Efficacy and safety outcomes from the MATISSE phase 3 trial of maternal bivalent RSVpreF
vaccination among pregnant women vaccinated at 32 to 36 weeks of gestation
DOI: https://doi.org/https://doi.org/10.57187/4711
Emma Shittua,
Barbara A. Pahudb,
David Radleyb,
Alejandra Gurtmanb,
Iona Munjalb
a Vaccine Research and Development,
Pfizer Ltd, Marlow, UK
b Vaccine Research and Development, Pfizer
Inc., Pearl River, NY, USA
Summary
AIMS OF THE STUDY: Marketing authorisation of the bivalent
respiratory syncytial virus prefusion F protein-based vaccine (RSVpreF) for maternal
vaccination was predominantly based on safety and efficacy findings from the pivotal
global phase 3 Maternal Immunization Study for Safety and Efficacy (MATISSE) trial
that included >7000 pregnant women and their infants. The aim of this post
hoc analysis was to evaluate clinical efficacy and safety endpoints within the subgroup
of participants from the MATISSE trial who received RSVpreF or placebo at 32−36 weeks
of gestation, which
is the indicated gestational age (GA) window for maternal RSVpreF vaccination in
Switzerland.
METHODS: Healthy pregnant women ≤49 years of age with uncomplicated,
singleton pregnancies were randomised 1:1 to receive a single dose of RSVpreF 120
μg or
placebo. Primary efficacy endpoints were vaccine efficacy against severe medically
attended RSV-associated lower respiratory tract illness and medically attended RSV-associated
lower respiratory tract
illness in infants occurring within 90 and 180 days after birth. This
is a descriptive post hoc analysis of efficacy and safety endpoints from MATISSE
among the subgroup of newborns whose mothers received RSVpreF or placebo at 32–36
weeks of gestation.
RESULTS: Of 7392 maternal participants included in the
primary MATISSE analysis, 3285 (RSVpreF: 1653; placebo: 1632) received vaccination
at a GA of 32–36 weeks and were included in this analysis. Efficacy was evaluated
in a total of 1628 and 1604 infants who were born to mothers
receiving RSVpreF or placebo, respectively. RSVpreF vaccine efficacy percentages against
severe medically attended RSV-associated lower respiratory tract illness were 91.1%
(95% CI: 38.8–99.8%) and 76.5% (95%
CI: 41.3–92.1%) within 90 and 180 days of birth, respectively. RSVpreF vaccine efficacy
percentages
against medically attended RSV-associated lower respiratory tract illness were 34.7%
(95% CI: –34.6–69.3%) and 57.3% (95%
CI: 29.8–74.7%) within 90 and 180 days of birth, respectively. Adverse event profiles
for maternal and infant participants were generally similar between RSVpreF and
placebo groups in this post hoc analysis; safety results were consistent with those
of the primary and final analyses.
CONCLUSIONS: Maternal vaccination with RSVpreF in pregnant women at 32−36 weeks of
gestation
is safe and efficacious against RSV-associated lower respiratory tract illness in
infants to 6 months of age,
aligning with the outcomes of the primary analysis.
ClinicalTrials.gov: NCT04424316
Introduction
Illness caused by respiratory syncytial virus (RSV) infection is an important cause
of infant morbidity and mortality worldwide, particularly in the first 6 months of
life and outside high-income regions [1]. Although >95% of episodes of RSV-associated
lower respiratory tract illness globally in 2019 occurred in low- and middle-income
countries, the hospital admission rate for RSV-associated lower respiratory tract
illness was high and comparable in lower-middle-, upper-middle- and high-income countries,
particularly for infants ≤3 months of age [1]. The age of onset of RSV and the potential
seriousness of related complications demonstrate the importance of maternal vaccination
against RSV.
Maternal vaccination with bivalent RSV prefusion F protein-based vaccine (RSVpreF;
Abrysvo®; Pfizer AG, Zürich, Switzerland) is a licenced indication in several countries for
passive protection against RSV-associated lower respiratory tract illness in infants
from birth to 6 months of age. The approval of RSVpreF for maternal vaccination was
predominantly based on the findings from the pivotal global phase 3 trial Maternal
Immunization Study for Safety and Efficacy (MATISSE) that included more than 7000
pregnant women and their infants [2]. Pregnant individuals were randomised to receive
RSVpreF (120 µg; 60 µg each of RSV-A and RSV-B antigens, which are the 2 cocirculating
strains causing disease) or placebo at 24−36 weeks of gestation. At the primary analysis,
RSVpreF was efficacious in preventing severe medically attended (i.e. infant participant was taken to or seen by a healthcare provider, which could
include outpatient, inpatient, emergency department, urgent care, or home visits)
RSV-associated lower respiratory tract illness within 90 days (vaccine efficacy: 81.8%)
and 180 days after birth (vaccine efficacy: 69.4%). RSVpreF had a favourable safety
profile both in pregnant women and their infants.
Licenced indications and recommendations for gestational age (GA) at administration
of maternal RSVpreF vaccination vary by country (i.e. 24‒36, 28‒36 or 32‒36 weeks)
[3–5]. Concern regarding preterm births occurring during the study, for which available
data from MATISSE were insufficient to exclude causal links with RSVpreF, was a consideration
for some regulatory and guideline committees in putting forth these recommendations
[3]. RSVpreF received marketing authorisation from Swissmedic on 23 August 2024, for
passive protection against RSV-associated lower respiratory tract illness in infants
from birth to 6 months of age after immunisation of pregnant women at 32−36 weeks
of gestation [6]. In Switzerland, it is recommended that pregnant women aged 18 years
or over be offered RSVpreF between 32 and 36 weeks of gestation from October to February
(i.e. the RSV season in Switzerland) if the expected delivery date is before the end
of March [7].
Because the indicated GA window for maternal RSVpreF vaccination in Switzerland is
shorter compared with that used in the pivotal MATISSE trial, a post hoc analysis
of clinical efficacy endpoints was conducted within the subgroup of participants from
MATISSE who received RSVpreF or placebo at 32−36 weeks of gestation (and at least
14 days before delivery) to demonstrate the clinical efficacy of RSVpreF in this subset
of pregnant women.
Materials and methods
The design of MATISSE (ClinicalTrials.gov Identifier: NCT04424316) has been described
previously [2]. Briefly, it was a double-blind, randomised, phase 3 trial conducted
in both the Northern Hemisphere (i.e. Canada, Denmark, Finland, the Gambia, Japan,
Republic of Korea, Mexico, the Netherlands, Spain, Taiwan and the United States) and
the Southern Hemisphere (i.e. Argentina, Australia, Brazil, Chile, New Zealand, the
Philippines and South Africa) over 4 RSV seasons (i.e. 2 seasons in each hemisphere).
Healthy pregnant women ≤49 years of age with uncomplicated, singleton pregnancies
were randomly assigned to receive a single dose of RSVpreF 120 μg or placebo in a
1:1 ratio. Inclusion/exclusion criteria are provided in the Supplementary Materials.
Before enrolment, all participants provided written informed consent for themselves
and their newborns for inclusion in the study and each site’s ethics committee approved
the study protocol. Study conduct complied with global ethical principles for medical
research involving human participants, good clinical practice guidelines and all applicable
laws and regulations. The study was conducted according to the guidelines of the Declaration
of Helsinki and approved by multiple institutional review boards, including the Central
Western Institutional Review Board (Puyallup, WA, USA) on 26 June 2020 (Protocol C3671008).
The two primary efficacy endpoints were vaccine efficacy against severe medically
attended and medically attended RSV-associated lower respiratory tract illness in
infants occurring within 90 and 180 days after birth. Secondary efficacy endpoints
included vaccine efficacy against medically attended RSV-associated lower respiratory
tract illness, RSV-associated hospitalisations and all-cause medically attended lower
respiratory tract illness in infants within 360 days after birth. Aligned with recommended
timeline thresholds for evaluation of the safety of vaccines used during pregnancy
[8], safety endpoints in maternal participants and their infants included adverse
events to 1 month after vaccination or birth, respectively, and serious adverse events
to 6 months after delivery (maternal participants) or 12 or 24 months after birth
(infant participants, depending on time of enrolment). Specific birth outcomes were
also collected for infant participants. Reported here is a descriptive post hoc analysis
of efficacy and safety endpoints from MATISSE among the subgroup of newborns whose
mothers received RSVpreF or placebo at 32–36 weeks of gestation.
The evaluation of RSVpreF efficacy in infants via maternal vaccination as per the
recommended gold standard [9], commenced with the surveillance of respiratory tract
illness in infants 72 hours after birth to 1 year (or 2 years for infants enrolled
in the first year of the trial) and visits were documented for all medically attended
respiratory tract illness to 6 months as described previously [2]. Vaccine efficacy
was estimated from the number of total disease cases and the number of disease cases
in the RSVpreF group to the placebo group. Vaccine efficacy was calculated as (1–RR)
× 100, with RR being the relative risk for the specific efficacy endpoint based on
the incidence in the RSVpreF group compared with that in the placebo group, where
RR = hP/(1–P); P is the number of cases in the RSVpreF group divided by the total
number of cases and h is the ratio of the number of participants at risk in the placebo
group to the number of participants at risk in the RSVpreF group. Vaccine efficacy
and associated 2-sided 95% CIs were estimated using the conditional exact method [10].
As a post hoc analysis, no formal statistical efficacy hypotheses were defined and
no adjustments for multiplicity were made. Safety data are presented descriptively
based on the final MATISSE analysis.
Results
Participants
The MATISSE trial was initiated in June 2020,
and the cutoff date for these post hoc infant efficacy analyses was 30 September
2022. Of the 7392 maternal participants included in the primary analysis of the
MATISSE trial, 3285 (RSVpreF: 1653; placebo: 1632) received vaccination at a GA
of 32–36 weeks and were included in this analysis. Efficacy was evaluated in a total
of 1628 and 1604 infants
who were born to mothers receiving RSVpreF or placebo, respectively. Reasons
infants born to maternal participants were not included in the efficacy analysis
included withdrawal before delivery (9 [0.3%]), continuing in the study but not
yet delivered (36 [1.1%]) and infants not enrolled (8 [0.2%]).
Demographic characteristics were similar across
the trial groups (table 1). Among maternal participants, 70% were White, 14% Black
and 12% Asian; 32% were Hispanic or Latina. Median age at vaccination was 30 years
(range: 16 to 47 years) and median GA was 34.1 weeks (range: 32.0–36.0 weeks). Among
infant participants, 51% were male (table 2).
Table 1Demographic characteristics of the
maternal participants in the post hoc analysis population.
| Characteristic |
RSVpreF (n = 1653*) |
Placebo (n = 1632*) |
| Female, n (%) |
1653 (100.0%) |
1632 (100.0%) |
| Age at vaccination, years |
Mean (SD) |
29.8 (5.56) |
29.7 (5.64) |
| Median (range) |
30.0 (16–45) |
30.0 (16–47) |
| Gestational age at vaccination,
weeks |
Mean (SD) |
34.1 (1.22) |
34.1 (1.22) |
| Median (range) |
34.1 (32.0–36.0) |
34.1 (32.0–36.0) |
| Race, n (%) |
White |
1155 (69.9%) |
1157 (70.9%) |
| Black |
240 (14.5%) |
228 (14.0%) |
| Asian |
207 (12.5%) |
195 (11.9%) |
| American Indian or
Alaska Native |
13 (0.8%) |
15 (0.9%) |
| Native Hawaiian or other Pacific
Islander |
7 (0.4%) |
6 (0.4%) |
| Multiracial |
9 (0.5%) |
10 (0.6%) |
| Not reported |
19 (1.1%) |
14 (0.9%) |
| Unknown |
3 (0.2%) |
7 (0.4%) |
| Ethnicity, n (%) |
Hispanic/Latina |
520 (31.5%) |
541 (33.1%) |
| Non-Hispanic/non-Latina |
1113 (67.3%) |
1076 (65.9%) |
| Not reported or unknown |
20 (1.2%) |
15 (0.9%) |
Table 2Demographic characteristics and
birth outcomes of the infant participants in the post hoc analysis population.
| Characteristic or birth
outcome |
RSVpreF (n = 1628*) |
Placebo (n = 1604*) |
| Sex, n (%) |
Male |
838 (51.5%) |
793 (49.4%) |
| Female |
790 (48.5%) |
811 (50.6%) |
| Race, n (%) |
White |
1116 (68.6%) |
1121 (69.9%) |
| Black |
234 (14.4%) |
222 (13.8%) |
| Asian |
196 (12.0%) |
186 (11.6%) |
| American Indian or
Alaskan Native |
18 (1.1%) |
16 (1.0%) |
| Native Hawaiian or other
Pacific Islander |
10 (0.6%) |
7 (0.4%) |
| Multiracial |
35 (2.1%) |
35 (2.2%) |
| Not reported |
13 (0.8%) |
13 (0.8%) |
| Unknown |
6 (0.4%) |
4 (0.2%) |
| Ethnicity, n (%) |
Hispanic/Latino |
524 (32.2%) |
523 (32.6%) |
| Non-Hispanic/non-Latino |
1087 (66.8%) |
1061 (66.1%) |
| Not reported or unknown |
17 (1.0%) |
20 (1.2%) |
| Gestational age at
birth, n (%) |
28 weeks to <34
weeks |
2 (0.1%) |
2 (0.1%) |
| 34 weeks to <37
weeks |
66 (4.1%) |
57 (3.6%) |
| 37 weeks to <42
weeks |
1547 (95.0%) |
1530 (95.4%) |
| ≥42 weeks |
13 (0.8%) |
14 (0.9%) |
| Apgar score at 5
minutes, n (%) |
n |
1620 |
1592 |
| <4 |
6 (0.4%) |
3 (0.2%) |
| 4 to <7 |
15 (0.9%) |
14 (0.9%) |
| 7 to 10 |
1599 (98.7%) |
1575 (98.9%) |
| Median (range) |
9.0 (1–10) |
9.0 (2–10) |
| Birth outcome, n (%) |
Normal |
1455 (89.4%) |
1426 (88.9%) |
| Congenital
malformation/anomaly |
85 (5.2%) |
93 (5.8%) |
| Other neonatal problem |
88 (5.4%) |
85 (5.3%) |
| Very low birthweight
(>1000 g to 1500 g) |
0 |
1 (<0.1%) |
| Low birthweight
(>1500 g to 2500 g) |
67 (4.1%) |
54 (3.4%) |
| Developmental delay** |
8 (0.5%) |
7 (0.4%) |
Efficacy
Based on data for the primary analysis (data
cutoff for infant efficacy: 30 September 2022), 12 cases of severe medically attended
RSV-associated lower respiratory tract illness accrued within 90 days of birth (RSVpreF:
1 [<0.1%]; placebo: 11 [0.7%])
and 31 cases within 180 days of birth (RSVpreF: 6 [0.4%]; placebo: 25 [1.6%]; figure
1A). RSVpreF vaccine efficacy against severe medically attended RSV-associated lower
respiratory tract illness was 91.1% (95% CI: 38.8–99.8%)
and 76.5% (95% CI: 41.3–92.1%) within 90 and 180 days of birth, respectively. Within
90 days of birth, 35 cases of medically attended RSV-associated lower respiratory
tract illness had accrued (RSVpreF:
14 [0.9%]; placebo: 21 [1.4%]), with 79 cases accruing within 180 days of birth
(RSVpreF: 24 [1.5%]; placebo: 55 [3.6%]; figure 1B). RSVpreF vaccine efficacy against
medically
attended RSV-associated lower respiratory tract illness was 34.7% (95% CI: –34.6–69.3%)
and 57.3% (95% CI: 29.8–74.7%)
within 90 and 180 days of birth, respectively.
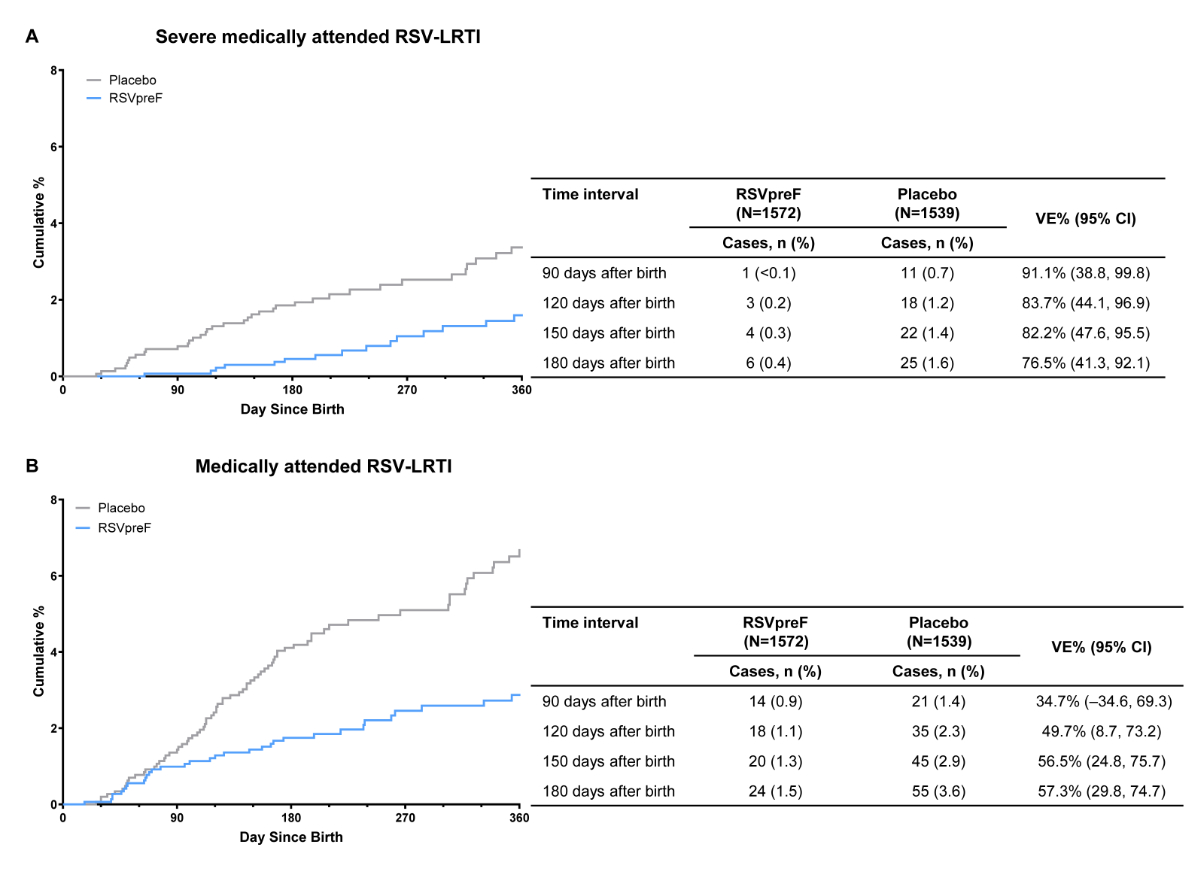
Figure 1Vaccine efficacy for (A) severe medically
attended RSV-associated lower respiratory tract illness and (B) medically attended
RSV-associated lower respiratory tract illness within 180 days after
birth in the efficacy-evaluable population. An endpoint adjudication committee
confirmed all cases. Vaccine efficacy was calculated as 1–(hP/[1–P]) and expressed
as a
percentage, where P is the number of cases in the RSVpreF group divided by the
total number of cases and h is the ratio of number of participants at risk in
the placebo group to the number of participants at risk in the RSVpreF group.
Severe medically attended RSV-associated lower respiratory tract illness included
infants with a medically attended
visit for a respiratory tract illness and an RSV-positive test result, and
either very fast breathing (RR ≥70 for <2 months of age [<60 days of
age], RR ≥60 for 2–<12 months of age or RR ≥50 for 12–24 months of age),
SpO2 <93%, high-flow nasal cannula or mechanical ventilation, intensive care
unit admission for >4 hours or failure to respond/unconsciousness. Medically
attended RSV-associated lower respiratory tract illness included infants with a medically
attended visit for a
respiratory tract illness and an RSV-positive RT-PCR or NAAT result, and either
fast breathing (RR ≥60 for <2 months of age [<60 days of age], RR ≥50 for
2–<12 months of age or RR ≥40 for 12–24 months of age), SpO2 <95% or
chest wall indrawing. NAAT: nucleic acid amplification
test; RR: respiratory rate; RSV-LRTI: lower respiratory tract illness
associated with respiratory syncytial virus; RSVpreF: respiratory syncytial
virus prefusion F protein−based vaccine; RT‑PCR: reverse
transcriptase-polymerase chain reaction; SpO2: oxygen saturation; VE: vaccine efficacy.
The RSVpreF vaccine efficacy against medically attended RSV-associated lower respiratory
tract illness
to 360 days after birth is presented in table 3; RSVpreF vaccine efficacy for medically
attended
RSV-associated lower respiratory tract illness occurring to 210 and 360 days after
birth was 59.9% (95% CI: 35.1–75.9%)
and 56.9% (95% CI: 34.3–72.3%), respectively.
Table 3Vaccine efficacy against medically
attended RSV-associated lower respiratory tract illness within 210 to 360 days after
birth for the evaluable efficacy
population.
| Time interval |
RSVpreF (n = 1572*), number
of cases (%) |
Placebo (n = 1539*), number
of cases (%) |
VE, % (95% CI)** |
| 210 days after birth |
25 (1.6%) |
61 (4.0%) |
59.9%
(35.1–75.9%) |
| 240 days after birth |
28 (1.8%) |
62 (4.0%) |
55.8%
(29.9–72.8%) |
| 270 days after birth |
30 (1.9%) |
64 (4.2%) |
54.1%
(28.1–71.3%) |
| 360 days after birth |
33 (2.1%) |
75 (4.9%) |
56.9%
(34.3–72.3%) |
Within 90 days of birth, 16 (RSVpreF: 4 [0.3%];
placebo: 12 [0.8%]) hospitalisations due to RSV occurred, with 26 (RSVpreF: 9 [0.6%];
placebo: 17 [1.1%]) hospitalisations occurring within 180 days of birth (table 4).
The RSVpreF vaccine efficacy against RSV-associated hospitalisations was 67.4% (95%
CI: –7.7–92.3%)
and 48.2% (–22.9–79.6%) within 90 and 180 days of birth, respectively.
Table 4Vaccine efficacy against hospitalisations
due to RSV occurring within 90 to 360 days after birth in the evaluable
efficacy population.
| Time interval |
RSVpreF (n = 1572*), number of cases (%) |
Placebo (n = 1539*), number of cases (%) |
VE, % (95% CI)** |
| 90 days after birth |
4 (0.3%) |
12 (0.8%) |
67.4%
(–7.7–92.3%) |
| 120 days after birth |
7 (0.4%) |
14 (0.9%) |
51.0%
(–29.6–83.3%) |
| 150 days after birth |
8 (0.5%) |
15 (1.0%) |
47.8%
(–31.2–80.8%) |
| 180 days after birth |
9 (0.6%) |
17 (1.1%) |
48.2%
(–22.9–79.6%) |
| 360 days after birth |
15 (1.0%) |
22 (1.4%) |
33.2%
(–34.6–67.8%) |
RSVpreF vaccination was not effective at preventing
all-cause medically attended lower respiratory tract illness; there were 142 cases
(RSVpreF: 70 [4.5%]; placebo:
72 [4.7%]) within 90 days of birth and 333 cases (RSVpreF: 162 [10.3%]; placebo:
171 [11.1%]) within 180 days of birth (table S1). The RSVpreF vaccine efficacy against
all-cause
medically attended lower respiratory tract illness occurring 0–90 and 0–180 days after
birth was 4.8% (95%
CI: –34.1–32.5%) and 7.3% (95% CI: –15.7–25.7%), respectively.
Safety
Safety analyses are based on data from the final
analysis (data cutoff for safety, 2 September 2022). Infant birth outcomes were
similar between RSVpreF and placebo groups (table 2). The adverse event profiles for
maternal
and infant participants were generally similar between RSVpreF and placebo groups.
Among maternal participants with a gestational age at vaccination of 32–36 weeks,
adverse events within 1 month after vaccination were reported by 18.0% of participants
in the
RSVpreF group and 16.9% in the placebo group (table 5). Corresponding rates for
infant participants within 1 month after birth were 38.0% and 34.7%, respectively.
The serious adverse events reported in >0.1% of maternal and infant participants are
shown in table
S3 and table S4; there were no notable differences between groups.
Table 5Adverse events by category within 1 month
after vaccination in maternal participants vaccinated at a gestational age of
32–36 weeks.
| Maternal participants |
RSVpreF (n = 1653) |
Placebo (n = 1632) |
| Any event, n (%) |
298 (18.0%) |
275 (16.9%) |
| Serious, n (%) |
107 (6.5%) |
98 (6.0%) |
| Immediate* |
0 |
0 |
| Severe, n (%) |
42 (2.5%) |
35 (2.1%) |
| Life-threatening, n (%) |
13 (0.8%) |
8 (0.5%) |
| Related, n (%) |
8 (0.5%) |
2 (0.1%) |
| Adverse
events of special interest, n (%) |
70 (4.2%) |
67 (4.1%) |
| Adverse events leading to withdrawal |
0 |
0 |
| Infant participants |
RSVpreF (n = 1628) |
Placebo (n = 1604) |
| Any event, n (%) |
618 (38.0%) |
557 (34.7%) |
| Serious, n (%) |
261 (16.0%) |
243 (15.1%) |
| Severe, n (%) |
67 (4.1%) |
55 (3.4%) |
| Life-threatening, n (%) |
17 (1.0%) |
17 (1.1%) |
| Related |
0 |
0 |
| Adverse
events of special interest, n (%) |
113 (6.9%) |
94 (5.9%) |
| Congenital anomalies, n (%) |
88 (5.4%) |
97 (6.0%) |
| Newly diagnosed chronic medical
conditions, n (%) |
4 (0.2%) |
1 (<0.1%) |
| Adverse events leading to withdrawal |
0 |
0 |
Discussion
This post hoc analysis of clinical efficacy for the subgroup of participants from
MATISSE who received RSVpreF or placebo at 32−36 weeks of gestation was conducted
in accordance with the indicated GA window for maternal RSVpreF vaccination in Switzerland.
In this descriptive post hoc analysis, vaccine efficacy against severe medically attended
RSV-associated lower respiratory tract illness within 90 days of birth was 91.1%,
which was consistent with the primary analysis (81.8%) that included participants
at 24−36 weeks of gestation [2]. Vaccine efficacy against severe medically attended
RSV-associated lower respiratory tract illness within 180 days of birth was also consistent
between this post hoc analysis and the primary analyses (76.5% and 69.4%, respectively)
[2]. For medically attended RSV-associated lower respiratory tract illness among infants,
vaccine efficacy percentages within 90 days of birth were 34.7% in this analysis and
57.1% in the primary analysis, while vaccine efficacy percentages within 180 days
of birth were 57.3% and 51.3%, respectively [2]. Vaccine efficacy against RSV-associated
hospitalisation within 90 days of birth was similar in the post hoc versus primary
analyses (67.4% and 67.7%, respectively) [2].
In the primary and final MATISSE analyses, RSVpreF was safe in maternal participants
and their infants with favourable adverse event profiles and pregnancy and birth outcomes
similar to those in the placebo group [2, 11]. Most local reactions and systemic events
in maternal participants were mild to moderate, and adverse event and serious adverse
event profiles were similar to those of placebo for maternal and infant participants
[2]. Additionally, a post hoc descriptive safety analysis found that maternal RSVpreF
vaccination was not associated with clinically significant increases in adverse events
of special interest, including preterm birth, low birthweight and neonatal hospitalisation
in the overall study population [12]. In this analysis of maternal participants from
MATISSE who received RSVpreF or placebo at 32−36 weeks of gestation, safety results
were consistent with those of the primary and final analyses.
Although immunogenicity was not assessed as part of the primary analysis, in the final
MATISSE analysis, RSVpreF elicited robust immune responses in maternal participants
and their infants compared with placebo regardless of GA at vaccination [11]. Among
infants of maternal participants in the RSVpreF group, RSV-A/RSV-B combined titres
at birth were well above the 50% RSV-neutralising titres of a serum palivizumab threshold
of 100 mg/ml (which is estimated to provide ≥97.5% protection from paediatric intensive
care unit admission for newborns and infants at high risk of severe RSV disease attributed
to RSV-A/RSV-B) [11].
Study limitations noted previously for the MATISSE trial include the exclusion of
individuals who had high-risk pregnancies and limited data from low-income countries
where RSVpreF may be most impactful in preventing severe disease and mortality in
infants [2]. Therefore, the findings reported herein may not be generalisable to all
populations. Additionally, in this post hoc analysis, vaccine efficacy calculations
are descriptive in nature and therefore should be interpreted with caution. Nevertheless,
it is encouraging that vaccine efficacy in this subgroup is consistent with that in
the primary analysis. It will be important to monitor real-world data after uptake
and implementation in Switzerland and any other countries, including those in Europe,
licencing RSVpreF for pregnant women to underscore these findings and enable ongoing
assessment of RSVpreF effectiveness.
In conclusion, this post hoc analysis of MATISSE trial data demonstrates that maternal
vaccination with RSVpreF in pregnant women at 32−36 weeks of gestation is safe and
efficacious against RSV-associated lower respiratory tract illness in infants up to
6 months of age, supporting the conclusions of the primary analysis.
Data sharing statement
Upon request, and subject to review, Pfizer
will provide the data that support the findings of this study. Subject to
certain criteria, conditions and exceptions, Pfizer may also provide access to
the related individual deidentified participant data. See
https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more
information.
Acknowledgments
We thank all MATISSE study sites and investigators
and all the participants and their infants who contributed to this trial. Medical
writing support was provided by Sheena Hunt, PhD, and Tricia Newell, PhD, of ICON
(Blue Bell, PA, USA) and was funded by Pfizer Inc.
Author contributions: All authors made substantial
contributions to the conception or design of the work, or acquisition, analysis
or interpretation of data for the work. All authors revised the work critically
for important intellectual content and approved the version to be published. Authors
agree to be accountable for all aspects of the work in ensuring that questions related
to the accuracy or integrity of any part of the work are appropriately investigated
and resolved.
Iona
Munjal, MD
Vaccine
Research and Development, Pfizer Inc.
Pearl
River, NY, USA
Iona.Munjal[at]pfizer.com
References
1. Li Y, Wang X, Blau DM, Caballero MT, Feikin DR, Gill CJ, et al.; Respiratory Virus
Global Epidemiology Network; RESCEU investigators. Global, regional, and national
disease burden estimates of acute lower respiratory infections due to respiratory
syncytial virus in children younger than 5 years in 2019: a systematic analysis. Lancet.
2022 May;399(10340):2047–64. doi: https://doi.org/10.1016/S0140-6736(22)00478-0
2. Kampmann B, Madhi SA, Munjal I, Simões EA, Pahud BA, Llapur C, et al.; MATISSE Study
Group. Bivalent prefusion F vaccine in pregnancy to prevent RSV illness in infants.
N Engl J Med. 2023 Apr;388(16):1451–64. doi: https://doi.org/10.1056/NEJMoa2216480
3. ABRYSVO (RSVpreF). Full Prescribing Information. New York (NY): Pfizer Inc; 2023.
4. ABRYSVO (RSVpreF). Summary of Product Characteristics. Bruxelles, Belgium: Pfizer
Europe MA EEIG; 2023.
5. Electronic Medicines Compendium. Package leaflet: Information for the user. Abrysvo®
powder and solvent for solution for injection. Available at: https://www.medicines.org.uk/emc/files/pil.15309.pdf. Accessed April 8, 2025.
6. Swiss Agency for Therapeutic Products. Swissmedic Journal 08/2024. Swissmedic. Available
at: https://www.swissmedic.ch/swissmedic/en/home/about-us/publications/swissmedic-journal/swissmedic-journal-2024.html. Accessed April 21, 2025.
7. Swiss Federal Office of Public Health. BAG-Bulletin 47/2024 (18 November 2024). Available
at: https://www.bag.admin.ch/dam/de/sd-web/g0ZwfShEIyXp/bu-47-24.pdf
8. Jones CE, Munoz FM, Spiegel HM, Heininger U, Zuber PL, Edwards KM, et al.; Brighton
Collaboration Immunization in Pregnancy Working Group. Guideline for collection, analysis
and presentation of safety data in clinical trials of vaccines in pregnant women.
Vaccine. 2016 Dec;34(49):5998–6006. doi: https://doi.org/10.1016/j.vaccine.2016.07.032
9. Roberts JN, Gruber MF. Regulatory considerations in the clinical development of vaccines
indicated for use during pregnancy. Vaccine. 2015 Feb;33(8):966–72. doi: https://doi.org/10.1016/j.vaccine.2014.12.068
10. Chan IS, Bohidar NR. Exact power and sample size for vaccine efficacy studies. Commun
Stat Theory Methods. 1998;27(6):1305–22. doi: https://doi.org/10.1080/03610929808832160
11. Simões EA, Pahud BA, Madhi SA, Kampmann B, Shittu E, Radley D, et al.; MATISSE (Maternal
Immunization Study for Safety and Efficacy) Clinical Trial Group. Efficacy, safety,
and immunogenicity of the MATISSE (Maternal Immunization Study for Safety and Efficacy)
maternal respiratory syncytial virus prefusion F protein vaccine trial. Obstet Gynecol.
2025 Feb;145(2):157–67. doi: https://doi.org/10.1097/AOG.0000000000005816
12. Madhi SA, Kampmann B, Simões EA, Zachariah P, Pahud BA, Radley D, et al. Preterm birth
frequency and associated outcomes from the MATISSE (Maternal Immunization Study for
Safety and Efficacy) maternal trial of the bivalent respiratory syncytial virus prefusion
F protein vaccine. Obstet Gynecol. 2025 Feb;145(2):147–56. doi: https://doi.org/10.1097/AOG.0000000000005817
Appendix
The appendix is available in the pdf version of the article at https://doi.org/10.57187/s.4711.