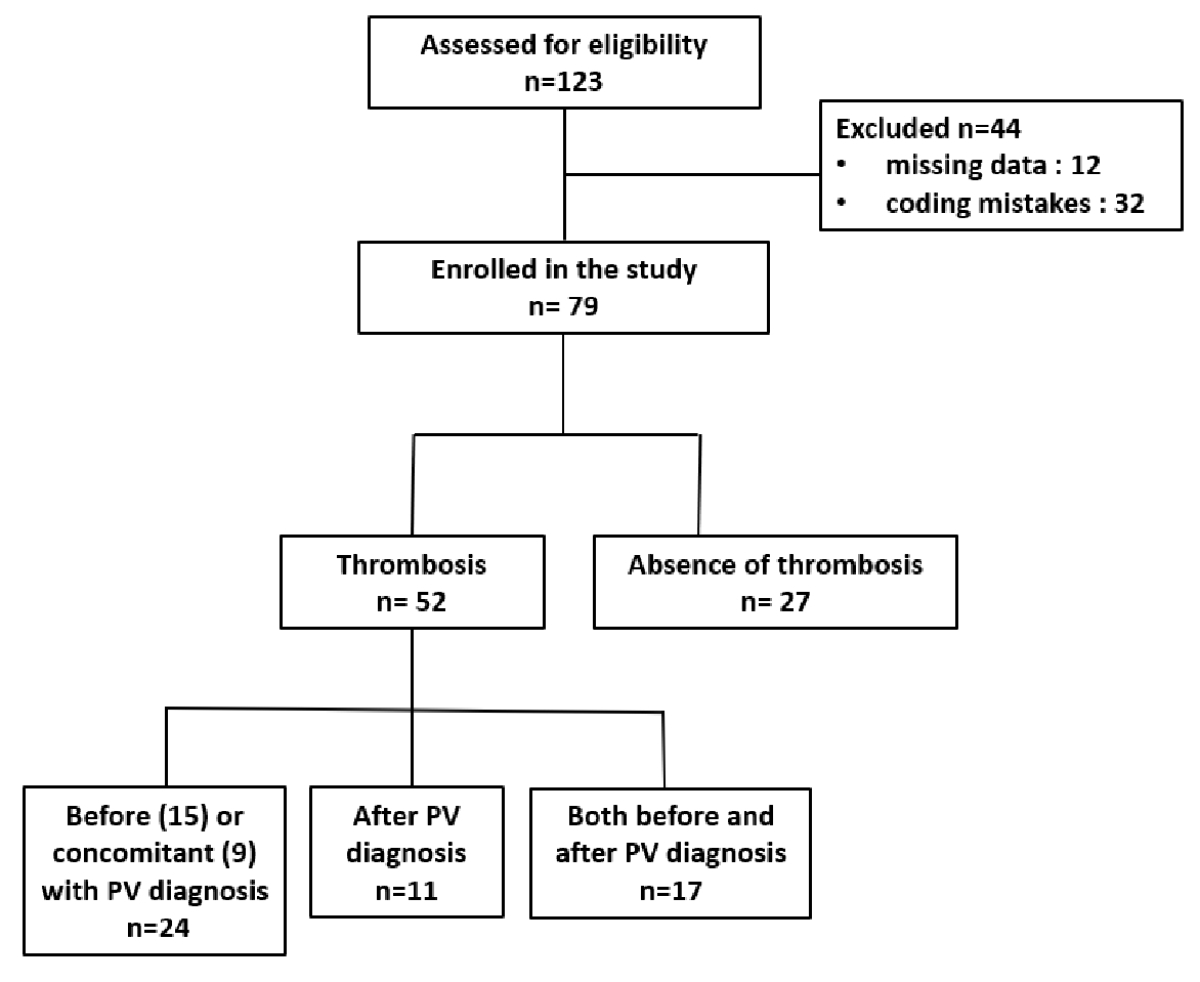
Figure 1Study cohort flow diagram. Flowchart illustrating patient identification, screening, eligibility assessment and inclusion in the final analysis set. Abbreviations: PV: polycythaemia vera.
DOI: https://doi.org/https://doi.org/10.57187/4669
Myeloproliferative neoplasms are recognised as one of several myeloid malignancies according to the 2022 World Health Organization (WHO) classification system and include three Philadelphia (Ph) chromosome-negative entities: polycythaemia vera, essential thrombocythaemia and primary myelofibrosis [1]. Polycythaemia vera is the most common myeloproliferative neoplasm and is characterised by stem cell-derived clonal erythrocytosis [2]. The estimated incidence of polycythaemia vera varies between 0.68 and 2.6 per 100,000 persons per year [3]. Generally, most patients with polycythaemia vera are discovered when an increased haemoglobin or haematocrit is found incidentally on a full blood count, which will prompt further investigations leading to the diagnosis of polycythaemia vera. Most patients with polycythaemia vera (96%) present a JAK2V617F (exon 14) mutation whereas 3% of patients harbour a JAK2 mutation in exon 12 [2]. Notably, JAK2V617F mutation also occurs in essential thrombocythaemia and primary myelofibrosis at a frequency of 55% and 65%, respectively [2]. In polycythaemia vera, a high allele burden of the JAK2V617F mutation does not appear to affect survival but increases the risk of thrombosis [2, 4] and is significantly associated with the risk of developing myelofibrosis [5]. Clinical polycythaemia vera manifestations include splenomegaly, microcirculatory symptoms and pruritus. There is a risk of leukaemic or fibrotic transformation, which is estimated to be 7.9% at 20 years and 34% at 10 years, respectively [6, 7].
Patients with myeloproliferative neoplasms are at a higher risk for both arterial and venous thrombosis: 4.9 to 6.7 times higher than in the general population [8]. Thromboembolic events remain a major cause of morbidity and mortality in the course of polycythaemia vera and their prevalence at diagnosis varies from 12% to 39% [8]. Thrombotic complications may occur before diagnosis or later during the disease. Age and prior thrombosis are considered the most consistent risk factors [8]. Additional risk factors such as cardiovascular risk factors, leukocytosis, high haematocrit and thrombocytosis have been reported to correlate with vascular events, but their causative effect is not equivocally established. Our clinical experience as well as published data indicate that thromboembolic events are more frequent at diagnosis but may also appear long before the presence of polycythaemia vera is recognised [9]. Furthermore, some data described isolated thrombocytosis as the initial manifestation of polycythaemia vera [10].
We aimed to analyse the frequency of thromboembolic events preceding the diagnosis of polycythaemia vera and the previous evolution of blood counts, particularly the presence of thrombocytosis and erythrocytosis in polycythaemia vera patients followed at our institution.
This monocentric, retrospective study was conducted at the Haematology Service and Central Laboratory of the Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland. The medical records of patients coded during the study period as D47.1 (Chronic Myeloproliferative Disease) or D45 (Polycythaemia Vera) according to the International Classification of Diseases, 10th version (ICD-10) were reviewed. The study met the regulatory requirements of the Swiss LRH (Loi relative à la recherche sur l'être humain) and the ORH (Ordonnance relative à la recherche sur l'être humain), and was approved by the institutional review board (CER-VD 2019-00023).
The primary outcome was the temporal sequence of blood-count abnormalities (especially thrombocytosis) and thromboembolic events prior to the date of polycythaemia vera recognition. Secondary outcomes included laboratory parameters at the time of polycythaemia vera diagnosis and at the time of thrombosis; distribution and characteristics of arterial/venous thromboembolic events; and driver-mutation status among those tested. Timing labels were defined as follows: events or laboratory abnormalities occurring more than 14 days before polycythaemia vera recognition were classified as before polycythaemia vera, those recorded within ±14 days of recognition as concomitant with polycythaemia vera and those occurring more than 14 days after recognition as after polycythaemia vera. Thrombocytosis was defined as platelet count ≥350×109/l and sustained thrombocytosis was defined as platelet count >450×109/l on at least two measurements obtained 14 days or more apart, while erythrocytosis followed contemporary WHO thresholds – haemoglobin >16.5 g/dl in men and >16.0 g/dl in women, or haematocrit >49% in men and >48% in women – confirmed on repeat testing. Mutation status included testing for JAK2V617F, JAK2 exon 12, BCR-ABL among those tested; patients with missing tests were included in the denominators.
Our study included patients who were aged at least 18 years, diagnosed with polycythaemia vera and who were seen as outpatients in the Haematology Service or hospitalised at our institution between January 2008 and December 2018. The thrombotic event was the principal reason for hospitalisation. During the diagnostic work-up at admission, erythrocytosis and/or thrombocytosis was detected, which led to additional haematological investigations and ultimately the diagnosis of polycythaemia vera. We included patients with sufficient clinical documentation to determine thromboembolic events and their timing relative to polycythaemia vera recognition. Polycythaemia vera was diagnosed according to the international criteria valid at the time of the initial clinical presentation. The polycythaemia vera recognition date was defined as the first entry of polycythaemia vera as confirmed by a haematologist or on the discharge summary. Exclusion criteria were: (1) erroneous or duplicate coding identified on manual verification; (2) insufficient documentation to classify outcomes or determine event timing; and (3) secondary causes of erythrocytosis or thrombocytosis in the absence of confirmed polycythaemia vera.
For each patient, the following information was recorded according to a pre-established labelled chart: baseline demographic data, polycythaemia vera diagnosis information (blood count at diagnosis, driver mutations), presence of other comorbidities (peripheral arteriopathy, cardiac arrhythmias, chronic obstructive pulmonary disease, obstructive sleep apnoea, chronic kidney insufficiency), presence of cardiovascular risk factors (dyslipidaemia, hypertension, diabetes, smoking, overweight), medications (antiplatelet, anticoagulant, antihypertensive, antidyslipidaemic or antidiabetic treatments), presence and type of thromboembolic manifestation (arterial: stroke, acute myocardial infarction, mesenteric ischaemia, acute limb ischaemia; venous: pulmonary embolism, deep-vein thrombosis, portal vein thrombosis, sub-hepatic vein thrombosis, cerebral vein thrombosis), blood counts at thromboembolic manifestation and/or polycythaemia vera diagnosis, and available blood counts before the thromboembolic manifestation.
The identification phase involved querying the hospital information systems for candidate polycythaemia vera cases using diagnostic codes and problem lists. During screening, demographic information, laboratory data, selected outpatient letters and discharge summaries were automatically retrieved. Chart eligibility was assessed independently by two reviewers (EL, GS) to confirm polycythaemia vera diagnosis and ensure minimal data sufficiency, including laboratory and narrative evidence for thromboembolic events and their timing. The final analysis set included all eligible patients after exclusion of cases with erroneous coding or missing key variables.
Categorical variables are described as counts and percentages. Continuous variables are described as medians and interquartile ranges (IQR). The following exploratory comparisons were performed: patients with vs without any thromboembolic event; patients with a thromboembolic event before vs after polycythaemia vera diagnosis; and arterial vs venous thromboembolic event. Categorical variables were compared using Fisher’s exact test and continuous variables with Wilcoxon rank-sum tests given non-normal distributions. All p-values were two-sided and considered statistically significant if the p-value was less than or equal to 0.05. These analyses were exploratory and not adjusted for multiplicity. All analyses were performed in R (version 4.x) using the standard, open-source packages stats, tidyverse and tableone, used respectively for descriptive and inferential statistics, data transformation and visualisation, and generation of summary tables.
In total, 123 patients were identified using the search strategy (figure 1). Forty-four (35%) were excluded because of erroneous coding or missing data. Ultimately, we included 79 patients, 27 (34%) without thrombosis and 52 (66%) with thrombosis.
Figure 1Study cohort flow diagram. Flowchart illustrating patient identification, screening, eligibility assessment and inclusion in the final analysis set. Abbreviations: PV: polycythaemia vera.
Overall, there was a slight male predominance, the median age at polycythaemia vera diagnosis was 69 years and the median BMI was 24.5 kg/m². The cohort showed a high prevalence of cardiovascular comorbidities, most commonly hypertension, followed by dyslipidaemia and diabetes. Antiplatelet and antihypertensive therapies were the most frequently prescribed medications, in line with the patients’ cardiovascular profiles. Detailed baseline characteristics, comorbidities and ongoing treatments are presented in table 1. There was no statistically significant difference between the population with and without thrombosis for these variables nor between those with thrombosis before or at polycythaemia vera diagnosis and those without thrombosis (table S1 in the appendix) for these different variables.
Table 1Baseline demographic and clinical characteristics of the study population at the time of polycythaemia vera diagnosis. Missing data are indicated in “n (%)” rows.
| Patient characteristic | All patients(n = 79) | Without thrombosis(n = 27) | With thrombosis (n = 52) | p-value | |
| Age at polycythaemia vera diagnosis in years, median (range) | 69 (20–89) | 68.5 (36–88) | 69 (20–89) | 0.829 | |
| Male sex | 45 (57%) | 12 (44%) | 33 (63%) | 0.284 | |
| BMI, median (range) | 24.5 (17–39) | 25 (18.5–35) | 24.5 (17–39) | 0.335 | |
| n (%) | 30 | 12 | 18 | ||
| Comorbidities | Peripheral arteriopathy | 14 (17.7%) | 2 (7.4%) | 12 (23%) | 0.124 |
| Rhythmic cardiopathy | 26 (32.9%) | 9 (33.3%) | 17 (32.6%) | 0.708 | |
| COPD | 6 (7.6%) | 3 (11.1%) | 3 (5.7%) | 0.320 | |
| Obstructive sleep apnoea | 8 (10.1%) | 4 (14.8%) | 4 (7.7%) | 0.244 | |
| Chronic kidney insufficiency | 19 (24%) | 5 (18.5%) | 14 (26.9%) | 0.563 | |
| Cardiovascular risk factors | Dyslipidaemia | 38 (48.1%) | 10 (37%) | 28 (53.8%) | 0.332 |
| Hypertension | 60 (75.9%) | 17 (63%) | 43 (82.6%) | 0.292 | |
| Diabetes | 14 (17.7%) | 5 (18.5%) | 9 (17.3%) | 0.728 | |
| Smoking | 5 (6.3%) | 1 (3.7%) | 4 (7.7%) | 0.560 | |
| n (%) | 0 | 0 | 0 | ||
| Medication | Antiplatelet | 60 (75.9%) | 22 (81.4%) | 38 (73%) | 0.234 |
| n (%) | 0 | 0 | 0 | ||
| Anticoagulant | 31 (39.2%) | 3 (11.1%) | 28 (53.8%) | 0.001 | |
| n (%) | 0 | 0 | 0 | ||
| Antihypertensive | 58 (73.4%) | 16 (59.2%) | 42 (80%) | 0.225 | |
| n (%) | 0 | 0 | 0 | ||
| Antidyslipidaemic | 35 (44.3%) | 10 (37%) | 25 (48%) | 0.597 | |
| n (%) | 0 | 0 | 0 | ||
| Antidiabetic | 14 (17.7%) | 5 (18.5%) | 9 (17.3%) | 0.728 | |
| n (%) | 0 | 0 | 0 | ||
Abbreviations: BMI: body mass index; COPD: chronic obstructive pulmonary disease.
The median platelet level at polycythaemia vera diagnosis was 479×109/l (table 2). This value was not statistically different between populations with and without thrombosis, nor were leukocytes, haemoglobin, haematocrit or other blood count parameters. Four patients of 79 (5%) had isolated thrombocytosis (without erythrocytosis) at polycythaemia vera diagnosis. As expected, JAK2V617F was the most prevalent polycythaemia vera driver mutation in the population: mutation status was available for 54 patients, of whom 53 were positive for the JAK2V617F mutation (98%) and 1 was double-negative (JAK2V617F and JAK2 exon 12). The distribution of mutation types was similar between patients with and without thrombosis. Unfortunately, we did not have information concerning the driver mutations at diagnosis for 25 patients: 18 patients were diagnosed before 2005, when the JAK2V617F mutation was first described, without any subsequent mutation search and data were missing for the other 7 patients.
Table 2Laboratory parameters at the time of polycythaemia vera diagnosis. Missing data are indicated in “n (%)” rows.
| Laboratory parameters | All patients (n = 79) | Without thrombosis (n = 27) | With thrombosis (n = 52) | p-value | |
| Blood count parameters, median [IQR] | Platelets in 109/l | 479 [318–627] | 483 [378–677] | 479 [296–599] | 0.417 |
| n (%) | 31 | 15 | 16 | ||
| Leukocytes in 109/l | 11.3 [9–13] | 11.2 [10–13] | 11.4 [9–14] | 0.955 | |
| n (%) | 31 | 15 | 16 | ||
| Haemoglobin in g/l | 180 [169–188] | 177 [168–183] | 182 [170–187] | 0.376 | |
| n (%) | 31 | 15 | 16 | ||
| Haematocrit in % | 54% [50–58%] | 53.5% [51–57%] | 53.5% [50–58%] | 0.684 | |
| n (%) | 31 | 15 | 16 | ||
| Erythrocytes in 1012/l | 6 [6–7] | 6 [6–7] | 6 [6–7] | 0.801 | |
| n (%) | 31 | 16 | 16 | ||
| MCV in fL | 85 [79–89] | 86 [79–90] | 85 [80–89] | 0.879 | |
| n (%) | 31 | 16 | 19 | ||
| MCH in pg | 29 [26–30] | 29 [25–30] | 29 [27–30] | 0.598 | |
| n (%) | 31 | 16 | 19 | ||
| MCHC in g/l | 331 [323–340] | 339 [327–341] | 330 [323–338] | 0.541 | |
| n (%) | 31 | 16 | 19 | ||
| Mutations | JAK2V617F | 53 (67%) | 17 (63%) | 36 (69.2%) | 0.364 |
| JAK2 exon 12 | 0 | 0 | 0 | - | |
| Double negative | 1 (1.2%) | 0 | 1 (1.9%) | - | |
| n (%) | 25 | 10 | 15 | ||
Abbreviations: IQR: interquartile range; MCH: mean corpuscular haemoglobin; MCHC: mean corpuscular haemoglobin concentration; MCV: mean corpuscular volume.
Fifty-two (66%) patients presented at least one thrombotic event (table 3). Among them, 24 (30%) experienced a thrombotic event before (15 patients) or concomitant with (9 patients) the polycythaemia vera diagnosis; 11 (14%) after the polycythaemia vera diagnosis; 17 (21%) presented a thrombotic event both before and after the diagnosis of polycythaemia vera.
Table 3Distribution of thromboembolic events by vascular territory.
| Thrombosis | All patients with thrombosis(n = 52) | Before or concomitant with polycythaemia vera diagnosis (n = 24) | After polycythaemia vera diagnosis (n = 11) | Before and after polycythaemia vera diagnosis (n = 17) |
| Arterial | 40 | 18 | 8 | 14 |
| ... Stroke | 22 | 13 | 3 | 6 |
| ... Acute myocardial infarction | 16 | 8 | 2 | 6 |
| ... Mesenteric ischaemia | 1 | 0 | 0 | 1 |
| ... Acute limb ischaemia | 5 | 1 | 2 | 2 |
| Venous | 31 | 12 | 5 | 14 |
| ... Pulmonary embolism | 14 | 6 | 1 | 7 |
| ... Deep-vein thrombosis | 14 | 5 | 4 | 5 |
| ... Portal vein thrombosis | 2 | 0 | 0 | 2 |
| ... Sub-hepatic vein thrombosis | 3 | 2 | 0 | 1 |
| ... Cerebral vein thrombosis | 2 | 0 | 0 | 2 |
Forty (87%) patients presented arterial thrombosis, 31 (60%) venous thrombosis and 19 (36%) both arterial and venous thrombotic events. Details of thrombotic manifestations are presented in table 3. Blood counts at the time of thrombotic manifestations showed median platelet values at 361×109/l. Other blood count parameters are summarised in table 4.
Table 4Laboratory parameters at the time of thrombotic manifestations. Missing data are indicated in “n (%)” rows.
| Laboratory parameters | All patients (n = 52) | Before or concomitant with polycythaemia vera diagnosis (n = 24) | After polycythaemia vera diagnosis (n = 11) | Thromboembolic events before and after polycythaemia vera diagnosis (n = 17) | p-value | |
| Before | After | |||||
| Platelets in 109/l, median [IQR] | 361 [261–495] | 360 [334–512] | 303 [192–396] | 422 [267–501] | 323 [285–432] | 0.151 |
| n (%) | 12 | 6 | 0 | 2 | 3 | |
| Leukocytes in 109/l, median [IQR] | 11 [8–13] | 12.5 [9–14] | 8.2 [8–10] | 11 [9–13] | 11.2 [7–13] | 0.930 |
| n (%) | 12 | 6 | 0 | 2 | 3 | |
| Haemoglobin in g/l, median [IQR] | 158 [134–182] | 178 [155–188] | 125 [109–156] | 158 [135–172] | 140 [101–155] | 0.571 |
| n (%) | 12 | 6 | 0 | 2 | 3 | |
| Haematocrit in %, median [IQR] | 47 [42–54] | 53 [48–58] | 38 [34–46] | 46 [33–43] | 43 [32–46] | 0.366 |
| n (%) | 12 | 6 | 0 | 2 | 3 | |
| Erythrocytes in 1012/l, median [IQR] | 6 [6–8] | 6 [6–7] | 4 [3–5] | 6 [5–6] | 4 [3–5] | 0.342 |
| n (%) | 12 | 6 | 0 | 2 | 3 | |
| MCV in fL | 92 [84–97] | 88 [83–93] | 94 [85–102] | 93 [88–99] | 99 [85–111] | 0.794 |
| n (%) | 13 | 7 | 0 | 2 | 3 | |
| MCH in pg, median [IQR] | 30 [28–32] | 30 [27–31] | 31 [29–33] | 31 [29–33] | 34 [27–38] | 0.708 |
| n (%) | 13 | 7 | 0 | 2 | 3 | |
| MCHC in g/l, median [IQR] | 330 [323–341] | 328 [323–340] | 328 [316–343] | 334 [330–342] | 336 [333–345] | 0.742 |
| n (%) | 13 | 7 | 0 | 2 | 3 | |
Abbreviations: IQR: interquartile range; MCH: mean corpuscular haemoglobin; MCHC: mean corpuscular haemoglobin concentration; MCV: mean corpuscular volume.
We collected blood counts at the time of diagnosis of polycythaemia vera for 53/79 patients (67%) and at the time of thrombosis for 45/52 patients (86%). Of the 41 patients with a thrombotic manifestation before or concomitant with the polycythaemia vera diagnosis, we collected 90 blood counts performed before diagnosis of polycythaemia vera in 17 (41%) patients. The median number of blood counts available before the diagnosis of polycythaemia vera was three and before the thromboembolic event was two.
Among patients with previous platelet counts available, the median time from the earliest available blood count to the date of the first thrombotic event was 29 months and the median time from the earliest available blood count showing platelets >350×109/l or >450×109/l to the diagnosis of polycythaemia vera was 36 months and 24 months, respectively. Of note, four patients had a thromboembolic event before the polycythaemia vera diagnosis with a normal platelet count. In these four patients, the median delay between the thromboembolic event and polycythaemia vera diagnosis was 32 months. In addition, all four presented elevated haemoglobin levels and two of them also had leucocytosis (>109/l) at that time. For the remaining 13 patients, the median time from the first blood count with platelets >350×109/l or >450×109/l and the thromboembolic event was 46 months and 10 months, respectively. Details of the evolution of the platelets for patients with a thromboembolic event before or at the time of polycythaemia vera diagnosis are shown in figure 2.
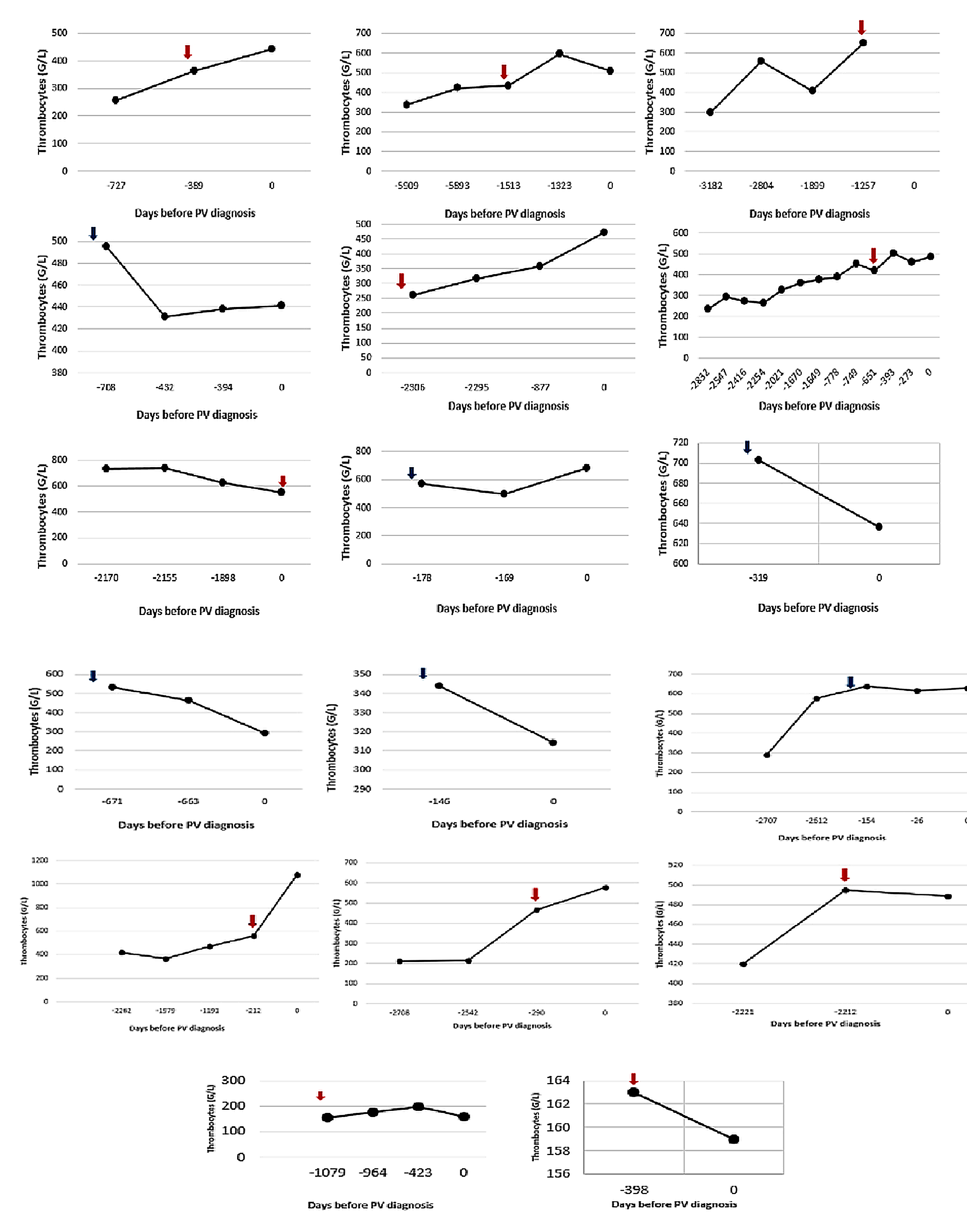
Figure 2Platelet count evolution up to the polycythaemia vera diagnosis in patients with thrombosis before or at the time of polycythaemia vera diagnosis. Blue arrow = venous thromboembolic event. Red arrow = arterial thromboembolic event. Day 0 = Day of polycythaemia vera diagnosis.
Regarding haemoglobin/haematocrit values among the 17 patients with previous blood counts available, the median time from the earliest available blood count showing Hb >16.5 g/dl (Hct >49%) for men or Hb >16.0 g/dl (Hct >48%) for women to the diagnosis of polycythaemia vera was 25 months and 24 months, respectively, and to the thromboembolic event was 12 months and 13 months, respectively. In the four patients who had a thromboembolic event prior to the polycythaemia vera diagnosis with a normal platelet count, an increased haematocrit was already present in all four of them.
Thrombotic events are the most prevalent complication in patients with polycythaemia vera and can occur before, coincident with or after diagnosis. In this study, we assessed the frequency of thromboembolic events preceding the diagnosis of polycythaemia vera and the previous evolution of blood counts, in particular the evolution of platelet counts and of Hb/Hct. Concerning the clinical characteristics of patients with polycythaemia vera, the median age at diagnosis is reported as 61 years, with males and females being equally affected [6]. In our cohort, median age at diagnosis was slightly higher (69 years), giving a possible explanation for the presence of multiple comorbidities and cardiovascular risk factors in these patients, most frequently hypertension. Regarding frequency of thromboembolic events, in our study 52 patients (66%) presented at least one arterial or venous thrombosis before, at or after diagnosis, among whom 24 (46%) presented a thromboembolic event before or concomitant with the diagnosis of polycythaemia vera and 17 (33%) presented a thromboembolic event before and after the polycythaemia vera diagnosis. This is higher than the risk of thrombosis in polycythaemia vera patients described in larger series, where the overall risk of arterial or venous thrombosis before the diagnosis of polycythaemia vera varies between 16% and 27% and between 8% and 12% for arterial and venous thrombosis, respectively [6, 11–14]. However, similar findings to ours were reported in the prospective European Collaboration on Low-dose Aspirin in Polycythemia (ECLAP) cohort study of 1638 patients, which indicated that 38.4% of patients experienced some thrombotic event before polycythaemia vera diagnosis [15]. This variation could be partly explained by the older age and the higher prevalence of cardiovascular risk factors in our population, and by the fact that a high fraction of patients with a thromboembolic event often benefit from watchful follow-up in tertiary care centres. Finally, these epidemiological differences at diagnosis reflect the heterogeneity of patient features that are found in the clinical practice of polycythaemia vera.
Of note, in our study, 11 (21%) of the 52 patients with a thromboembolic event developed it after the polycythaemia vera diagnosis. These findings are consistent with data from larger studies; in the real-world analysis of the MPN registry of the Study Alliance Leukemia, one third of all thromboembolic events occurred after the date of diagnosis [9]. Similarly, in a study from Mayo Clinic including 665 patients with polycythaemia vera, 24% presented a thrombosis after the polycythaemia vera diagnosis was established [16]. These findings highlight the importance of treating cardiovascular risk factors besides cytoreductive therapy for high-risk polycythaemia vera, point out the fact that treatment of polycythaemia vera patients does not always reflect current clinical guidelines and suggest that not all prothrombotic mechanisms can be controlled with available drugs.
To our knowledge, very few studies have systematically analysed the temporal sequence of thrombocytosis, erythrocytosis, thrombotic events and polycythaemia vera diagnosis; our work adds a single‑centre perspective with detailed timing rules.
We started our investigation based on our clinical experience of several patients diagnosed with polycythaemia vera at the time of a thrombotic event (or long afterwards), in whom a thrombocytosis and/or increased Hb/Hct had already existed for several months or even years. While in the majority of cases, thrombocytosis coexists with polyglobulia at the time of the polycythaemia vera diagnosis, there is evidence that some patients with polycythaemia vera may initially present with isolated thrombocytosis; Maslah et al. demonstrated that masked polycythaemia vera frequently presents with an essential thrombocythaemia-like profile, characterised by isolated or predominant thrombocytosis and only subtly increased red-cell indices, underscoring that polycythaemia vera may exist even in the absence of overt erythrocytosis [17]. In a retrospective study by Thiele et al. including 44 patients with early polycythaemia vera, 23 (52%) presented initially with an elevated platelet count and an increased Hb [10]. In another study by Abdulkarim and colleagues, significant thrombocytosis (median of 700×109/l) in combination with polyglobulia (median Hb of 17.1 g/dl) was noted at the polycythaemia vera diagnosis [18]. In agreement with these findings, Gugliotta et al. reported in their cohort that the median platelet count at diagnosis was 783×109/l [19]. Likewise, in our cohort platelet and haemoglobin (haematocrit) levels at polycythaemia vera diagnosis were elevated with median values of 479×109/l and 179 g/l (53%), respectively. In a monocentric, retrospective study by Lakhwani et al., 17 of 63 patients with polycythaemia vera (27%) experienced thrombosis prior to diagnosis [20]. The median time between detection of polyglobulia and thrombosis was 8.2 months and those patients with thrombosis prior to polycythaemia vera diagnosis had significantly lower overall survival, highlighting that early diagnosis can help reduce morbidity associated with thromboembolic events.
In our study, a threshold of 350×109/l was used for considering the presence of a potential thrombocytosis. According to the study of Biino et al., factors such as sex, age and origin may influence the number of circulating platelets [21]. Thus, in this study extended reference intervals for platelet count were reported.
In our cohort, the median interval between the first documentation of thrombocytosis and polycythaemia vera diagnosis was 36 months for platelet counts >350×109/l and 24 months for counts >450×109/l, indicating a substantial diagnostic delay. Thrombocytosis often represented the earliest haematological abnormality, preceding increased Hb/Hct values, in patients who later developed polycythaemia vera and experienced a thrombotic event. These findings underscore the importance of investigating unexplained, sustained thrombocytosis with JAK2V617F testing as this may facilitate earlier detection of polycythaemia vera.
In polycythaemia vera, in addition to age >60 years and/or thrombosis, new features such as cardiovascular risk factors and JAK2V617F allele burden have been proposed as candidate biomarkers for predicting risk of thrombosis [2, 4]. The role of thrombocytosis as a causative factor for thrombosis has been the object of numerous investigations, but to date no definitive conclusion has been drawn on this matter.
On the one hand, thrombocytosis was associated with a better survival in polycythaemia vera patents according to the study by Tefferi et al. [6]. In the large prospective polycythaemia vera cohort of Berk et al. (PV Study Group-PVSG), platelet counts measured at the closest observation prior to the thrombotic event did not correlate with thrombosis [22]. Moreover, studies have indicated that high platelet counts correlate more with a higher risk of bleeding, mainly due to an acquired von Willebrand syndrome [23, 24]. In a 2015 series of 142 consecutive patients with polycythaemia vera, concomitant acquired von Willebrand syndrome was identified in 17 (12%) and most of them (58.8%) presented with typical signs of bleeding disorder and higher platelet counts (389×109/l versus 284×109/l, p <0.01) [25]. However, the true co-prevalence of polycythaemia vera and acquired von Willebrand syndrome remains unknown.
On the other hand, it has been shown that polycythaemia vera patients have an increased thromboxane synthesis, suggesting that platelet activation, and not necessarily thrombocytosis, may contribute to the increased risk of thrombosis [6]. In a Spanish retrospective study by Alvarez-Larran et al., in patients with polycythaemia vera treated with hydroxyurea, a lack of platelet response (> 400×109/l), which may suggest a strong megakaryopoietic drive, was associated with an increased risk of thrombosis [26]. The association between thrombotic risk, iron deficiency anaemia and secondary thrombocytosis is controversial. It is known that a high phlebotomy rate (>4 per year) in polycythaemia vera patients may facilitate propensity to thrombosis [22], but whether this is due to worsening iron deficiency or to the underlying disease dynamics remains poorly understood [27].
Taking together all the above, it remains unclear whether platelet count plays a causal role in thrombotic complications in patients with polycythaemia vera or it has to be considered as an epiphenomenon. Our study cannot evaluate a prognostic value of thrombocytosis in polycythaemia vera thrombotic complications, but it clearly indicates that thrombocytosis can precede thrombotic events in myeloproliferative neoplasms [28].We underline the fact that early diagnosis of polycythaemia vera is clinically relevant given the high mortality associated with thrombotic events in these patients. As already mentioned, an isolated thrombocytosis can be present for several months or even years before the polycythaemia vera diagnosis. Therefore, persistent, unexplained thrombocytosis after exclusion of secondary causes, warrants stepwise molecular testing (JAK2/CALR/MPL); awareness that polycythaemia vera and other Ph-negative myeloproliferative neoplasms may present with elevated platelet counts long before the onset of other symptoms would lead to their recognition. Early diagnosis of polycythaemia vera can guide therapy and has relevant treatment implications. According to current guidelines, treatment of polycythaemia vera is dependent on age and history of thrombosis [2]. All patients should be treated with low-dose aspirin and periodic phlebotomies, whereas high-risk patients (age >60 years, history of thrombosis) should be considered for cytoreductive drugs (hydroxyurea and recombinant interferon alpha) as first-line therapies [2]. Lastly, an earlier diagnosis of polycythaemia vera could lead to the identification and modification of cardiovascular risk factors to further minimise the risk of thrombosis [29].
Limitations of our study include the relatively small sample size and its retrospective, single-centre design. Consequently, some data are incomplete and certain clinical details may be missing. Although we had full access to all medical data available within our tertiary hospital, access to laboratory results and records from external physicians (e.g. general practitioners) was limited. As a result, early laboratory abnormalities or thromboembolic events that occurred outside our institution may not have been captured. This undercapture could lead to underestimation of the true duration between the onset of haematological abnormalities and polycythaemia vera diagnosis, as well as underreporting of thromboembolic events: the first captured abnormality in our system may occur after the true onset, biasing timing estimates towards later onset and shorter intervals. Another limitation concerns diagnostic accuracy, as molecular data (JAK2V617F mutation status) were missing for a subset of patients, which may have led to potential misclassification or underestimation of confirmed polycythaemia vera cases. Furthermore, the monocentric setting may limit the generalisability of our findings to other healthcare environments with different referral patterns or diagnostic workflows. Despite these limitations, the observed variability in timing remains clinically relevant, as it underscores the potential for earlier recognition of polycythaemia vera through careful interpretation of laboratory abnormalities.
With the increasing availability of routine blood cell counts and the high prevalence of myeloproliferative disorders, polycythaemia vera (along with other Ph-negative myeloproliferative neoplasms) should always be considered in the differential diagnosis not only of erythrocytosis but also of isolated otherwise unexplained thrombocytosis. The early recognition of polycythaemia vera is crucial as it may spare the patients serious complications, notably thanks to timely implementation of prophylaxis with aspirin and/or treatment with cytoreductive drugs in high-risk patients, as well as more aggressive management of cardiovascular risk factors. Timely recognition of polycythaemia vera and initiation of appropriate therapy may contribute to a reduction in thrombotic risk, although thromboembolic events can still occur despite adequate management.
Deidentified study data can be shared upon reasonable request to the corresponding author. The code can be made available upon reasonable request to the corresponding author.
We thank the physicians and nurses of the Haematology Service (CHUV, Lausanne) for their dedicated patient care and the staff of the Haematology Laboratory (CHUV, Lausanne) for the accuracy of their analytical work.
The authors did not receive any financial support for the preparation of this manuscript.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. No potential conflict of interest related to the content of this manuscript was disclosed.
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May;127(20):2391–405. doi: https://doi.org/10.1182/blood-2016-03-643544
2. Tefferi A, Barbui T. Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023 Sep;98(9):1465–87. doi: https://doi.org/10.1002/ajh.27002
3. Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol. 2014 Apr;92(4):289–97. doi: https://doi.org/10.1111/ejh.12256
4. Guglielmelli P, Loscocco GG, Mannarelli C, Rossi E, Mannelli F, Ramundo F, et al. JAK2V617F variant allele frequency >50% identifies patients with polycythemia vera at high risk for venous thrombosis. Blood Cancer J. 2021 Dec;11(12):199. doi: https://doi.org/10.1038/s41408-021-00581-6
5. Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010 Sep;24(9):1574–9. doi: https://doi.org/10.1038/leu.2010.148
6. Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013 Sep;27(9):1874–81. doi: https://doi.org/10.1038/leu.2013.163
7. Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014 Oct;124(16):2507–13. doi: https://doi.org/10.1182/blood-2014-05-579136
8. Hultcrantz M, Björkholm M, Dickman PW, Landgren O, Derolf ÅR, Kristinsson SY, et al. Risk for Arterial and Venous Thrombosis in Patients With Myeloproliferative Neoplasms: A Population-Based Cohort Study. Ann Intern Med. 2018 Mar;168(5):317–25. doi: https://doi.org/10.7326/M17-0028
9. Kaifie A, Kirschner M, Wolf D, Maintz C, Hänel M, Gattermann N, et al.; Study Alliance Leukemia (SAL). Bleeding, thrombosis, and anticoagulation in myeloproliferative neoplasms (MPN): analysis from the German SAL-MPN-registry. J Hematol Oncol. 2016 Mar;9(1):18. doi: https://doi.org/10.1186/s13045-016-0242-9
10. Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol. 2005;113(4):213–9. doi: https://doi.org/10.1159/000084673
11. Barbui T, Carobbio A, Rumi E, Finazzi G, Gisslinger H, Rodeghiero F, et al. In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. Blood. 2014 Nov;124(19):3021–3. doi: https://doi.org/10.1182/blood-2014-07-591610
12. Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med. 1995 Nov;123(9):656–64. doi: https://doi.org/10.7326/0003-4819-123-9-199511010-00003
13. Grunwald MR, Stein BL, Boccia RV, Oh ST, Paranagama D, Parasuraman S, et al. Clinical and disease characteristics from REVEAL at time of enrollment (baseline): prospective observational study of patients with polycythemia vera in the United States. Clin Lymphoma Myeloma Leuk. 2018 Dec;18(12):788–795.e2. doi: https://doi.org/10.1016/j.clml.2018.08.009
14. Cerquozzi S, Barraco D, Lasho T, Finke C, Hanson CA, Ketterling RP, et al. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017 Dec;7(12):662. doi: https://doi.org/10.1038/s41408-017-0035-6
15. Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, et al.; European Collaboration on Low-Dose Aspirin in Polycythemia Vera Investigators. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004 Jan;350(2):114–24. doi: https://doi.org/10.1056/NEJMoa035572
16. Szuber N, Mudireddy M, Nicolosi M, Penna D, Vallapureddy RR, Lasho TL, et al. 3023 Mayo Clinic Patients With Myeloproliferative Neoplasms: Risk-Stratified Comparison of Survival and Outcomes Data Among Disease Subgroups. Mayo Clin Proc. 2019 Apr;94(4):599–610. doi: https://doi.org/10.1016/j.mayocp.2018.08.022
17. Maslah N, Soret J, Dosquet C, Vercellino L, Belkhodja C, Schlageter MH, et al. Masked polycythemia vera: analysis of a single center cohort of 2480 red cell masses. Haematologica. 2020 Mar;105(3):e95–7. doi: https://doi.org/10.3324/haematol.2018.215582
18. Abdulkarim K, Ridell B, Johansson P, Kutti J, Safai-Kutti S, Andréasson B. The impact of peripheral blood values and bone marrow findings on prognosis for patients with essential thrombocythemia and polycythemia vera. Eur J Haematol. 2011 Feb;86(2):148–55. doi: https://doi.org/10.1111/j.1600-0609.2010.01548.x
19. Gugliotta L, Iurlo A, Gugliotta G, Tieghi A, Specchia G, Gaidano G, et al.; Registro Italiano Trombocitemie RIT. Unbiased pro-thrombotic features at diagnosis in 977 thrombocythemic patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Res. 2016 Jul;46:18–25. doi: https://doi.org/10.1016/j.leukres.2016.04.004
20. Lakhwani S, Pardina-Echevarría M, Arcas-Vega R, Díaz-Sánchez OR, Hernández-García MT, Raya JM. Relevance of early diagnosis in polycythemia vera and essential thrombocythemia: A single center’s experience. Rev Clin Esp (Barc). 2022 Mar;222(3):169–73. doi: https://doi.org/10.1016/j.rceng.2021.07.003
21. Biino G, Santimone I, Minelli C, Sorice R, Frongia B, Traglia M, et al. Age- and sex-related variations in platelet count in Italy: a proposal of reference ranges based on 40987 subjects’ data. PLoS One. 2013;8(1):e54289. doi: https://doi.org/10.1371/journal.pone.0054289
22. Berk PD, Goldberg JD, Donovan PB, Fruchtman SM, Berlin NI, Wasserman LR. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol. 1986 Apr;23(2):132–43.
23. Tartaglia AP, Goldberg JD, Berk PD, Wasserman LR. Adverse effects of antiaggregating platelet therapy in the treatment of polycythemia vera. Semin Hematol. 1986 Jul;23(3):172–6.
24. Passamonti F. How I treat polycythemia vera. Blood. 2012 Jul;120(2):275–84. doi: https://doi.org/10.1182/blood-2012-02-366054
25. Mital A, Prejzner W, Świątkowska-Stodulska R, Hellmann A. Factors predisposing to acquired von Willebrand syndrome during the course of polycythemia vera - retrospective analysis of 142 consecutive cases. Thromb Res. 2015 Oct;136(4):754–7. doi: https://doi.org/10.1016/j.thromres.2015.07.029
26. Alvarez-Larrán A, Pereira A, Cervantes F, Arellano-Rodrigo E, Hernández-Boluda JC, Ferrer-Marín F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012 Feb;119(6):1363–9. doi: https://doi.org/10.1182/blood-2011-10-387787
27. Song J, Kim SJ, Thiagarajan P, Prchal JT; Iron Deficiency in Polycythemia Vera Increases HIF Activity and Transcription of Prothrombotic Genes. Iron Deficiency in Polycythemia Vera Increases HIF Activity and Transcription of Prothrombotic Genes. Blood. 2021;138 Supplement 1:2549. doi: https://doi.org/10.1182/blood-2021-153221
28. Stalder G, Da Silva WR, Segot A, Blum S, Grandoni F, Alberio L. Long-standing thrombocytosis often precedes thromboembolic complications heralding the diagnosis of essential thrombocythemia. Eur J Intern Med. 2023 Jan;107:110–2. doi: https://doi.org/10.1016/j.ejim.2022.09.002
29. Benevolo G, Marchetti M, Melchio R, Beggiato E, Sartori C, Biolé CA, et al. Diagnosis and Management of Cardiovascular Risk in Patients with Polycythemia Vera. Vasc Health Risk Manag. 2023 Nov;19:765–78. doi: https://doi.org/10.2147/VHRM.S429995
The appendix is available in the pdf version of the article at https://doi.org/10.57187/4669.