematology and Medical Oncology (Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie), in cooperation with several societies and groups from Austria, Germany and Switzerland, including the Swiss Society for Medical Oncology (Schweizerische Gesellschaft für Medizinische Onkologie) [28].
Two guidelines to help clinicians interpret DPYD genotypes and to adjust the starting FP dose accordingly have been published by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG) [2, 29]. These two guidelines, in particular those by the CPIC, form the basis of the recent EMA recommendations. However, neither includes 5-FU TDM. A separate guideline addressing the issue of 5-FU-TDM was recently published by Beumer et al. [21].
In some European countries, pre-emptive DPD testing (genetic and/or phenotypic) prior to therapy start is already established in clinical medicine: For example, the Netherlands included DPYD genotyping in its “national guidelines for colorectal carcinoma” in 2017. A recently published study reported that by the end of 2018, 87% of FP-treated patients in Amsterdam were tested for DPD deficiency using genotypic and/or phenotypic tests prior to the start of FP therapy [30]. In France, DPD testing is recommended by the National Agency for the Safety of Medicines and Health Products (Agence national de sécurité du médicament et des produits de santé) and laboratory assays (genetic and/or phenotypic) are routinely implemented in 17 laboratories across the country [31].
Drug labels are only slowly being updated regarding recent insights into the safety of FP chemotherapy and the benefits of genotype-guided FP dosing, and are currently incomplete: although most current 5-FU and Cap drug labels in Switzerland include and recommend DPYD genotyping, they lack information regarding genotype-guided dose adjustments or 5-FU TDM, and do not include the above-mentioned guidelines. Notably, these drug labels also do not fully reflect the current state of the literature regarding other pharmacogenetic markers that are still under investigation in a research context. Specifically, all 5-FU labels contain the outdated information that TYMS genotyping (i.e., testing for variants in the gene encoding for thymidylate synthase, the primary therapeutic target of 5-FU chemotherapy) could be beneficial in predicting toxicity to 5-FU-based chemotherapy. However, no prospective evidence for the clinical relevance of these pharmacogenetic markers with regard to 5-FU toxicity currently exits [32] (table 1). Such outdated information may lead to confusion and hamper the clinical implementation of DPYD genotyping and TDM in order to further individualise FP dosing.
Table 1 Overview of information on fluoropyrimidine pharmacogenetics and therapeutic drug monitoring in Swiss 5-fluorouracil and capecitabine drug labels.
| Drug | Update | Includes DPD deficiency | Includes DPYD genetic variation | Recommends testing for DPYD risk variants | Provides dosing recommendation | Includes therapeutic drug monitoring | Includes TYMS genotyping* |
|---|---|---|---|---|---|---|---|
| Fluorouracil Accord† | Jul 20 | + | + | + | – | – | + |
| Fluorouracil Labatec‡ | Apr 19 | + | + | + | – | – | + |
| Fluorouracil Sandoz§ | Jul 20 | + | + | + | – | – | + |
| Fluorouracil Teva¶ | Jul 20 | + | + | + | – | – | + |
| Xeloda‖ | Aug 20 | + | + | + | – | – | – |
* TYMS encodes for thymidylate synthase enzyme, which is the primary target of 5-FU chemotherapy. Several studies showed only a moderate effect of TYMS variants on the development of fluoropyrimidine-related toxicities. No prospective study has shown the usefulness of TYMS markers as toxicity predictors. † Accord Healthcare AG; ‡ Labatec Pharma SA; § Sandoz Pharmaceuticals AG; ¶ Teva Pharma AG; ‖ Roche Pharma (Schweiz) AG Drug labels were retrieved from https://www.swissmedicinfo.ch/?Lang=EN and last accessed on 19 Sept 2020.
Both available guidelines (CPIC and DPWG) highlight the four DPYD risk variants (c.1905+1G>A (rs3918290), c.1679T>G (rs55886062), c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, c.1236G>A/HapB3)), as these have the strongest evidence regarding their impact on DPD activity and FP toxicity risk [2, 29] (table 2). All four variants have been consistently associated with severe FP-related toxicity and impaired DPD activity. Two of these variants (c.1905+1G>A (rs3918290), c.1679T>G (rs55886062)) lead to alleles producing an enzyme with only minimal or no residual activity, termed “no function” alleles according to the CPIC functional classification. The other two variants (c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, c.1236G>A/HapB3)) have a less severe effect on DPD activity and are termed “decreased function” alleles. In patients carrying one copy of these two variants, DPD activity in peripheral blood cells is reduced by 31-34% and 20-35% respectively, whereas this reduction in activity is approximately 45-68% for the two “no function” variants [3, 33]. The combined carrier frequency of DPYD risk variants in the Swiss population is about 6.5%, which means that one out of every 15 patients carries at least one such variant [4].
Table 2 DPYD risk variants.
| DPYD variant | Carrier frequencies | Biological effect | |
|---|---|---|---|
| Swiss population | EU population | ||
| c.1129-5923C>G (rs75017182, c.1236G>A/HapB3) | 4.7% | 4.1% | Decreased function allele (affects mRNA splicing) |
| c.2846A>T (rs67376798) | 0.6% | 1.0% | Decreased function allele (affects co-factor binding Asp949Val) |
| c.1679T>G (rs55886062) | 0.4% | 0.1% | Nonfunctional allele (affects protein stability Ile560Ser) |
| c.1905+1G>A (rs3918290) | 0.8% | 1.0% | Nonfunctional allele (affects mRNA splicing) |
Frequencies for Swiss population are from Froehlich et al [4]. Frequencies for the EU population were retrieved from gnomAD database (European non-Finnish).
Both guidelines translate genotypes into an enzymatic activity score to derive dosing recommendations based on the DPYD genotype. Briefly, normal function alleles are assigned a value of one, “decreased function” alleles a value of 0.5, and “no function” alleles a value of zero. The DPD phenotype is subsequently assigned using a gene activity score, calculated as the sum of the two lowest allele function scores. For heterozygous carriers of the four DPYD risk variants described above, the gene activity score is thus 1.5 for c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, c.1236G>A/HapB3) and one for c.1905+1G>A (rs3918290) and c.1679T>G (rs55886062).
In spite of the varying severity of their impacts on DPD activity and the resulting differing gene activity scores, both guidelines currently recommend a 50% dose reduction for all heterozygous carriers of one of these variants. This recommendation is primarily based on a recent prospective study where a 25% reduction in the initial FP dose in heterozygous carriers of the “decreased function” variants was not sufficient to reduce the rate of early-onset FP-related toxicity to that observed in patients with no risk variants, while a 50% dose reduction achieved this goal for the two “no function variants” [3, 23].
While the guidelines are in agreement with respect to dose recommendations for heterozygous carriers of a single DPYD risk variant, which constitute the vast majority (>98%) of risk variant carriers, they differ for rare homozygous or compound heterozygous carriers (table 3). The CPIC guidelines recommend a 50% dose reduction in patients homozygous or compound heterozygous for any of the two “decreased function” alleles and the use of alternative, non-FP-based chemotherapy in patients compound heterozygous for different combinations of a “no function” and a “decreased function” allele (table 3). The DPWG guidelines, on the other hand, recommend the measurement of DPD activity in peripheral blood cells in these patients and an FP dose adjustment corresponding to the measured enzyme activity. Both guidelines recommend against the use of FP-based chemotherapy in patients who are homozygous or compound heterozygous for the two “no function” alleles (table 3). It is important to note that among Swiss patients, carriers of more than one of the four risk variants are expected to be very rare (∼1 in 1000), and clinical data on patients carrying these genotypes is very limited. For this reason, activity scores for combinations of multiple DPYD risk variants are mostly extrapolated from data based on carriers of a single risk variant.
Table 3 Fluoropyrimidine dosing recommendations according to DPYD genotype.
| DPYD genotype | Recommended starting dose in % | |||
|---|---|---|---|---|
| CPIC | DPWG | SPT | ||
| Heterozygous for a single DPYD risk allele | e.g., c.2846A>T/= or c.1905+1G>A/= | 50% | 50% | 50%† |
| Two decreased function alleles | e.g., c.2846A>T/c.2846A>T or c.1129-5923C>G/c.2846A>T |
50% | PHENO* | 25%† |
| One decreased function -and one nonfunctional allele | e.g., c.1905+1G>A/c.1129-5923C>G or c.2846A>T/c.1679T>G |
0% | PHENO* | 0% |
| Two nonfunctional alleles | e.g., c.1905+1G>A/c.1905+1G>A or c.1679T>G/c.1905+1G>A |
0% | 0% | 0% |
5-FU = 5-fluorouracil; CPIC = Clinical Pharmacogenetics Implementation Consortium; DPWG = Dutch Pharmacogenetics Working Group; SPT = Swiss Group of Pharmacogenomics and Personalised Therapy; TDM = therapeutic drug monitoring * Requires further assessment by DPD phenotyping; † use infusional 5-FU followed by TDM-based dose titration
Another discrepancy between these guidelines is that the CPIC does not give any recommendation on tegafur due to limited evidence, whereas the DPWG includes dose recommendations for tegafur. Since tegafur is not available in Switzerland, we do not consider it in this review.
Currently, all existing guidelines advise against FP treatment in homozygous carriers of no function DPYD alleles due to a lack of data. Although it was shown for a single case that treatment with extremely low FP doses in is principle feasible, the therapeutic effectiveness of such a low-dose treatment is unknown [34].
A common criticism of preemptive DPYD genotyping is its low sensitivity, i.e. the considerable inter-individual variability in FP exposure and FP toxicity that is not fully explained by the four well-studied DPYD risk variants [2, 29]. These variants explain only approximately 20% of the severe toxicities (grade ≥3) in patients of Caucasian origin. However, when considering life-threatening toxicities (grade 4–5 toxicity within the first 12 weeks of treatment), a systematic review of 6403 patients revealed that 29.3% of the 518 severe toxicity cases were carriers of one of the four DPYD risk variants [35]. Importantly, preemptive genotyping in the Netherlands has been shown to reduce overall treatment costs due to reduced costs related to the management of adverse events even when only a proportion of severe toxicities can be prevented with this pharmacogenetic test [36].
A second concern regarding the implementation of preemptive DPYD pharmacogenetic testing is the potential risk of underdosing patients when using reduced initial doses, and thereby impairing the treatment efficacy of these drugs. This concern is based on the observation that about half of patients carrying DPYD risk variants tolerate standard doses [3, 23]. To address this concern, the CPIC guidelines recommend dose titration based on TDM or tolerable toxicities in order to maintain treatment efficacy [2, 37].
There are also some concerns regarding potential treatment delays caused by pre-treatment DPYD genotyping. As neither 5-FU nor Cap is used for oncological emergencies, a turnaround time of five working days is adequate. Targeted genotyping of the four well-established DPYD risk variants can be performed sufficiently quickly with standard equipment available in diagnostic genetic laboratories in Switzerland. Furthermore, delays due to prolonged sample transport time are of no concern in Switzerland given the highly developed transportation services which allow the overnight delivery of samples if necessary.
Furthermore, genotyping only these four most common DPYD risk variants means that other, rarer DPYD variants cannot be ruled out. However, the cumulative frequency of all other deleterious DPYD mutations known to date is <0.1% in Europeans. Thus, it is estimated that these extremely rare deleterious variants can only explain a small additional fraction of all occurrences of severe toxicity [38], making targeted genotyping a pragmatic approach in these populations. Importantly, most data currently available on DPYD variation and FP-related toxicity is based on studies in Caucasian populations, and additional variants may occur at higher frequencies in patients of other ethnicities. Indeed, an additional “decreased function” variant, c.557 A>G (rs115232898, p.Y186C), has a frequency of 2.1% in African and African American populations [39]. Unfortunately, due to its size, current technologies do not allow resequencing of the entire DPYD gene in a time- and cost-efficient manner for wide clinical implementation. However, resequencing all of DPYD’s protein-coding sequences might become feasible in the near future, and can already be used today in special cases. This has the potential to further increase the sensitivity of DPYD genotyping [40], albeit only to a limited extent and particularly in patients of non-Caucasian or mixed ancestry [2]. For such cases, the CPIC guidelines include curated allele function definitions for many rare and non-Caucasian variants to enable the calculation of activity scores and dosing recommendations. It is worth noting that genetic variants in other FP-metabolising genes have also been shown to be associated with FP-related SAEs, which might explain further toxicity cases. However, these variants require further research in order to be implemented as clinical markers [41].
Given the substantial residual variability in DPD activity even among patients with identical risk variant genotypes, several approaches for direct phenotyping of DPD activity have been investigated as potential predictors for FP-related toxicities [42]. As mentioned above, the DPWG guidelines recommend the measurement of DPD activity in peripheral blood cells in patients with certain DPYD genotype combinations (table 3). However, none of the phenotyping methods evaluated so far have a comparable strength of evidence supporting their predictive power for severe FP-related toxicity as is available for DPYD genotyping. The sensitivity and specificity of these assays have not been clearly established, they are not as easily standardised between laboratories as a genotyping test, and some are also labor-intensive and logistically challenging. For this reason, no general cut-offs or dosing recommendations based on DPD phenotyping have been established, and to our knowledge no clinically validated phenotyping assay is currently available in Switzerland. While such assays may become important in the future as complementary methods to DPYD genotyping, no phenotyping approach can be recommended at this point [41].
In a majority of patients, BSA-based FP dosing results in drug concentrations below the therapeutic range, suggesting significant potential for added therapeutic benefit with improved FP dosing tailored towards the individual requirements of each patient. The traditional practice of escalating the dose of 5-FU in patients experiencing no or low toxicity in the previous cycle [43] has been largely abandoned in recent times because this clinician-based dosing is difficult to reproduce and could not be implemented in the latest generation of trials. Indeed, several studies have demonstrated the superiority of pharmacokinetics-guided 5-FU-dosing both in relation to treatment effectiveness and toxicity [16–20]. In the light of this strong evidence, the recently published comprehensive review on 5-FU TDM by Beumer et al. concludes that TDM is strongly recommended in all patients treated with various 5-FU treatment regimens [21]. The established target range for continuous 5-FU infusion, determined based on a balance between therapeutic benefit and toxicity, is an AUC of 20–30 mg×h/l. However, this target range is not recommended for bolus only regimens and infusion durations of over 120h. The review concludes that the following 5-FU regimens should include TDM: “FOLFOX4, FOLFOX6, FOLFOX7, FOLFIRI, LV5FU, FUFOX, AIO, weekly 1.5g/m2/8 hours for CRC and 1.0g/m2/ day D1–4 or 1.0g/m2/day D1–5 for SCCHN” [21]. Importantly, this recommendation only applies to therapies with infusional 5-FU, whereas the utility of TDM in Cap-based regimens is not clearly established. Cap is converted to 5-FU in cancer and some non-neoplastic cells; consequently, the plasma exposure of 5-FU is very low [44]. At present, the correlation of plasma concentrations of Cap or its metabolites with clinical outcomes (effectiveness, toxicity) is unclear, and therapeutic target ranges have not been defined. As a result, TDM cannot be used to adjust dosing for Cap-based regimens.
Assays for measuring 5-FU plasma concentrations to determine the AUC of infusional 5-FU therapies are available in Switzerland. For example, a high-performance liquid chromatography–mass spectrometry method for measuring 5-FU in plasma was developed by Büchel et al. [26]. Commercial assays are also available and have been implemented in diagnostic laboratories [25]. However, 5-FU TDM is currently hampered by pre-analytical challenges [21]. 5-FU is rapidly catabolised after blood collection by DPD enzyme, which is present in blood cells. Therefore, rapid centrifugation or the immediate addition of a stabilising agent, a DPD inhibitor, is required. Improper sample preservation can cause falsely low 5-FU concentration measurements, which could lead to overdosing of the patient in the following cycles. In the case of a suspected false low result, a repeated measurement in the next therapy cycle is therefore suggested prior to any dose-adjustment. Another limitation is the inaccuracy of the run time of infusion pumps for 5-FU. This was illustrated by a study reporting high variability in 5-FU drug delivery using elastomeric pumps [45]. While electronic infusion systems might improve the accuracy of drug delivery, they carry the risks of false programming and free flow. They are also more labour- and cost-intensive [46, 47]. But most importantly, the electronic devices are less convenient and reassuring for the patient than the “connect and forget” elastomeric pumps. In 2010, the FDA launched the Infusion Pump Improvement Initiative, recognising the need to address issues associated with the different types of pumps [48]. Calculation of the 5-FU AUC based on a single time point requires a steady-state drug concentration measurement, taken when the pump is delivering the drug at a constant rate during the continuous infusion. If the blood draw is taken too close to the expected end of the infusion time, it is not uncommon that the pump is already empty, and thus the measured drug concentration no longer reflects the steady state, making any PK assessment impossible. To avoid uninterpretable measurements, it is therefore recommended that the blood collection is scheduled with sufficient time prior to the planned end of the infusion (e.g., after 24–36 hours for 48-hour infusions), which in many cases requires an additional office visit by the patient. An early blood draw close to the therapy start is not recommended since it has been shown that the steady-state level of 5-FU is not reached after two hours of infusion [49]. Importantly, blood must be drawn from a separate venipuncture and not from the infusion port, as this will lead to falsely high results, even if the port is washed several times [21]. Based on current evidence, we agree with the guideline recommendation [21] that 5-FU TDM would be beneficial in all patients treated with this drug. However, the limitations of the infusion pumps in current use and pre-analytical difficulties may impair the general implementation of 5-FU TDM in clinical routine. Therefore, we recommend 5-FU TDM specifically in patients carrying a DPYD risk variant and in whom starting doses are reduced based on this genotype so as to minimise the risk of underdosing. We provide a dosing scheme in table 4, which was adapted from Kaldate et al. [49]. In order to fully recommend 5-FU TDM in every patient, infusion pumps with more accurate run times and less error-prone pre-analytical procedures are required.
Table 4 5-fluorouracil TDM dosing scheme.
| 5-FU AUC (mg×h/l) | Dose adjustment in the next cycle in % |
|---|---|
| >40 | 30% lower |
| 37–39 | 25% lower |
| 34–36 | 20% lower |
| 31–33 | 10% lower |
| 20–30 | No change required |
| 17–19 | 10% higher |
| 14–16 | 20% higher |
| 8–13 | 25% higher |
| <8 | Repeat the previous dose to exclude possible pre-analytical errors. If repeated AUC <8, dose adjustment: 30% higher |
5-FU = 5-fluorouracil; AUC = area under curve; TDM = therapeutic drug monitoring This dosing scheme is adaptated from Kaldate et al. [46]
In brief, we strongly recommend DPYD genotyping of the four DPYD risk variants (c.1905+1G>A (rs3918290), c.1679T>G (rs55886062), c.2846A>T (rs67376798) and c.1129-5923C>G (rs75017182, c.1236G>A/HapB3)) prior to the start of therapy, followed by a reduction of the initial dose to 50% of the standard dose in patients who are heterozygous carriers of one of these variants, as recommended in the CPIC guidelines [2]. In order avoid unnecessary treatment delays, it is advisable to include the date of treatment start when submitting samples for DPYD genotyping; genotyping results should be reported within five working days after receipt of the samples by the laboratories. Our recommendations are summarised in figure 1. A more detailed scheme is shown in figure S1 in appendix 1. While we generally recommend following the CPIC guidelines, we propose the following adjustments or additions:
Figure 1 Simplified scheme for fluoropyrimidine dosing. 5-FU = 5-fluorouracil.
Figure S1: SPT guideline for fluoropyrimidine dosing.
The appendix is available as a separate file at: https://smw.ch/article/doi/smw.2020.20375.
The authors declare no potential conflicts of interest.
1 Carrillo E , Navarro SA , Ramírez A , García MÁ , Griñán-Lisón C , Perán M , et al. 5-Fluorouracil derivatives: a patent review (2012 - 2014). Expert Opin Ther Pat. 2015;25(10):1131–44. doi:.https://doi.org/10.1517/13543776.2015.1056736
2 Amstutz U , Henricks LM , Offer SM , Barbarino J , Schellens JHM , Swen JJ , et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther. 2018;103(2):210–6. doi:.https://doi.org/10.1002/cpt.911
3 Henricks LM , Lunenburg CATC , de Man FM , Meulendijks D , Frederix GWJ , Kienhuis E , et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459–67. doi:.https://doi.org/10.1016/S1470-2045(18)30686-7
4 Froehlich TK , Amstutz U , Aebi S , Joerger M , Largiadèr CR . Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int J Cancer. 2015;136(3):730–9. doi:.https://doi.org/10.1002/ijc.29025
5 Longley DB , Harkin DP , Johnston PG . 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. doi:.https://doi.org/10.1038/nrc1074
6 Amstutz U , Froehlich TK , Largiadèr CR . Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics. 2011;12(9):1321–36. doi:.https://doi.org/10.2217/pgs.11.72
7 Petrelli F , Cabiddu M , Barni S . 5-Fluorouracil or capecitabine in the treatment of advanced colorectal cancer: a pooled-analysis of randomized trials. Med Oncol. 2012;29(2):1020–9. doi:.https://doi.org/10.1007/s12032-011-9958-0
8 Primrose JN , Fox RP , Palmer DH , Malik HZ , Prasad R , Mirza D , et al.; BILCAP study group. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019;20(5):663–73. doi:.https://doi.org/10.1016/S1470-2045(18)30915-X
9 Barin-Le Guellec C , Lafay-Chebassier C , Ingrand I , Tournamille JF , Boudet A , Lanoue MC , et al. Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: A real-life evaluation in France. Eur J Cancer. 2020;124:37–46. doi:.https://doi.org/10.1016/j.ejca.2019.09.028
10 Gusella M , Crepaldi G , Barile C , Bononi A , Menon D , Toso S , et al. Pharmacokinetic and demographic markers of 5-fluorouracil toxicity in 181 patients on adjuvant therapy for colorectal cancer. Ann Oncol. 2006;17(11):1656–60. doi:.https://doi.org/10.1093/annonc/mdl284
11 Di Paolo A , Danesi R , Falcone A , Cionini L , Vannozzi F , Masi G , et al. Relationship between 5-fluorouracil disposition, toxicity and dihydropyrimidine dehydrogenase activity in cancer patients. Ann Oncol. 2001;12(9):1301–6. doi:.https://doi.org/10.1023/A:1012294617392
12 Etienne MC , Milano G , Renée N , Lagrange JL , Dassonville O , Thyss A , et al. [Population study of dihydropyrimidine dehydrogenase in cancer patients]. Bull Cancer. 1995;82(9):705–10. Article in French.
13 Baker SD , Verweij J , Rowinsky EK , Donehower RC , Schellens JH , Grochow LB , et al. Role of body surface area in dosing of investigational anticancer agents in adults, 1991-2001. J Natl Cancer Inst. 2002;94(24):1883–8. doi:.https://doi.org/10.1093/jnci/94.24.1883
14 Gamelin E , Boisdron-Celle M , Guérin-Meyer V , Delva R , Lortholary A , Genevieve F , et al. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: A potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol. 1999;17(4):1105–10. doi:.https://doi.org/10.1200/JCO.1999.17.4.1105
15 Gamelin E , Delva R , Jacob J , Merrouche Y , Raoul JL , Pezet D , et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(13):2099–105. doi:.https://doi.org/10.1200/JCO.2007.13.3934
16 Saam J , Critchfield GC , Hamilton SA , Roa BB , Wenstrup RJ , Kaldate RR . Body surface area-based dosing of 5-fluoruracil results in extensive interindividual variability in 5-fluorouracil exposure in colorectal cancer patients on FOLFOX regimens. Clin Colorectal Cancer. 2011;10(3):203–6. doi:.https://doi.org/10.1016/j.clcc.2011.03.015
17 Patel JN , O’Neil BH , Deal AM , Ibrahim JG , Sherrill GB , Olajide OA , et al. A community-based multicenter trial of pharmacokinetically guided 5-fluorouracil dosing for personalized colorectal cancer therapy. Oncologist. 2014;19(9):959–65. doi:.https://doi.org/10.1634/theoncologist.2014-0132
18 Capitain O , Asevoaia A , Boisdron-Celle M , Poirier AL , Morel A , Gamelin E . Individual fluorouracil dose adjustment in FOLFOX based on pharmacokinetic follow-up compared with conventional body-area-surface dosing: a phase II, proof-of-concept study. Clin Colorectal Cancer. 2012;11(4):263–7. doi:.https://doi.org/10.1016/j.clcc.2012.05.004
19 Saif MW , Choma A , Salamone SJ , Chu E . Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes. J Natl Cancer Inst. 2009;101(22):1543–52. doi:.https://doi.org/10.1093/jnci/djp328
20 Kline CLB , Schiccitano A , Zhu J , Beachler C , Sheikh H , Harvey HA , et al. Personalized dosing via pharmacokinetic monitoring of 5-fluorouracil might reduce toxicity in early- or late-stage colorectal cancer patients treated with infusional 5-fluorouracil-based chemotherapy regimens. Clin Colorectal Cancer. 2014;13(2):119–26. doi:.https://doi.org/10.1016/j.clcc.2013.11.001
21 Beumer JH , Chu E , Allegra C , Tanigawara Y , Milano G , Diasio R , et al. Therapeutic Drug Monitoring in Oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology Recommendations for 5-Fluorouracil Therapy. Clin Pharmacol Ther. 2019;105(3):598–613.
22 Meulendijks D , Henricks LM , Sonke GS , Deenen MJ , Froehlich TK , Amstutz U , et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(16):1639–50. doi:.https://doi.org/10.1016/S1470-2045(15)00286-7
23 Deenen MJ , Meulendijks D , Cats A , Sechterberger MK , Severens JL , Boot H , et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: A safety and cost analysis. J Clin Oncol. 2016;34(3):227–34. doi:.https://doi.org/10.1200/JCO.2015.63.1325
24Analysenliste (AL). Available at: https://www.bag.admin.ch/bag/de/home/versicherungen/krankenversicherung/krankenversicherung-leistungen-tarife/Analysenliste.html [accessed 2020 April 9].
25 Büchel B , Sistonen J , Joerger M , Aebi Y , Schürch S , Largiadèr CR . Comparative evaluation of the My5-FU™ immunoassay and LC-MS/MS in monitoring the 5-fluorouracil plasma levels in cancer patients. Clin Chem Lab Med. 2013;51(8):1681–8. doi:.https://doi.org/10.1515/cclm-2012-0641
26 Büchel B , Rhyn P , Schürch S , Bühr C , Amstutz U , Largiadèr CR . LC-MS/MS method for simultaneous analysis of uracil, 5,6-dihydrouracil, 5-fluorouracil and 5-fluoro-5,6-dihydrouracil in human plasma for therapeutic drug monitoring and toxicity prediction in cancer patients. Biomed Chromatogr. 2013;27(1):7–16. doi:.https://doi.org/10.1002/bmc.2741
27Fluorouracil and fluorouracil related substances (capecitabine, tegafur and flucytosine) containing medicinal products. European Medicines Agency. Available at: https://www.ema.europa.eu/en/medicines/human/referrals/fluorouracil-fluorouracil-related-substances-capecitabine-tegafur-flucytosine-containing-medicinal [accessed 2020 May18].
28Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie. Positionspapier: Dihydropyrimidin-Dehydrogenase (DPD) -Testung vor Einsatz von 5-Fluorouracil, Capecitabin und Tegafu. 2020.
29 Lunenburg CATC , van der Wouden CH , Nijenhuis M , Crommentuijn-van Rhenen MH , de Boer-Veger NJ , Buunk AM ,, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines. Eur J Hum Genet. 2020;28(4):508–17. doi:.https://doi.org/10.1038/s41431-019-0540-0
30 Martens FK , Huntjens DW , Rigter T , Bartels M , Bet PM , Cornel MC . DPD Testing Before Treatment With Fluoropyrimidines in the Amsterdam UMCs: An Evaluation of Current Pharmacogenetic Practice. Front Pharmacol. 2020;10:1609. doi:.https://doi.org/10.3389/fphar.2019.01609
31Agence nationale de sécurité du médicament et des produits de santé (ANSM). Prévention des effets indésirables graves liés à un déficit en dihydropyrimidine déshydrogénase (DPD) lors de traitement par fluoropyrimidines (5-fluorouracile et capécitabine) - Point d’information actualisé au 28 février 2018. Available at: https://ansm.sante.fr/S-informer/Points-d-information-Points-d-information/Prevention-des-effets-indesirables-graves-lies-a-un-deficit-en-dihydropyrimidine-deshydrogenase-DPD-lors-de-traitement-par-fluoropyrimidines-5-fluorouracile-et-capecitabine-Point-d-information-actualise-au-28-fevrier-2018 [accessed 2020 April 19].
32 Hamzic S , Kummer D , Froehlich TK , Joerger M , Aebi S , Palles C , et al. Evaluating the role of ENOSF1 and TYMS variants as predictors in fluoropyrimidine-related toxicities: An IPD meta-analysis. Pharmacol Res. 2020;152:104594. doi:.https://doi.org/10.1016/j.phrs.2019.104594
33 Nie Q , Shrestha S , Tapper EE , Trogstad-Isaacson CS , Bouchonville KJ , Lee AM , et al. Quantitative Contribution of rs75017182 to Dihydropyrimidine Dehydrogenase mRNA Splicing and Enzyme Activity. Clin Pharmacol Ther. 2017;102(4):662–70. doi:.https://doi.org/10.1002/cpt.685
34 Henricks LM , Siemerink EJM , Rosing H , Meijer J , Goorden SMI , Polstra AM , et al. Capecitabine-based treatment of a patient with a novel DPYD genotype and complete dihydropyrimidine dehydrogenase deficiency. Int J Cancer. 2018;142(2):424–30. doi:.https://doi.org/10.1002/ijc.31065
35 Etienne-Grimaldi M-C , Cozic N , Boyer J-C , Boige V , Diasio RB , Taieb J , et al. FUSAFE individual patient data meta-analysis (MA) to assess the performance of dihydropyrimidine dehydrogenase (DPD) gene polymorphisms for predicting grade 4-5 fluoropyrimidine (FP) toxicity. Ann Oncol. 2019;30:v214. doi:.https://doi.org/10.1093/annonc/mdz246.046
36 Henricks LM , Lunenburg CATC , de Man FM , Meulendijks D , Frederix GWJ , Kienhuis E , et al. A cost analysis of upfront DPYD genotype-guided dose individualisation in fluoropyrimidine-based anticancer therapy. Eur J Cancer. 2019;107:60–7. doi:.https://doi.org/10.1016/j.ejca.2018.11.010
37 Henricks LM , van Merendonk LN , Meulendijks D , Deenen MJ , Beijnen JH , de Boer A , et al. Effectiveness and safety of reduced-dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: A matched pair analysis. Int J Cancer. 2019;144(9):2347–54. doi:.https://doi.org/10.1002/ijc.32022
38CPIC® Guideline for Fluoropyrimidines and DPYD – CPIC. Available at: https://cpicpgx.org/guidelines/guideline-for-fluoropyrimidines-and-dpyd/ [accessed 2020 September 9].
39 Offer SM , Lee AM , Mattison LK , Fossum C , Wegner NJ , Diasio RB . A DPYD variant (Y186C) in individuals of african ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther. 2013;94(1):158–66. doi:.https://doi.org/10.1038/clpt.2013.69
40 Etienne-Grimaldi M-C , Boyer JC , Beroud C , Mbatchi L , van Kuilenburg A , Bobin-Dubigeon C , et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One. 2017;12(5):e0175998. doi:.https://doi.org/10.1371/journal.pone.0175998
41 Hamzic S , Amstutz U , Largiadèr CR . Come a long way, still a ways to go: from predicting and preventing fluoropyrimidine toxicity to increased efficacy? Pharmacogenomics. 2018;19(8):689–92. doi:.https://doi.org/10.2217/pgs-2018-0040
42 Meulendijks D , Cats A , Beijnen JH , Schellens JHM . Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity - Ready for clinical practice? Cancer Treat Rev. 2016;50:23–34. doi:.https://doi.org/10.1016/j.ctrv.2016.08.002
43 de Gramont A , Krulik M , Cady J , Lagadec B , Maisani JE , Loiseau JP , et al. High-dose folinic acid and 5-fluorouracil bolus and continuous infusion in advanced colorectal cancer. Eur J Cancer Clin Oncol. 1988;24(9):1499–503. doi:.https://doi.org/10.1016/0277-5379(88)90341-0
44 Miwa M , Ura M , Nishida M , Sawada N , Ishikawa T , Mori K , et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34(8):1274–81. doi:.https://doi.org/10.1016/S0959-8049(98)00058-6
45 Salman D , Biliune J , Kayyali R , Ashton J , Brown P , McCarthy T , et al. Evaluation of the performance of elastomeric pumps in practice: are we under-delivering on chemotherapy treatments? Curr Med Res Opin. 2017;33(12):2153–9. doi:.https://doi.org/10.1080/03007995.2017.1374936
46 Hobbs JG , Ryan MK , Mohtar A , Sluggett AJ , Sluggett JK , Ritchie B , et al. Flow rate accuracy of ambulatory elastomeric and electronic infusion pumps when exposed to height and back pressures experienced during home infusion therapy. Expert Rev Med Devices. 2019;16(8):735–42. doi:.https://doi.org/10.1080/17434440.2019.1632187
47Which Ambulatory Infusion Pump Is Best for 5-FU? ONS Voice 2019. Available at: https://voice.ons.org/news-and-views/which-ambulatory-infusion-pump-is-best-for-5-fu [accessed 2020 April 19].
48FDA. Infusion Pump Improvement Initiative. Available at: https://www.fda.gov/medical-devices/infusion-pumps/white-paper-infusion-pump-improvement-initiative [accessed 2020 May1].
49 Kaldate RR , Haregewoin A , Grier CE , Hamilton SA , McLeod HL . Modeling the 5-fluorouracil area under the curve versus dose relationship to develop a pharmacokinetic dosing algorithm for colorectal cancer patients receiving FOLFOX6. Oncologist. 2012;17(3):296–302. doi:.https://doi.org/10.1634/theoncologist.2011-0357
50 Henricks LM , Kienhuis E , de Man FM , van der Veldt AAM , Hamberg P , van Kuilenburg ABP , et al. Treatment Algorithm for Homozygous or Compound Heterozygous DPYD Variant Allele Carriers With Low-Dose Capecitabine. JCO Precis Oncol. 2017. doi:.https://doi.org/10.1200/po.17.00118
51 Blasco H , Boisdron-Celle M , Bougnoux P , Calais G , Tournamille JF , Ciccolini J , et al. A well-tolerated 5-FU-based treatment subsequent to severe capecitabine-induced toxicity in a DPD-deficient patient. Br J Clin Pharmacol. 2008;65(6):966–70. doi:.https://doi.org/10.1111/j.1365-2125.2008.03106.x
The authors declare no potential conflicts of interest.