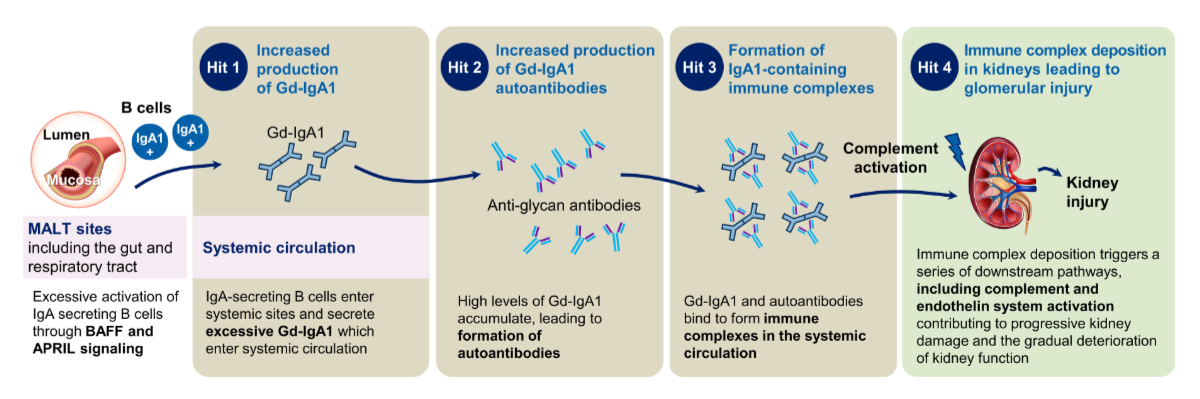
Figure 1“Multi-hit” model of pathogenesis of immunoglobulin A (IgA) nephropathy. Adapted with permission courtesy of Novartis. APRIL: A proliferation-inducing ligand; BAFF: B-cell activating factor; Gd-IgA1: galactose-deficient immunoglobulin A1.
DOI: https://doi.org/https://doi.org/10.57187/s.4435
antineutrophil cytoplasmic antibody
body mass index
serine protease domain
Chronic Kidney Disease Epidemiology Collaboration
European Medicines Agency
U.S. Food and Drug Administration
estimated glomerular filtration rate (calculated by the CKD-EPI formula; in ml/min/1.73 m2 of body surface)
immunoglobulin A
galactose-deficient immunoglobulin A1
urine protein-creatinine ratio
sodium-glucose cotransporter-2
Immunoglobulin (Ig) A nephropathy is the most common primary glomerulonephritis worldwide, with a global incidence of about 2.5 per 100,000 individuals per year [1, 2]. Usually, IgA nephropathy is found in adults aged around 20 to 30 years and has a high lifetime risk of disease progression [3]. It is frequently associated with severe loss of kidney function, leading to kidney failure, especially in those with elevated protein excretion [4–6]. Currently, approved disease-specific therapies for patients with IgA nephropathy in Switzerland are scarce. The KDIGO 2024 Clinical Practice Guideline for the management of IgA nephropathy was recently available for public review and will be published soon. According the guideline, current treatments for IgA nephropathy focus on reducing proteinuria and nephron loss with nephroprotective regimens consisting of renin-angiotensin system blockade, the use of sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and the dual endothelin and angiotensin receptor antagonist sparsentan, which has been approved for IgA nephropathy treatment by the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA) and recently, by Swissmedic [7, 8]. Historically, treatments for glomerular inflammation as a main cause of nephron loss in patients with IgA nephropathy were limited to systemic glucocorticoids [9]. Furthermore, a so called targeted-release formulation of budesonide (also known as Nefecon®), another therapeutic option to reduce IgA nephropathy-specific drivers of nephron loss (i.e., reducing the production of pathogenic immunoglobulin A [IgA] by the mucosal immune system) was the first drug approved by the FDA (Tarpeyo®, Calliditas Therapeutics AB, Sweden) and EMA (Kinpeygo®, STADA Arzneimittel AG, Germany) to treat IgA nephropathy, but it has not yet been approved by Swissmedic [10, 11]. However, the use of these therapies has been associated with adverse effects, even with the targeted-release formulation of budesonide, and their benefit remains to be weighed against the risk of treatment-emergent toxicity. This highlights the ongoing need to identify more effective and safer therapies for the treatment of IgA nephropathy. Deposition of immune complexes containing galactose-deficient IgA1 (GD-IgA1), which is an important “hit” within the “multi-hit” model of IgA nephropathy pathogenesis [12], triggers a series of downstream pathways, including activation of the alternative complement pathway [13, 14] and endothelin system [15], as shown in figure 1. Additionally, over the last few years, increasing understanding of the pathogenetic role of alternative complement pathway dysregulation in the onset and progression of IgA nephropathy has led to the development of new complement-targeting therapies [16].
Figure 1“Multi-hit” model of pathogenesis of immunoglobulin A (IgA) nephropathy. Adapted with permission courtesy of Novartis. APRIL: A proliferation-inducing ligand; BAFF: B-cell activating factor; Gd-IgA1: galactose-deficient immunoglobulin A1.
Iptacopan (Fabhalta®, Novartis Pharma AG, Switzerland) is an oral, first-in-class, highly potent inhibitor of complement factor B that effectively blocks the alternative complement pathway [17]. Complement factor B includes a serine protease domain (Bb), which is the proteolytically active component of the alternative complement pathway C3 (C3bBb) and C5 (C3bBb3b) convertases [17]. Consequently, iptacopan-mediated inhibition of complement factor B suppresses C3 convertase activity, blocking the cleavage of C3 and activation of the so-called amplification loop [17]. Further, this results in the prevention of the downstream generation of the C5 convertase complex, as well as the terminal complement cascade with its effector consequences [17]. The activation of the alternative complement pathway, as well as blockade by iptacopan, is summarised in figure 2.
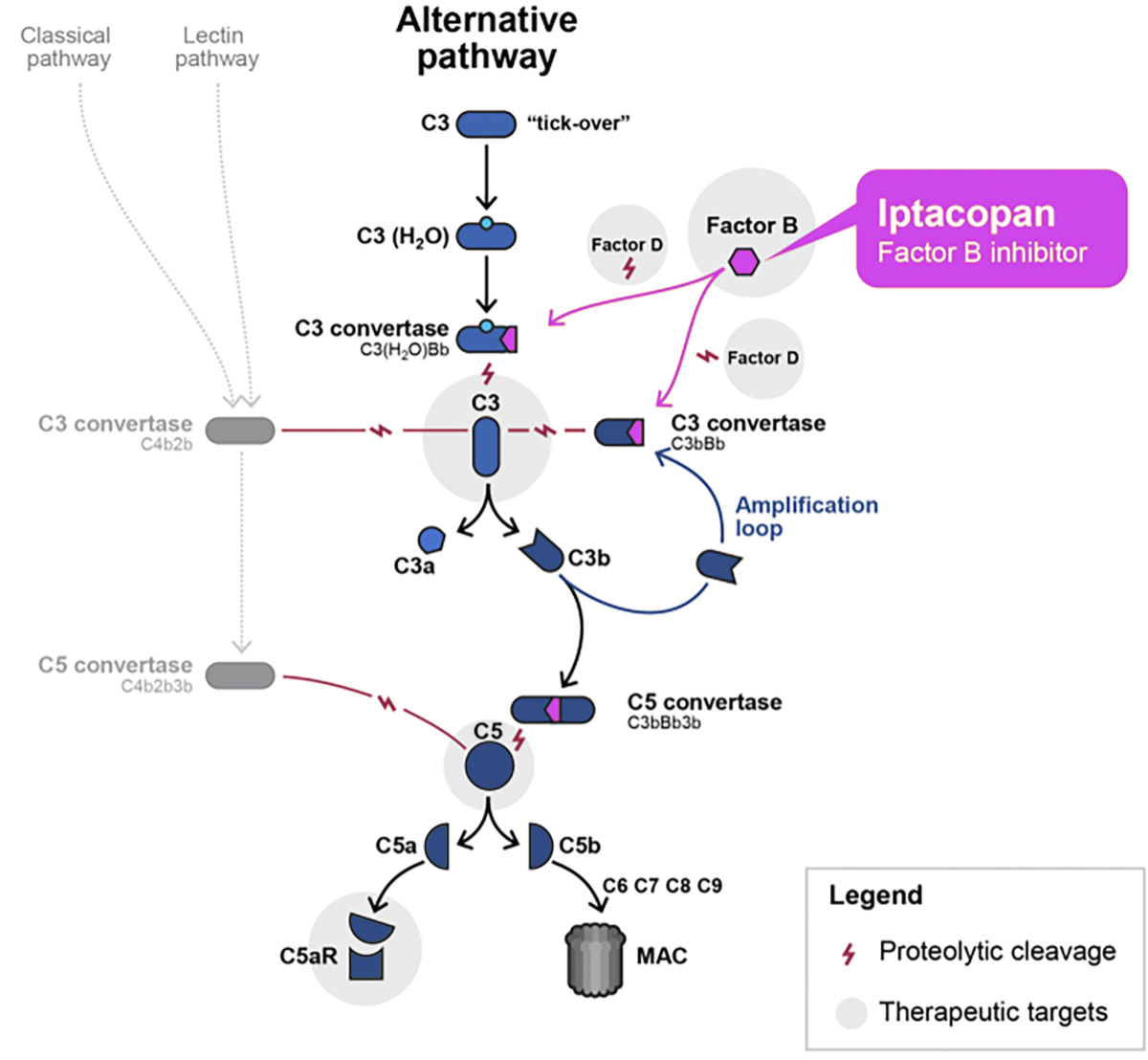
Figure 2Targeting of the alternative complement pathway by the complement factor B inhibitor iptacopan. Adapted with permission courtesy of Novartis. Bb: serine protease domain; MAC: membrane attack complex.
To date, iptacopan has been investigated in several diseases associated with alternative complement pathway dysregulation, like paroxysmal nocturnal haemoglobinuria, for which indication Swissmedic recently granted approval [18–20]. Further, iptacopan was investigated in a phase 2 clinical trial in patients with C3 glomerulopathy [21] and is under investigation in a phase 3 double-blind, placebo-controlled clinical trial for the treatment of C3 glomerulopathy (Clinical.Trials.gov number NCT04817618) [22]. In addition, iptacopan is being investigated in a single-arm, open-label, phase 3 study to evaluate its efficacy and safety in patients with complement-mediated atypical haemolytic uremic syndrome (aHUS) (ClinicalTrials.gov number NCT04889430) [23]. Moreover, the results of a randomised phase 2 study including patients with IgA nephropathy demonstrated that iptacopan reduces proteinuria in a dose-dependent manner, with a 23% (80% confidence interval 8–34%) reduction form baseline in the urine protein-creatinine ratio (UPCR) from 24-hour urine collection achieved with iptacopan 200 mg twice a day at three months [24]. UPCR decreased further across six months, and biomarker data confirmed the mechanism of action of iptacopan [24]. The prespecified interim analysis of a phase 3 double-blind, randomised, placebo-controlled trial (APPLAUSE-IgA nephropathy study, ClinicalTrials.gov number NCT04578834), including 250 patients with IgA nephropathy (i.e., 125 in the iptacopan group and 125 in the placebo group), has just been published, showing a 38.3% reduction in the 24-hour UPCR at 9 months in patients treated with iptacopan compared to placebo [25]. To the best of our knowledge, we herewith report the first successful treatment of a female patient with IgA nephropathy with iptacopan in Switzerland, which has not been published before. Furthermore, the latest innovative therapies for treating IgA nephropathy are summarised within the discussion section.
In February 2023, a 40-year-old female patient with recurrent episodes of macrohaematuria during infectious episodes since childhood, but otherwise healthy status, was referred to our outpatient clinic for a diagnostic work-up. The patient was of Asian origin and had no risk behaviour. Approximately two weeks before the initial consultation, she suffered from an upper respiratory tract infection and noticed again that her urine was very dark in colour. At that time, she had no flank pain or dysuria. Her medical history was negative for recurrent urinary tract infections, as well as any kind of joint or skin problems. Further, the patient did not take any regular medication, including any over-the-counter drugs. Family history was negative for known renal diseases; however, her father suffered from an unspecified “nephritis”. At initial consultation, she presented with a good general condition and did not complain about any pain or discomfort. Her nutrition status was within the lower range (body mass index [BMI] 17 kg/m2). Blood pressure (108/75 mm Hg) and pulse rate (80/min) were both normal, with unremarkable cardiopulmonary auscultation. In addition, the patient had no lower leg oedema, no skin lesions, as well as an unremarkable abdominal examination. Laboratory tests confirmed normal kidney function, with a serum creatinine value of 60 µmol/l (reference range 45–84 µmol/l), corresponding to an estimated glomerular filtration rate (eGFR) according to the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula of 110 ml/min/1.73 m2. However, UPCR was 98 mg/mmol (reference range <11 mg/mmol), which peaked at 186 mg/mmol in November 2023. Urine sediment examination showed a nephritic sediment with glomerular microhaematuria and cellular casts, including erythrocytes. Ultrasonography of both kidneys was normal. Blood and urine tests were repeated within four weeks after the patient’s initial assessment (in March 2023) at our outpatient clinic and showed similar results (i.e., normal kidney function, glomerular microhaematuria and a UPCR of 77 mg/mmol). The clinical course of kidney function, proteinuria and established treatment is illustrated in figure 3.
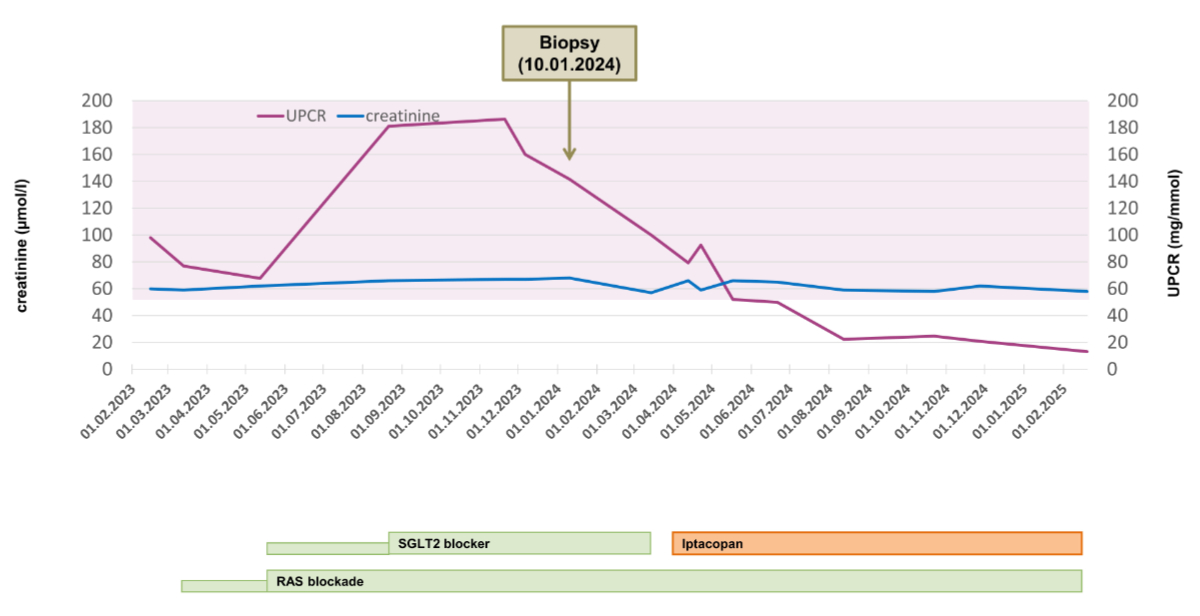
Figure 3Course of successful treatment of a 40-year-old woman suffering from biopsy-confirmed immunoglobulin A nephropathy (IgAN) with iptacopan. The purple area indicates proteinuria >500 mg/day (i.e. urine protein-creatinine ratio (UPCR) >50 mg/mmol). For the dosage of medication, please refer to the text. RAS: renin-angiotensin system; SGLT2: sodium-glucose cotransporter-2.
Given a highly suspected diagnosis of IgA nephropathy, the patient was first treated with conservative therapy, which was started in March 2023 with renin-angiotensin system blockade and completed in May 2023 with an SGLT-2 inhibitor (nephroprotective medication consisted of ramipril at a maximum tolerated dosage of 2.5 mg per day in combination with dapagliflozin 10 mg per day). Despite the patient’s excellent compliance concerning the established conservative, antiproteinuric therapy and a systolic blood pressure between 100 and 110 mm Hg, proteinuria ranged between 1 and 1.8 g/day, and glomerular microhaematuria persisted, with preserved renal function (figure 3). Therefore, the patient agreed to a kidney biopsy, which was performed in January 2024. Histological work-up of the kidney biopsy followed standard procedures (of 2 biopsy cores obtained with a 16-gauge needle), including evaluation by light microscopy, immunofluorescence (staining for immunoglobulins, complement factors and light chain components, as well as fibrinogen), and electron microscopy. A detailed description of the histological findings is given in table 1 and illustrated in figure 4. Briefly, the biopsy processed for light microscopy contained only three glomeruli, compared to six glomeruli investigated by immunofluorescence and one glomerulus by transmission electron microscopy. While the findings allowed a definitive diagnosis of IgA nephropathy, giving a meaningful MEST-C score was not possible.
Table 1Kidney biopsy findings.
| Light microscopy | Marginal biopsy with mainly renal medulla (approximately 80% of the biopsy). In the cortex, 3 glomeruli, 2 of them globally obliterated, the last one with a slightly widened mesangium without hypercellularity, tender capillary loops with one small capillary adhesion (black arrow) without an elevated number of granulocytes and with a free capsular space. |
| Tubular apparatus of the cortex well preserved, <5% tubular atrophy. Tender interstitium without inflammatory infiltrates. One well-preserved small interlobular artery. No preglomerular arterioles. No increased inflammatory infiltrates in the medulla. | |
| Immunofluorescence | Immunofluorescence examinations were performed on cryosections. Six assessed glomeruli with pronounced granular mesangial deposits of IgA and C3c, moderate kappa- and lambda-light chain and IgM deposits, low C5b-9 and fibrinogen, as well as minimal IgG deposits. No glomerular evidence of C1q and C4. One globally obliterated glomerulus with IgA deposits. Preglomerular vessels with low deposits of C5b-9. |
| Electron microscopy | One glomerulus was examined by electron microscopy. The glomerular capillary loops have a regular structure. No widening of the mesangium. The mesangium cells are activated. Several irregular, non-structured, electron-dense deposits of various size are found in the mesangium. Peripheral glomerular basement membranes partially thinned out and splittered in an arcade-shape. Otherwise, normal structure and size of the lamina densa. Podocyte foot processes overall well-preserved. Unremarkable endothelium. No subepithelial or subendothelial deposits. No fibrils. |
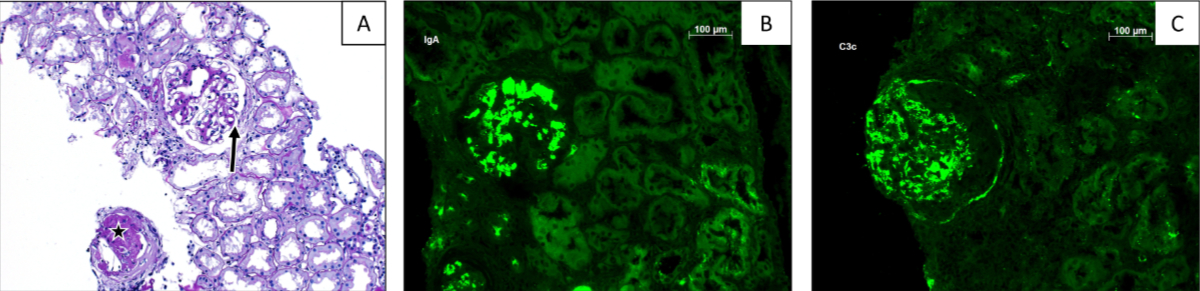
Figure 4Histopathological findings. (A) Light microscopy section showing one globally obliterated glomerulus (asterisk) and one with a slightly widened mesangium without hypercellularity, tender capillary loops with one small capillary adhesion (arrow), without an elevated number of granulocytes and with a free capsular space. Overall, minimal interstitial fibrosis, and tubular atrophy of less than 5%, PAS staining, 100×. Immunofluorescence showed pronounced granular mesangial deposits of IgA (B) and C3c (C) in all glomeruli (immunofluorescence, 400×).
Additionally, complement activity screening was performed by measuring serum levels of C3 and C4 (Siemens Healthcare) and testing the activity of all three complement pathways (i.e., the classical, lectin and alternative cascades) with a commercially available enzyme immunoassay (COMPL300, Complement System Screen kit, Wieslab®), showing normal activity within all three pathways.
As established nephroprotective/antiproteinuric therapies failed to control the IgA nephropathy, with persistently high proteinuria, there was an urgent need for expansion of the treatment targeting the (complement-mediated) inflammatory disease process as a main trigger of nephron loss (figure 1). Due to the dreaded side effects of systemic corticosteroids in this young, slender patient and the promising preliminary results from treating patients with IgA nephropathy with iptacopan, a request was made to the company (Novartis Pharma AG, Switzerland) to obtain iptacopan timely within a Managed Access Programme (MAP) for which the patient gave written informed consent. At that time, the patient could no longer be included in the ongoing clinical phase 3 trial of iptacopan for IgA nephropathy because the screening/inclusion phase of the APLLAUSE-IgA nephropathy study had already been closed for a while. Iptacopan was started at the end of March 2024 (200 mg twice a day). Four weeks before starting the treatment with iptacopan, the patient received meningococcal and pneumococcal vaccination, and two weeks before, dapagliflozin was stopped. Just two months after starting iptacopan, a reduction in proteinuria was evident, with a decrease of the UPCR to 50 mg/mmol (figure 3). In the further course of treatment with iptacopan, proteinuria decreased impressively to a target level of <0.3 g/day (i.e., the UPCR was repeatedly about 20 mg/mmol) (figure 3). Complete blockade of the alternative complement pathway in our case was confirmed and monitored after starting treatment with iptacopan (with COMPL300, Complement System Screen kit, Wieslab®). The iptacopan treatment is being continued and is well-tolerated, with no adverse effects. It is worth mentioning that kidney function is still normal, and proteinuria remains at a very low level since the start of the therapy 12 months ago. Furthermore, the steroid-sparing therapy with iptacopan is highly appreciated by the patient herself.
The research was conducted in accordance with the World Medical Association Declaration of Helsinki (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/). The retrospective review of patient data did not require ethical approval in accordance with local guidelines.
We report the first successful treatment of a 40-year-old female patient suffering from IgA nephropathy with iptacopan in Switzerland. It should be emphasised that this case supports the concept that the complement system, particularly the alternative complement pathway, plays an important role in the pathogenesis of IgA nephropathy. This is of great interest, as approved disease-specific therapies for patients with IgA nephropathy in Switzerland are currently scarce. Furthermore, complement-targeting therapies are generally well-tolerated.
Since the first description of IgA nephropathy in 1968 by Berger and Hinglais [26], dramatic improvements in the understanding of its pathogenesis have paved the way for the investigation of new therapeutic approaches, targeting different activation pathways. These pathways play an important role in mediating cellular inflammation and tissue damage, for example, the complement and endothelin systems [13–15]. Now, the understanding of the pathogenesis of IgA nephropathy is reflected in a model called the “multi-hit” model. To be more precise, the currently accepted “multi-hit” model of the pathogenesis of IgA nephropathy is a “four-hit” hypothesis, as genetically predisposed patients are assumed to suffer from pathophysiological changes, commonly referred to as “hits”, that induce disease development and progression, as summarised in [16, 27]. The “four-hit” model starts with the generation of GD-IgA1 (the first “hit”) within the mucosa-associated lymphoid tissue (MALT) of the gut and respiratory tract [28, 29], followed by the overproduction of autoantibodies that specifically recognise GD-IgA1 (the second “hit”) [30] and the formation of circulating immune complexes composed of GD-IgA1 and anti-GD-IgA1 autoantibodies, which deposit in the mesangium of the glomeruli (the third “hit”) [30, 31]. Then, these immune complexes can generate cytokine-, chemokine-, and complement-mediated glomerular inflammation and tissue destruction (the fourth “hit”), as illustrated in figure 1 [28, 30]. Over time, continued tissue injury maintained by the above-described processes leads to the characteristic findings of haematuria and proteinuria, as well as glomerular and tubulointerstitial scarring (i.e., sclerosis), ultimately causing kidney dysfunction and failure.
Local and systemic complement activation in patients with IgA nephropathy was first described decades ago; however, the significance of its pathogenesis was not recognised immediately [32]. Although this process is not yet fully understood, it is assumed that the deposition of complexes composed of GD-IgA1 and anti-GD-IgA1 autoantibodies can trigger local complement activation [33, 34]. Furthermore, several studies have provided evidence that activation of the alternative complement pathway is the “key driver” of the complement cascade in IgA nephropathy, as the hallmark of complement activation in IgA nephropathy is the co-localisation of C3 deposits with IgA, which is observed in about 90% of patients, including our reported case, as shown in figure 4B and 4C [33, 35, 36]. Nevertheless, in several patients, the co-deposition of C4d and C3 (without the deposition of C1q) suggests the potential involvement of the lectin pathway as the local initiator of complement cascade activation (figure 2) [13, 37, 38]. Interestingly, glomerular C3 deposits correlate with disease progression and may help to differentiate IgA nephropathy cases from individuals found to have asymptomatic deposits of IgA (i.e., 4–16% of the population [39]), as well as to decide which therapeutic strategy should be pursued. Finally, serum C3 levels are characteristically normal in patients suffering from IgA nephropathy, although studies have shown that elevated plasma levels of C3 activation markers, such as C3b, among others, are associated with worse kidney prognosis [40, 41]. In addition, a high serum C3/IgA ratio is correlated with poorer kidney outcome [42, 43]. In our case, serum C3 levels were repeatedly normal. Further, we tested the activity of all three complement pathways with a commercially available enzyme immunoassay before starting a complement-inhibiting therapy, noting normal activity within all three pathways. Thus, our case report supports the hypothesis that local alternative complement pathway activation is an important pathogenetic trigger in IgA nephropathy.
By targeting complement factor B, iptacopan suppresses the activation of the amplification loop of the alternative complement pathway, as highlighted in figure 2. It also blocks the amplification of classical and lectin pathway-dependent C5 activation, although it does not impact amplification-independent complement activation via these pathways. This may potentially lead to a lower infection risk in vaccinated patients compared with other proximal complement inhibitors [44], like pegcetacoplan (Aspaveli®, Apellis Pharmaceuticals, USA), which is approved by the FDA, EMA and Swissmedic for the treatment of patients with paroxysmal nocturnal haemoglobinuria [45]. Pegcetacoplan has been investigated in a phase 2 discovery study (ClinicalTrials.gov number NCT03453619) in patients with IgA nephropathy, but the results are still pending. The first approved complement inhibitor was eculizumab, which is a highly effective C5 inhibitor. Today, C5 blockade is the first-line treatment for patients with complement-mediated atypical haemolytic uremic syndrome. While the effects of eculizumab in IgA nephropathy have been inconsistent, a newer long-acting C5 inhibitor named ravulizumab is currently being evaluated in IgA nephropathy in a phase 2 study (ClinicalTrials.gov number NCT04564339), as summarised in [34]. Further, the C5a receptor 1 (C5aR1) inhibitor avacopan (Tavneos®, CLS Vifor, Switzerland) has recently been approved by the FDA, EMA and Swissmedic for the treatment of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Activation of the alternative complement pathway, which results in terminal C5a production, is a key component of the pathogenesis of ANCA-associated vasculitis [46]. Avacopan selectively suppresses the effects of C5a through C5aR1 blockade, including impeding neutrophil chemoattraction and activation [47]. Therefore, avacopan mainly acts as an anti-inflammatory agent preventing organ inflammation and damage [47]. The deleterious effects of kidney expression of C3aR and C5aR1 in patients suffering from IgA nephropathy, both in terms of disease activity and kidney damage severity, provide a rationale to investigate the pharmacological blockade of C5aR1 with avacopan [48, 49]. Thus, the potential efficacy of C5aR1 blockade in IgA nephropathy has also been evaluated in an open-label phase 2 pilot study [50]. Numerical reduction in the UPCR was observed in 6/7 IgA nephropathy patients at week 12 (about 50% in 3/7 patients), and 5/7 participants showed persistent improvements in the UPCR at week 24 [50]. Although the results of this small pilot study were significant, they were more modest than those found in ANCA-associated vasculitis [47]. Another reason not to treat our patient with avacopan was the fact that MAPs from companies for the off-label use of therapies must be closed by law upon approval of a therapeutic agent for any indication. This makes innovative therapies difficult to access timely, since applications processed by health insurance companies are time-consuming, including in Switzerland. Narsoplimab is a human monoclonal antibody targeting mannan-binding lectin-associated serine protease-2, the effector enzyme of the lectin pathway of the complement system, and was evaluated in a phase 3 randomised, double-blind, placebo-controlled trial in IgA nephropathy (ARTEMIS-IgA nephropathy, ClinicalTrials.gov number NCT03608033). However, the trial was discontinued, as treatment with narsoplimab did not result in a statistically significant reduction in proteinuria compared to placebo in the interim analysis [34]. Additional complement-targeting therapies under investigation aim to inhibit complement factor D and the expression of factor B and C3 genes and accelerate the decay of C3 convertase, and are summarised in [27].
What about other novel treatment options besides complement system blockade? Animal models have demonstrated that activation of the endothelin A receptor by endothelin-1 contributes to endothelial injury, inflammation, mesangial proliferation, glomerulosclerosis and tubulointerstitial fibrosis in chronic kidney disease [51]. Clinical trials evaluating endothelin receptor antagonists (ERAs) showed decreased proteinuria and improved kidney outcomes in diabetic and non-diabetic kidney diseases, as summarised in [52]. However, fluid retention needs to be taken into account when treating patients with ERAs [52]. An interesting approach is to add an SGLT-2 inhibitor to ERAs, which may help mitigate fluid retention and enhance the reduction of proteinuria [53]. Sparsentan is a dual endothelin and angiotensin receptor antagonist with high selectivity for the endothelin receptor type A and angiotensin II receptor type 1 (AT1-R) [54]. The nephroprotective effect of sparsentan in patients with IgA nephropathy was investigated in a phase 3 trial (PROTECT, ClinicalTrials.gov number NCT03762850) [8]. For this trial, 404 patients were randomised to receive sparsentan versus an active control arm treated with the AT1-R blocker irbesartan. It is worth mentioning that > 90% of the patients assigned to the irbesartan arm of the study reached the maximum allowed dose of 300 mg per day. The primary endpoint was change in proteinuria between the treatment groups at 36 weeks. Secondary endpoints included eGFR slope, changes in proteinuria, a composite of kidney failure, and safety and tolerability up to 110 weeks from randomisation [8]. Based on the positive interim analysis, sparsentan received accelerated approval from the FDA, EMA, and recently from Swissmedic for the treatment of proteinuria in patients with IgA nephropathy [7]. Meanwhile, the 2-year results of the PROTECT study have become available [8]. Patients in the sparsentan group had a slower rate of eGFR decline compared to the irbesartan group. Furthermore, the significant reduction of proteinuria from baseline at 36 weeks with sparsentan was maintained over the whole study period of 110 weeks. Importantly, treatment-emergent adverse events were balanced between sparsentan and irbesartan, with no unexpected safety problems [8]. Thus, the authors of the phase 3 trial stated that sparsentan is an effective and safe treatment for IgA nephropathy, with meaningful clinical benefit beyond renin-angiotensin system inhibition [8]. However, because of high-grade granular mesangial deposits of IgA and C3 in the kidney biopsy of our patient, we consciously chose a complement-blocking therapy with iptacopan and decided against sparsentan. Atrasentan is another ERA currently under investigation in a placebo-controlled, randomised phase 3 trial (ALIGN, Clinical.Trials.gov number NCT04573478) in IgA nephropathy patients [15]. The full phase 3 trial is still ongoing and will assess the effect of atrasentan versus placebo in patients with IgA nephropathy taking the maximum tolerated doses of renin-angiotensin system blockers on proteinuria at week 36 from baseline and change in eGFR at 136 weeks. Recently, the results of the interim analysis among the first 270 included patients who completed the week 36 visit were published [55]. There was a significantly greater percentage reduction in the UPCR relative to baseline with atrasentan compared to placebo [55], and based on the positive results of the interim analysis, atrasentan has been submitted to the FDA and granted accelerated review but has not yet been approved. Interestingly, an exploratory separate stratum of patients with IgA nephropathy using an SGLT-2 inhibitor is being enrolled to document the long-term effects of atrasentan compared to placebo in patients under an SGLT-2 inhibitor, but the results are pending [15]. As our patient was concerned about the onset of oedema and experienced skin problems after several weeks on the SGLT-2 inhibitor, treating IgA nephropathy with ERAs was not the preferred option in this case. To conclude the topic of further therapeutic options, another approach worth mentioning is the inhibition of B lymphocyte maturation to produce antibodies, including GD-IgA1 and anti-GD-IgA1 autoantibodies. There are different options to block this process in IgA nephropathy [56, 57]. One such option is to administer anti-CD19 antibodies. However, although a small series of IgA vasculitis patients seemed to have a response [58], a small, randomised trial of patients with IgA nephropathy demonstrated no impact on serum levels of GD-IgA1 or anti-GD-IgA1 autoantibodies, and adding rituximab to standard supportive therapy with renin-angiotensin system blockers did not significantly improve renal function or proteinuria assessed over one year [59]. It is worth mentioning at this point that the search for other B-cell inhibitors is the subject of current research, with programmes underway with proteosome inhibition, efforts to block receptor ligands such as the B lymphocyte stimulator (BLyS), also known as B-cell activating factor (BAFF), and A proliferation-inducing ligand (APRIL) or plasma cell receptors, as summarised in [16]. Today, reports from two different phase 2 studies suggest some efficacy in reducing GD-IgA1 levels and proteinuria in IgA nephropathy by atacicept which is a first-in-class fusion protein that can bind and neutralise BAFF and APRIL [60–62]. Furthermore, these results provided evidence to conduct a phase 3 study of atacicept in IgA nephropathy, which is ongoing (ORIGIN 3, Clinical.Trials.gov number NCT04716231).
Despite revolutionising advances in our understanding of the pathogenesis of IgA nephropathy, which directed us to more disease-specific treatment options as summarised above, there is still an urgent need for timely access to these new targeted treatments to move the management of IgA nephropathy towards a more personalised approach. This approach must slow the rate of disease progression, with the ultimate goal of preventing kidney failure during the lifespan of a patient suffering from IgA nephropathy. One important task for the future is to determine which therapy or combination of therapeutic options is most suitable for a given patient.
As there was no possibility to include our patient in the ongoing APPLAUSE-IgA nephropathy trial, the patient was included within the MAP of Novartis for iptacopan in IgA nephropathy. Nevertheless, it is important to highlight that today, this MAP is closed because the drug has been approved in the meanwhile by Swissmedic for the treatment of paroxysmal nocturnal haemoglobinuria. Nowadays, requests for iptacopan for the treatment of IgA nephropathy in Switzerland must be submitted to health insurance providers by the off-label use process (via the so-called article 71a–d of the health insurance ordinance [KVV]). Together with data from published clinical trials, case reports provide important information about the efficacy and safety of treating IgA nephropathy with iptacopan in Switzerland. Of course, we must note that clinical information obtained from a case report will not have the same level of evidence as a thoroughly conducted randomised, placebo-controlled clinical trial. Nevertheless, this information may help to locally prove the concept of a novel therapeutic option and support application processes to health insurance providers.
To the best of our knowledge, our case report is the first in Switzerland to show that selective inhibition of the alternative complement pathway in IgA nephropathy results in the significant and ongoing reduction of proteinuria, as well as preservation of kidney function, and therefore, supports the innovative concept of targeting the alternative complement pathway with iptacopan to treat IgA nephropathy.
All the data analysed for this case report are included in this article. Further enquiries can be directed to the corresponding author.
Author contributions: All authors contributed equally to the literature review and the text of the manuscript. LI, MD and PHM were responsible for the patient’s care and treatment. TM and HH were responsible for the correct interpretation of the biopsy results.
The patient gave written informed consent for the publication of this case report, including details of the medical case and any accompanying images.
The patient was provided with iptacopan therapy via the Managed Access Programme (MAP) of Novartis Pharma AG, Basel, Switzerland. Novartis was not involved in the case study design, data collection, analysis and interpretation, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests. Additionally, no further funding was necessary.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. PHM took part in an IgA nephropathy Advisory Board from Vifor on 9 September 2024. No other potential conflicts of interest related to the content of this manuscript were disclosed.
1. McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol Dial Transplant. 2011 Feb;26(2):414–30. doi: https://doi.org/10.1093/ndt/gfq665
2. Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013 Jun;368(25):2402–14. doi: https://doi.org/10.1056/NEJMra1206793
3. Pitcher D, Braddon F, Hendry B, Mercer A, Osmaston K, Saleem MA, et al. Long-Term Outcomes in IgA Nephropathy. Clin J Am Soc Nephrol. 2023 Jun;18(6):727–38. doi: https://doi.org/10.2215/CJN.0000000000000135
4. Reich HN, Troyanov S, Scholey JW, Cattran DC; for the Toronto Glomerulonephritis Registry. Remission of proteinuria improves prognosis in IgA nephropathy. J Am Soc Nephrol. 2007 Dec;18(12):3177–83. doi: https://doi.org/10.1681/ASN.2007050526
5. Suzuki H, Kikuchi M, Koike K, Komatsu H, Matsuzaki K, Takahashi K, et al.; Committee of Clinical Practical Guideline for IgA Nephropathy 2020. A digest from evidence-based clinical practice guideline for IgA nephropathy 2020. Clin Exp Nephrol. 2021 Dec;25(12):1269–76. doi: https://doi.org/10.1007/s10157-021-02095-8
6. Zhang H, Barratt J. Is IgA nephropathy the same disease in different parts of the world? Semin Immunopathol. 2021 Oct;43(5):707–15. doi: https://doi.org/10.1007/s00281-021-00884-7
7. Heerspink HJ, Radhakrishnan J, Alpers CE, Barratt J, Bieler S, Diva U, et al.; PROTECT Investigators. Sparsentan in patients with IgA nephropathy: a prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial. Lancet. 2023 May;401(10388):1584–94. doi: https://doi.org/10.1016/S0140-6736(23)00569-X
8. Rovin BH, Barratt J, Heerspink HJ, Alpers CE, Bieler S, Chae DW, et al.; DUPRO steering committee and PROTECT Investigators. Efficacy and safety of sparsentan versus irbesartan in patients with IgA nephropathy (PROTECT): 2-year results from a randomised, active-controlled, phase 3 trial. Lancet. 2023 Dec;402(10417):2077–90. doi: https://doi.org/10.1016/S0140-6736(23)02302-4
9. Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. Executive summary of the KDIGO 2021 Guideline for the Management of Glomerular Diseases. Kidney Int. 2021 Oct;100(4):753–79. doi: https://doi.org/10.1016/j.kint.2021.05.015
10. Barratt J, Lafayette R, Kristensen J, Stone A, Cattran D, Floege J, et al.; NefIgArd Trial Investigators. Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int. 2023 Feb;103(2):391–402. doi: https://doi.org/10.1016/j.kint.2022.09.017
11. Lafayette R, Kristensen J, Stone A, Floege J, Tesař V, Trimarchi H, et al.; NefIgArd trial investigators. Efficacy and safety of a targeted-release formulation of budesonide in patients with primary IgA nephropathy (NefIgArd): 2-year results from a randomised phase 3 trial. Lancet. 2023 Sep;402(10405):859–70. doi: https://doi.org/10.1016/S0140-6736(23)01554-4
12. Cheung CK, Alexander S, Reich HN, Selvaskandan H, Zhang H, Barratt J. The pathogenesis of IgA nephropathy and implications for treatment. Nat Rev Nephrol. 2025 Jan;21(1):9–23. doi: https://doi.org/10.1038/s41581-024-00885-3
13. Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current Understanding of the Role of Complement in IgA Nephropathy. J Am Soc Nephrol. 2015 Jul;26(7):1503–12. doi: https://doi.org/10.1681/ASN.2014101000
14. Rizk DV, Maillard N, Julian BA, Knoppova B, Green TJ, Novak J, et al. The Emerging Role of Complement Proteins as a Target for Therapy of IgA Nephropathy. Front Immunol. 2019 Mar;10:504. doi: https://doi.org/10.3389/fimmu.2019.00504
15. Kohan DE, Barratt J, Heerspink HJ, Campbell KN, Camargo M, Ogbaa I, et al. Targeting the Endothelin A Receptor in IgA Nephropathy. Kidney Int Rep. 2023 Aug;8(11):2198–210. doi: https://doi.org/10.1016/j.ekir.2023.07.023
16. Caster DJ, Lafayette RA. The Treatment of Primary IgA Nephropathy: Change, Change, Change. Am J Kidney Dis. 2024 Feb;83(2):229–40. doi: https://doi.org/10.1053/j.ajkd.2023.08.007
17. Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc Natl Acad Sci USA. 2019 Apr;116(16):7926–31. doi: https://doi.org/10.1073/pnas.1820892116
18. Risitano AM, Röth A, Soret J, Frieri C, de Fontbrune FS, Marano L, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol. 2021 May;8(5):e344–54. doi: https://doi.org/10.1016/S2352-3026(21)00028-4
19. Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study. Blood Adv. 2022 Aug;6(15):4450–60. doi: https://doi.org/10.1182/bloodadvances.2022006960
20. Peffault de Latour R, Röth A, Kulasekararaj AG, Han B, Scheinberg P, Maciejewski JP, et al. Oral Iptacopan Monotherapy in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2024 Mar;390(11):994–1008. doi: https://doi.org/10.1056/NEJMoa2308695
21. Wong E, Nester C, Cavero T, Karras A, Le Quintrec M, Lightstone L, et al. Efficacy and Safety of Iptacopan in Patients With C3 Glomerulopathy. Kidney Int Rep. 2023 Sep;8(12):2754–64. doi: https://doi.org/10.1016/j.ekir.2023.09.017
22. Bomback AS, Kavanagh D, Vivarelli M, Meier M, Wang Y, Webb NJ, et al. Alternative Complement Pathway Inhibition With Iptacopan for the Treatment of C3 Glomerulopathy-Study Design of the APPEAR-C3G Trial. Kidney Int Rep. 2022 Aug;7(10):2150–9. doi: https://doi.org/10.1016/j.ekir.2022.07.004
23. Kavanagh D, Greenbaum LA, Bagga A, Karki RG, Chen CW, Vasudevan S, et al. Design and Rationale of the APPELHUS Phase 3 Open-Label Study of Factor B Inhibitor Iptacopan for Atypical Hemolytic Uremic Syndrome. Kidney Int Rep. 2023 Apr;8(7):1332–41. doi: https://doi.org/10.1016/j.ekir.2023.04.029
24. Zhang H, Rizk DV, Perkovic V, Maes B, Kashihara N, Rovin B, et al. Results of a randomized double-blind placebo-controlled Phase 2 study propose iptacopan as an alternative complement pathway inhibitor for IgA nephropathy. Kidney Int. 2024 Jan;105(1):189–99. doi: https://doi.org/10.1016/j.kint.2023.09.027
25. Perkovic V, Barratt J, Rovin B, Kashihara N, Maes B, Zhang H, et al.; APPLAUSE-IgAN Investigators. Alternative Complement Pathway Inhibition with Iptacopan in IgA Nephropathy. N Engl J Med. 2025 Feb;392(6):531–43. doi: https://doi.org/10.1056/NEJMoa2410316
26. Berger J, Hinglais N. [Intercapillary deposits of IgA-IgG]. J Urol Nephrol (Paris). 1968 Sep;74(9):694–5.
27. Duval A, Caillard S, Frémeaux-Bacchi V. The complement system in IgAN: mechanistic context for therapeutic opportunities. Nephrol Dial Transplant. 2023 Nov;38(12):2685–93. doi: https://doi.org/10.1093/ndt/gfad140
28. Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, et al. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front Immunol. 2016 Apr;7:117. doi: https://doi.org/10.3389/fimmu.2016.00117
29. Gesualdo L, Di Leo V, Coppo R. The mucosal immune system and IgA nephropathy. Semin Immunopathol. 2021 Oct;43(5):657–68. doi: https://doi.org/10.1007/s00281-021-00871-y
30. Boyd JK, Cheung CK, Molyneux K, Feehally J, Barratt J. An update on the pathogenesis and treatment of IgA nephropathy. Kidney Int. 2012 May;81(9):833–43. doi: https://doi.org/10.1038/ki.2011.501
31. Matsumoto Y, Aryal RP, Heimburg-Molinaro J, Park SS, Wever WJ, Lehoux S, et al. Identification and characterization of circulating immune complexes in IgA nephropathy. Sci Adv. 2022 Oct;8(43):eabm8783. doi: https://doi.org/10.1126/sciadv.abm8783
32. Evans DJ, Williams DG, Peters DK, Sissons JG, Boulton-Jones JM, Ogg CS, et al. Glomerular deposition of properdin in Henoch-Schönlein syndrome and idiopathic focal nephritis. BMJ. 1973 Aug;3(5875):326–8. doi: https://doi.org/10.1136/bmj.3.5875.326
33. Poppelaars F, Faria B, Schwaeble W, Daha MR. The Contribution of Complement to the Pathogenesis of IgA Nephropathy: Are Complement-Targeted Therapies Moving from Rare Disorders to More Common Diseases? J Clin Med. 2021 Oct;10(20):4715. doi: https://doi.org/10.3390/jcm10204715
34. Caravaca-Fontán F, Gutiérrez E, Sevillano AM, Praga M. Targeting complement in IgA nephropathy. Clin Kidney J. 2023 Dec;16 Suppl 2:ii28–39. doi: https://doi.org/10.1093/ckj/sfad198
35. Schmitt R, Ståhl AL, Olin AI, Kristoffersson AC, Rebetz J, Novak J, et al. The combined role of galactose-deficient IgA1 and streptococcal IgA-binding M Protein in inducing IL-6 and C3 secretion from human mesangial cells: implications for IgA nephropathy. J Immunol. 2014 Jul;193(1):317–26. doi: https://doi.org/10.4049/jimmunol.1302249
36. Chiu YL, Lin WC, Shu KH, Fang YW, Chang FC, Chou YH, et al. Alternative Complement Pathway Is Activated and Associated with Galactose-Deficient IgA1 Antibody in IgA Nephropathy Patients. Front Immunol. 2021 Jun;12:638309. doi: https://doi.org/10.3389/fimmu.2021.638309
37. Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006 Jun;17(6):1724–34. doi: https://doi.org/10.1681/ASN.2005090923
38. Barratt J, Lafayette RA, Zhang H, Tesar V, Rovin BH, Tumlin JA, et al. IgA nephropathy: the lectin pathway and implications for targeted therapy. Kidney Int. 2023 Aug;104(2):254–64. doi: https://doi.org/10.1016/j.kint.2023.04.029
39. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003 Jun;63(6):2286–94. doi: https://doi.org/10.1046/j.1523-1755.63.6s.2.x
40. Zwirner J, Burg M, Schulze M, Brunkhorst R, Götze O, Koch KM, et al. Activated complement C3: a potentially novel predictor of progressive IgA nephropathy. Kidney Int. 1997 Apr;51(4):1257–64. doi: https://doi.org/10.1038/ki.1997.171
41. Janssen U, Bahlmann F, Köhl J, Zwirner J, Haubitz M, Floege J. Activation of the acute phase response and complement C3 in patients with IgA nephropathy. Am J Kidney Dis. 2000 Jan;35(1):21–8. doi: https://doi.org/10.1016/S0272-6386(00)70296-4
42. Mizerska-Wasiak M, Małdyk J, Rybi-Szumińska A, Wasilewska A, Miklaszewska M, Pietrzyk J, et al. Relationship between serum IgA/C3 ratio and severity of histological lesions using the Oxford classification in children with IgA nephropathy. Pediatr Nephrol. 2015 Jul;30(7):1113–20. doi: https://doi.org/10.1007/s00467-014-3024-z
43. Kawasaki Y, Maeda R, Ohara S, Suyama K, Hosoya M. Serum IgA/C3 and glomerular C3 staining predict severity of IgA nephropathy. Pediatr Int. 2018 Feb;60(2):162–7. doi: https://doi.org/10.1111/ped.13461
44. Ispasanie E, Muri L, Schubart A, Thorburn C, Zamurovic N, Holbro T, et al. Alternative Complement Pathway Inhibition Does Not Abrogate Meningococcal Killing by Serum of Vaccinated Individuals. Front Immunol. 2021 Oct;12:747594. doi: https://doi.org/10.3389/fimmu.2021.747594
45. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021 Mar;384(11):1028–37. doi: https://doi.org/10.1056/NEJMoa2029073
46. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009 Feb;20(2):289–98. doi: https://doi.org/10.1681/ASN.2008050497
47. Jayne DR, Merkel PA, Bekker P. Avacopan for the Treatment of ANCA-Associated Vasculitis. Reply [Reply]. N Engl J Med. 2021 May;384(21):e81.
48. Liu L, Zhang Y, Duan X, Peng Q, Liu Q, Zhou Y, et al. C3a, C5a renal expression and their receptors are correlated to severity of IgA nephropathy. J Clin Immunol. 2014 Feb;34(2):224–32. doi: https://doi.org/10.1007/s10875-013-9970-6
49. Zhang Y, Yan X, Zhao T, Xu Q, Peng Q, Hu R, et al. Targeting C3a/C5a receptors inhibits human mesangial cell proliferation and alleviates immunoglobulin A nephropathy in mice. Clin Exp Immunol. 2017 Jul;189(1):60–70. doi: https://doi.org/10.1111/cei.12961
50. Bruchfeld A, Magin H, Nachman P, Parikh S, Lafayette R, Potarca A, et al. C5a receptor inhibitor avacopan in immunoglobulin A nephropathy-an open-label pilot study. Clin Kidney J. 2022 Jan;15(5):922–8. doi: https://doi.org/10.1093/ckj/sfab294
51. Kohan DE, Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014 Nov;86(5):896–904. doi: https://doi.org/10.1038/ki.2014.143
52. Smeijer JD, Kohan DE, Webb DJ, Dhaun N, Heerspink HJ. Endothelin receptor antagonists for the treatment of diabetic and nondiabetic chronic kidney disease. Curr Opin Nephrol Hypertens. 2021 Jul;30(4):456–65. doi: https://doi.org/10.1097/MNH.0000000000000716
53. Heerspink HJ, Kohan DE, de Zeeuw D. New insights from SONAR indicate adding sodium glucose co-transporter 2 inhibitors to an endothelin receptor antagonist mitigates fluid retention and enhances albuminuria reduction. Kidney Int. 2021 Feb;99(2):346–9. doi: https://doi.org/10.1016/j.kint.2020.09.026
54. Kowala MC, Murugesan N, Tellew J, Carlson K, Monshizadegan H, Ryan C, et al. Novel dual action AT1 and ETA receptor antagonists reduce blood pressure in experimental hypertension. J Pharmacol Exp Ther. 2004 Apr;309(1):275–84. doi: https://doi.org/10.1124/jpet.103.055855
55. Heerspink HJ, Jardine M, Kohan DE, Lafayette RA, Levin A, Liew A, et al.; ALIGN Study Investigators. Atrasentan in Patients with IgA Nephropathy. N Engl J Med. 2025 Feb;392(6):544–54. doi: https://doi.org/10.1056/NEJMoa2409415
56. Gutiérrez E, Carvaca-Fontán F, Luzardo L, Morales E, Alonso M, Praga M. A Personalized Update on IgA Nephropathy: A New Vision and New Future Challenges. Nephron J. 2020;144(11):555–71. doi: https://doi.org/10.1159/000509997
57. Knoppova B, Reily C, King RG, Julian BA, Novak J, Green TJ. Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment. J Clin Med. 2021 Sep;10(19):4501. doi: https://doi.org/10.3390/jcm10194501
58. Fenoglio R, Sciascia S, Naretto C, De Simone E, Del Vecchio G, Ferro M, et al. Rituximab in severe immunoglobulin-A vasculitis (Henoch-Schonlein) with aggressive nephritis. Clin Exp Rheumatol. 2020;38 Suppl 124(2):195-200.
59. Lafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J Am Soc Nephrol. 2017 Apr;28(4):1306–13. doi: https://doi.org/10.1681/ASN.2016060640
60. Barratt J, Tumlin J, Suzuki Y, Kao A, Aydemir A, Pudota K, et al.; JANUS study investigators. Randomized Phase II JANUS Study of Atacicept in Patients With IgA Nephropathy and Persistent Proteinuria. Kidney Int Rep. 2022 May;7(8):1831–41. doi: https://doi.org/10.1016/j.ekir.2022.05.017
61. Lafayette R, Barbour S, Israni R, Wei X, Eren N, Floege J, et al. A phase 2b, randomized, double-blind, placebo-controlled, clinical trial of atacicept for treatment of IgA nephropathy. Kidney Int. 2024 Jun;105(6):1306–15. doi: https://doi.org/10.1016/j.kint.2024.03.012
62. Barratt J, Barbour SJ, Brenner RM, Cooper K, Wei X, Eren N, et al.; ORIGIN Phase 2b Investigators. Long-Term Results from an Open-Label Extension Study of Atacicept for the Treatment of IgA Nephropathy. J Am Soc Nephrol. 2025 Apr;36(4):679–87. doi: https://doi.org/10.1681/ASN.0000000541