From gaps to compliance: a 12-year retrospective cohort study of trends in mismatch
repair protein testing and Lynch syndrome identification in colorectal cancer in Central
Switzerland
DOI: https://doi.org/https://doi.org/10.57187/s.4345
Bettina Marturet Fendta,
Celina
Geissb,
Muriel Elhaib,
Joachim Diebolda,
Alexander
Vogetsedera
a Institute of Pathology, Cantonal
Hospital Lucerne, Lucerne, Switzerland
b University Hospital Zurich,
University of Zurich, Department of Rheumatology, Centre of Experimental
Rheumatology, Zurich, Switzerland
Summary
STUDY AIM: Alongside an analysis of
incidence trends in colorectal cancer and Lynch syndrome over time, the study sought
to evaluate the implementation and trends of reflex testing for mismatch
repair proteins and key mutations in relevant genes (BRAF, KRAS, NRAS) in
colorectal cancer in Central Switzerland from 2011 to 2022, specifically assessing
adherence to the Swiss Academy for Quality in Medicine (SAQM) guidelines, in
order to identify any gaps or inconsistencies in testing practices that may
hinder the diagnosis of Lynch syndrome or microsatellite instability,
highlighting areas requiring improvements for optimal patient care.
METHODS: This retrospective study enrolled
2602 patients with 2673 histologically confirmed colorectal cancers. Data
collection from the Central Switzerland Cancer Registry included demographic,
molecular and immunohistochemical profiles of all histologically confirmed colorectal
cancers over the analysed 12-year period. Statistical analyses were
performed using R (v4.3.1) with the tidyverse package. Normality was
assessed with the Shapiro-Wilk test and non-parametric comparisons were made
using the Wilcoxon rank-sum test. Chi-square and Fisher’s exact tests were used
for categorical variables, while Poisson and binomial regression models were
used to evaluate temporal trends.
RESULTS: Of 2673 tumours analysed, 76% were
tested for mismatch repair proteins, with testing rates improving significantly
from 58% in 2011 to >99% in 2022. Among these, 14% showed a mismatch repair
protein deficiency, with 77% being MLH1-related and 23% non-MLH1-related,
categorising them as Lynch-suspected. 73% (n = 257) of the MLH1-deficient tumours
underwent further molecular testing for BRAF mutations. Among these, 33%
showed no mutation, also categorising them as Lynch-suspected, while the remaining
67% were categorised as sporadic. In total, 6% of the tested tumours were
categorised as Lynch-suspected and required further testing and/or genetic counselling.
Statistical estimates suggest that among the non-tested tumours, 88 cases could
potentially harbour a microsatellite instability, including approximately 5
Lynch-suspected cases. Additionally, in 44 cases, incorrect mismatch repair
proteins were tested, potentially leading to missed microsatellite instability.
Among the 59 tumours that did not undergo BRAF testing, approximately 20
may have been Lynch-suspected and missed due to insufficient testing. Tumour
incidence and the proportion of Lynch-suspected tumours among all tumours
remained stable over time, without cantonal hotspots.
CONCLUSIONS: Remarkable progress in colorectal
cancer diagnostics across Central Switzerland could be demonstrated, leading to
a near-complete compliance with guidelines for mismatch repair proteins and
molecular testing by 2022. This high adherence to guidelines provides a solid
foundation for better personalised surveillance and treatment, ultimately
improving the quality of care for colorectal cancer patients in the region.
However, during the early years of the study some gaps existed, particularly in
testing practices for rectal cancers and incomplete molecular follow-up,
potentially missing some patients with a microsatellite instability, who could
have benefited from different therapies, and Lynch syndrome patients, who
together with their families could have benefited from tighter surveillance.
Abbreviations
- MRPd
-
mismatch repair protein-deficient
- MRPp
-
mismatch repair protein-proficient
- MSI-H
-
high microsatellite instability
- MSI-L
-
low microsatellite instability
Introduction
Lynch syndrome, also known as hereditary
non-polyposis colorectal cancer (HNPCC), is the most common hereditary cause of
colorectal cancer, responsible for approximately 3% of all colorectal cancer
cases [1, 2]. It
is caused by inherited autosomal dominant germline mutations in genes encoding mismatch
repair proteins (MRPs), which are involved in the repair of deoxyribonucleic
acid (DNA) replication errors. Its malfunction results in an accumulation of
such errors in areas prone to replication slippage, particularly in repetitive
DNA sequences known as microsatellites, resulting in variations in their length.
This phenomenon is termed microsatellite instability (MSI) [3]. Rarely,
alternative mechanisms can also lead to Lynch syndrome, such as the
inactivation of MSH2 after the 3’ terminal deletion of the gene or the constitutive hypermethylation
syndrome [4–6].
Microsatellite instability is observed in
nearly 15% of colorectal carcinomas and nearly all colorectal tumours
associated with Lynch syndrome [7]. Based
on the degree of instability, microsatellite status can be classified into
three categories: high microsatellite instability (MSI-H), low microsatellite
instability (MSI-L) and microsatellite stability. MSI-H tumours typically show significant
instability in more than 30% of tested microsatellite markers, depending on the
test, with the Bethesda panel being the most commonly used [8]. MSI-H
is strongly associated with defective mismatch repair proteins and is
frequently observed in Lynch syndrome-associated tumours. MSI-L tumours exhibit
a lesser number of instability events, below the 30% threshold, are less
clearly associated with Lynch syndrome and their clinical implications are less
well-defined in terms of prognosis and treatment response. Microsatellite-stable
tumours display no detectable microsatellite instability, indicating an intact mismatch
repair mechanism, and represent the majority of colorectal cancer cases.
In daily practice, colorectal cancer can be
categorised as either mismatch repair protein-proficient (MRPp) or mismatch
repair protein-deficient (MRPd), using immunohistochemistry as a surrogate
marker for microsatellite stability or instability. The four main mismatch
repair (MMR) genes associated with Lynch syndrome are MLH1, MSH2,
MSH6 and PMS2. Their protein products (MLH1, MSH2, MSH6 and PMS2)
can be assessed by immunohistochemistry. Mismatch repair protein-deficienttumours
can be further categorised into likely somatic or likely syndromic.
Somatic tumours arise mainly due to silencing of the MLH1 gene through methylation
of its promoter and are strongly associated with a BRAF mutation. Both BRAF
mutation and MLH1 methylation analyses are routinely used to confirm the
somatic nature of the tumour and rule out Lynch syndrome. Additionally, microsatellite
instability can also result as a secondary event due to a DNA polymerase ε
(POLE) mutation [9]. Occasionally
some patients show a so-called “Lynch-like phenotype”, which is characterised
by a mismatch repair protein deficiency without BRAF mutations or
promoter hypermethylation, but with somatic double hit mutations instead of a
germline mutation [10, 11].
Identifying tumours with a microsatellite
instability and individuals with Lynch syndrome is crucial, since the latter show
an earlier age of cancer onset and a higher risk for malignancy across multiple
organs, mainly colon and endometrium. Additionally, Lynch-associated cancers
exhibit distinct prognostic features compared to sporadic cancers [12]. So,
these individuals benefit from earlier and more frequent cancer screening and
surveillance protocols [13–15].
Some microsatellite-instable tumours belong
to the recently introduced hypermutated molecular subtype [16]. The
hypermutated nature of these tumours leads to the formation of neoantigens,
which in turn trigger an immune response, a feature exploited by immune
checkpoint therapies such as pembrolizumab and nivolumab, as they can stimulate
the immune system to target cancer cells [17, 18]. Lately,
impressive results have been achieved in the treatment of locally advanced
rectal cancer [19]; therefore
it is now increasingly essential to assess for microsatellite instability, as
this can guide an appropriate individualised treatment plan for each patient.
Given the clinical implications of microsatellite
instability and Lynch syndrome, the Swiss Academy for Quality in Medicine (SAQM)
introduced guidelines in 2011 to improve the detection of mismatch repair
protein-deficient tumours and Lynch syndrome in newly diagnosed colorectal
cancer [20]. Initially,
microsatellite instability testing was recommended only for patients meeting
the Revised Bethesda criteria, which included at least one of the following: colorectal
cancer before age 50; multiple hereditary non-polyposis colorectal cancer-related
cancers; colorectal cancer with microsatellite instability-associated histology
before age 60; colorectal cancer / hereditary non-polyposis colorectal cancer
in a first-degree relative before age 50; colorectal cancer / hereditary
non-polyposis colorectal cancer in at least two first- or second-degree
relatives at any age [21]. Testing
could be performed either by immunohistochemistry for MLH1, PMS2, MSH2 and MSH6
or by PCR-based microsatellite instability analysis using the Bethesda panel
(BAT-25, BAT-26, D2S123, D5S346, D17S250). However, adherence to these criteria
was inconsistent, leading to potential underdiagnosis.
To address these limitations, the SAQM updated
its guidelines in 2019, implementing universal reflex testing for all newly
diagnosed colorectal cancer cases, regardless of clinical criteria [22]. The
revised protocol maintained both immunohistochemistry and PCR-based microsatellite
instability analysis as standard testing methods.
The Lucerne Cantonal Hospital (LUKS) takes
care of a population of around 700,000 [23]. In
arithmetical terms, the LUKS therefore manages about 2300 people with Lynch
syndrome.
At the Institute of Pathology LUKS,
colorectal carcinomas are tested for microsatellite instability according to a
standardised algorithm. Immunohistochemistry for mismatch repair protein (MLH1,
MSH2, MSH6 and PMS2) is primarily performed on the biopsy specimen. If
insufficient or inconclusive, the resection specimen is used. Tumours with
retained expression of all mismatch repair protein are classified as MRPp, indicative
of microsatellite stability. MRPp carcinomas requiring chemotherapy may be
further tested for BRAF, KRAS and NRAS mutations upon
clinical request. In contrast, tumours with loss of expression of any mismatch
repair protein are classified as MRPd, suggestive of microsatellite instability.
Cases with MLH1-loss are reflex-tested mainly for BRAF mutation and/or MLH1
methylation. If a BRAF mutation or MLH1 promoter hypermethylation
is identified, a sporadic microsatellite-instable tumour is diagnosed, since
these are both strong predictors of sporadic origin [24]. If
neither a BRAF mutation nor MLH1 promoter hypermethylation is
detected, the patient is referred for genetic counselling to rule out Lynch
syndrome. Younger patients, typically under 50 years old, are recommended for
genetic counselling regardless of BRAF status [25].
Germline mutation analysis is conducted by
geneticists at external institutions, such as the University of Basel or the University
of Zurich, or at companies, such as Genetica. The Lucerne Cantonal Hospital
offers limited family counselling in cases of diagnosed Lynch syndrome.
The aim of the present study was to
evaluate the implementation of reflex testing for mismatch repair protein and mutations
in key genes such as BRAF, KRAS and RAS in colorectal
cancer in Central Switzerland. It set out to identify any gaps or
inconsistencies in testing practices, especially regarding reflex molecular
testing for diagnosing or ruling out Lynch syndrome and trends in the
implementation of such testing over the 12-year period. The study also compared
clinical and demographic characteristics between different cohorts (MRPd vs
MRPp, Lynch-suspected vs sporadic) and identified trends in the incidence of colorectal
cancer and Lynch syndrome over the years. We expected that the study findings would
help to identify any gaps or inconsistencies in testing practices that may
hinder the diagnosis of Lynch syndrome or microsatellite instability,
highlighting areas requiring improvements for optimal patient care.
Methods
Study setting and design
In total, 2602 patients with 2673
histologically confirmed colorectal carcinomas were enrolled in this
retrospective study. A new database was created representing the first
comprehensive repository of patients with colorectal carcinomas in Central Switzerland,
encompassing not only demographic
data but also the molecular and immunohistochemical profiles of the tumours.
Most cases were diagnosed at the Lucerne
Cantonal Hospital, for which full access to histological and molecular
pathological examination reports were available through the “Pathowin” archive.
Pathowin serves as the primary data repository for all histopathological and
molecular analyses conducted within the Lucerne Cantonal Hospital Group
catchment area. For patients residing in Central Switzerland but treated
outside their home canton, data availability relied on mandatory reporting of
cancer cases by external clinicians to the Cancer Registry of Central
Switzerland.
Inclusion criteria and data collection
The inclusion criteria consisted of
individuals residing in Central Switzerland at the time of diagnosis –
encompassing the cantons of Lucerne, Obwalden, Nidwalden and Uri – with
histologically confirmed adenocarcinomas of the colon, all under ICD-O codes
C18.0, C18.2, C18.3, C18.4, C18.5, C18.6, C18.7, C18.8, C18.9, C19.9, C20.9,
C21.1 and C21.8 ranging from the caecal pole to the rectoanal junction, as
defined by ICD-10 versions 3.1 and 3.2. Metachronous and synchronous carcinomas
were included. Only invasive adenocarcinomas stage pT1 or higher
according to the definition of the UICC TNM classification were considered. Tumours
of
the left flexure, descending colon, sigmoid and rectosigmoid carcinomas were
classified as left-sided; those of the caecum, ascending colon, right flexure
and transverse colon as right-sided; those of the rectum were considered a
separate category. Missing or ambiguous data in any category was classified as
unknown and excluded from the pertinent calculations.
Data retrieval was conducted both manually
and through automated integration from the cancer registry. Manual data extraction
from Pathowin provided information on the expression status of the mismatch
repair proteins MLH1, PMS2, MSH2 and MSH6 (expressed, not expressed, not
tested, unknown); the mutation status of the genes KRAS, BRAF, NRAS
(mutated, not mutated, not tested, unknown); the methylation status of the MLH1
promotor (methylated, not methylated, not tested, unknown); microsatellite instability
PCR results (microsatellite stability, MSI-low, MSI-high, not tested, unknown)
and type of resected specimen (biopsy, resection specimen). Additionally, data
from the cancer registry was automatically pooled including tumour
identification number, number of tumours per patient, sex (male, female),
incidence age, canton of residence at the time of incidence (Lucerne, Uri,
Obwalden, Nidwalden), year of occurrence, ICD-10 and ICD-O location code, ICD-O
morphology codes and tumour grade (G1, G2, G3, unknown).
Each registry entry underwent meticulous
verification against the original pathological reports to ensure data accuracy.
Cases diagnosed within the Lucerne Cantonal Hospital Group allowed for direct
cross-validation using Pathowin, ensuring data completeness. However, for
externally diagnosed cases, the analysis was restricted to information
available from the cancer registry. If histological reports with mismatch
repair protein status or molecular reports on mutation status were incomplete
or not transmitted by external institutions, these data were not accessible.
This limitation primarily affected manually retrieved variables, whereas
automated data from the cancer registry remained complete.
To address missing data, we systematically
reviewed all available sources, including pathological reports, molecular
testing reports, tumour board summaries and oncological records. If data remained
unavailable despite these efforts, the respective data fields were classified
as “unknown” and excluded from the affected analyses.
The Cancer Registry of Central Switzerland,
which provided part of the data, has systematically documented patient data of colorectal
cancer since 2010, with validated data extending only until mid-2023 at the
time of writing. So, the study analysed the data of all 12 years available –
from 2011 to 2022 – in full.
Laboratory methods
At the Lucerne Cantonal Hospital, immunohistochemistry
was performed on FFPE samples on a Bond III machine (Leica) following
internally established and validated protocols. The following antibodies were
used: MLH1 (NCL-L-MLH1, Novocastra), PMS2 (BSB-BSB 2124, BioSB), MSH6
(AC-0047EU, Epitomics) and MSH2 (286M-16, Cell Marque). The absence of nuclear
staining in the presence of internal controls, as well as abnormal staining
patterns like dotted patterns or cytoplasmic staining, were interpreted as
negative. Mutational analysis for BRAF, KRAS and NRAS was
performed using mainly Sanger sequencing (home-made assay) and recently with
the Oncomine Precision Assay v3 GX (Thermo Fisher Scientific). The microsatellite
instability was analysed with a multiplex PCR assay targeting the Bethesda
markers [26]. MLH1
promoter methylation was assessed with Pyromark Gold Q24 Reagents 5x24 (971802,
Quiagen).
Statistical analysis
All statistical analyses were conducted
using R (v4.3.1), using the tidyverse framework, including dplyr
(v1.1.4) for data wrangling and ggplot2 (v3.5.1) for data visualisation.
Normality of data was assessed using the Shapiro-Wilk test and a non-parametric
test, the Wilcoxon rank-sum test with continuity correction, was employed for
non-normally distributed data. A chi-square test was used to compare the
distribution of categorical variables between two cohorts, with Fisher’s exact
test employed for smaller sample sizes. Poisson and binomial regression models
were used to evaluate temporal trends in tumour incidence, Lynch tumour
frequency and tumour testing proportions. Models estimated yearly changes with
95% confidence intervals; binomial confidence intervals for observed data were
calculated using the Wilson method. Data on the permanent resident population
by canton per year, as published by the Federal Statistical Office (Bundesamt
für Statistik), was used. [27]
Statistical significance was defined as p <0.05.
Where not specified, results can be assumed to have reached a significance
level of p <0.01.
Ethics approval
The study was approved by the Ethics Committee
Northwestern and Central Switzerland (EKNZ).
Results
General patient and tumour characteristics
Cohort characteristics are summarised in table
1. During the analysed period from 2011 to 2022, a total of 2673 tumours were
identified in 2602 patients (1533 men and 1069 women). The median age of
incidence was 71 (range: 24 to 101). There was no significant difference in tumour
incidence between the sexes. No statistically significant difference in
incidence was found between cantons.
Table 1Cohort characteristics. All values represent the number
of cases (n); unless otherwise specified, percentages in brackets (%) are based
on the total number of available cases per category. Missing data is indicated
at the bottom of the table.
| Descriptive data |
Category |
n (% of total) |
| Patients (n = 2602) |
Patient age of incidence |
Median (range) |
71 (24–101) |
| <50 years |
188 (7%) |
| Patient sex |
Male |
1533 (59%) |
| Female |
1069 (41%) |
| Patient canton of residence |
Lucerne |
2035 (78%) |
| Nidwalden |
212 (8%) |
| Obwalden |
175 (7%) |
| Uri |
180 (7%) |
| Patient tumour count |
Single tumour |
2537 (98%) |
| Multiple tumours |
65 (2%) |
| Tumour count (n = 2673) |
Tumour location* |
Right colon |
923 (35%) |
| Left colon |
981 (37%) |
| Rectum |
743 (28%) |
| Tumour grading** |
Grade 1 (WHO low-grade) |
133 (5%) |
| Grade 2 (WHO low-grade) |
1672 (65%) |
| Grade 3 (WHO high-grade) |
749 (29%) |
General testing behaviour and outcomes of mismatch
repair protein testing and mutation analysis
Testing behaviour is outlined in figure 1. Among
the 2651 tumours with available data, 76% (n = 2017) were tested for at least
one mismatch repair protein, leaving 24% (n = 634) that were not tested for mismatch
repair proteins. There was no significant difference in patient age between the
tested and non-tested tumours.
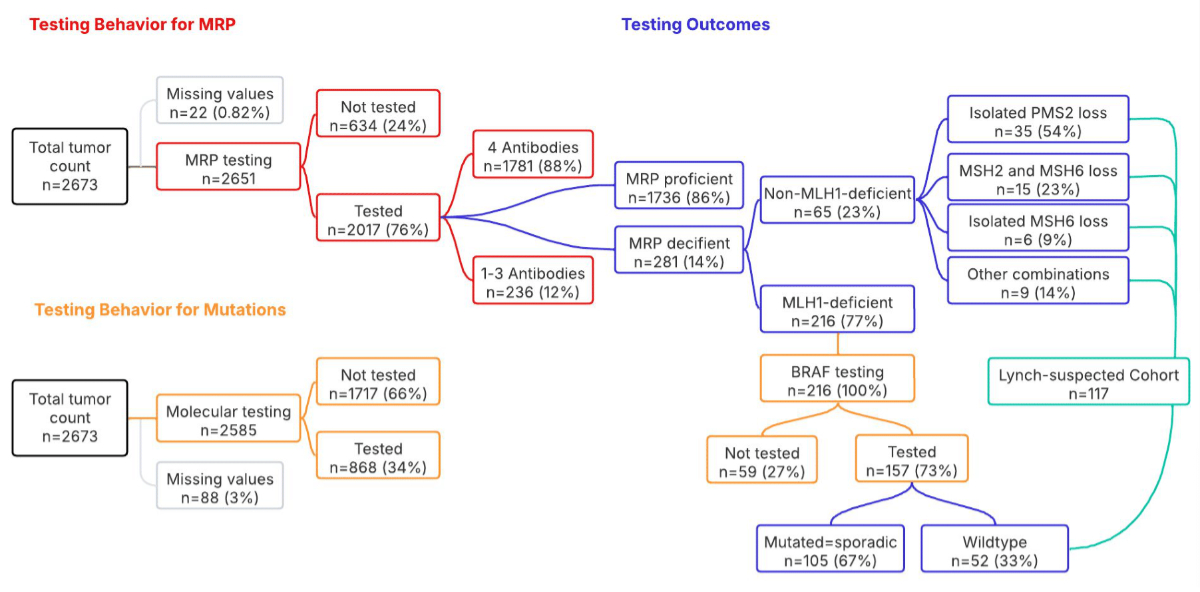
Figure 1Testing behaviour and outcomes.This
flowchart provides an overview of the key results of testing behaviour for
mismatch repair protein (MRP) status and mutation analysis of all diagnosed
colorectal cancers from 2011 to 2022 in Central Switzerland, along with the
main testing outcomes. n represents the number of tumours and the percentage
(%) indicates the proportion within the specific parent category.
14% of the tested tumours showed a mismatch
repair protein deficiency (MRPd) on immunohistochemistry. Among these, 77% showed
loss of MLH1, with concurrent PMS2 loss assumed in cases where only MLH1 was
tested, while 23% of cases showed a non-MLH1-related deficiency (figure 1).
Regarding molecular testing, of the 2585 tumours
with available data, 34% underwent molecular testing (figure 1): 785 tumours underwent
BRAF testing, with 22% showing mutations; 734 tumours underwent KRAS
testing, with 51% showing mutations; and 646 tumours underwent NRAS
testing, with 3% showing mutations. Interestingly, five MRPp tumours showed
concurrent mutations: two had both BRAF and KRAS mutations, one
had BRAF and NRAS mutations and two had mutations in BRAF,
KRAS and NRAS.
Testing behaviour among the MRPd cohort
Among the 281 MRPd tumours, data was
unavailable for 4 cases and excluded from the calculations. Of the remaining
277 tumours, 66% underwent molecular testing. All but one underwent BRAF
testing, with 64% showing a BRAF mutation; among the latter group, 95% had
a p.V600E mutation in exon 15, while the remaining mutations were p.D594G and
p.K601E, also in exon 15, or remained unknown. Additional KRAS testing
was carried out for 54 tumours, independently of BRAF status, with 19%
showing an isolated KRAS mutation. Finally, 52 tumours underwent NRAS
testing, independently of BRAF status, with 4% showing an isolated NRAS
mutation. No concurrent mutations were observed for any of the three analysed
genes.
Of the 216 tumours with lost MLH1
expression, only 73% were further tested for BRAF. One third of them (n
= 52 or 33%) showed no mutation, categorising them as Lynch-suspected, while the
remaining two thirds (n = 105 or 67%) showed a BRAF mutation, categorising them
as sporadic (figure 1).
The remaining 65 cases showed a
non-MLH1-related MRPd and were classified as Lynch-suspected (figure 1).
In total, 6% (n = 117) of the 2017 tested tumours
were considered Lynch-suspected and required further testing and/or genetic counselling.
Trends over time in tumour incidence, testing
rates and Lynch-suspected tumours
Temporal trends in tumour incidence,
testing rates and the proportion of Lynch-suspected tumours were assessed over
the study period (2011–2022) (figure 2).
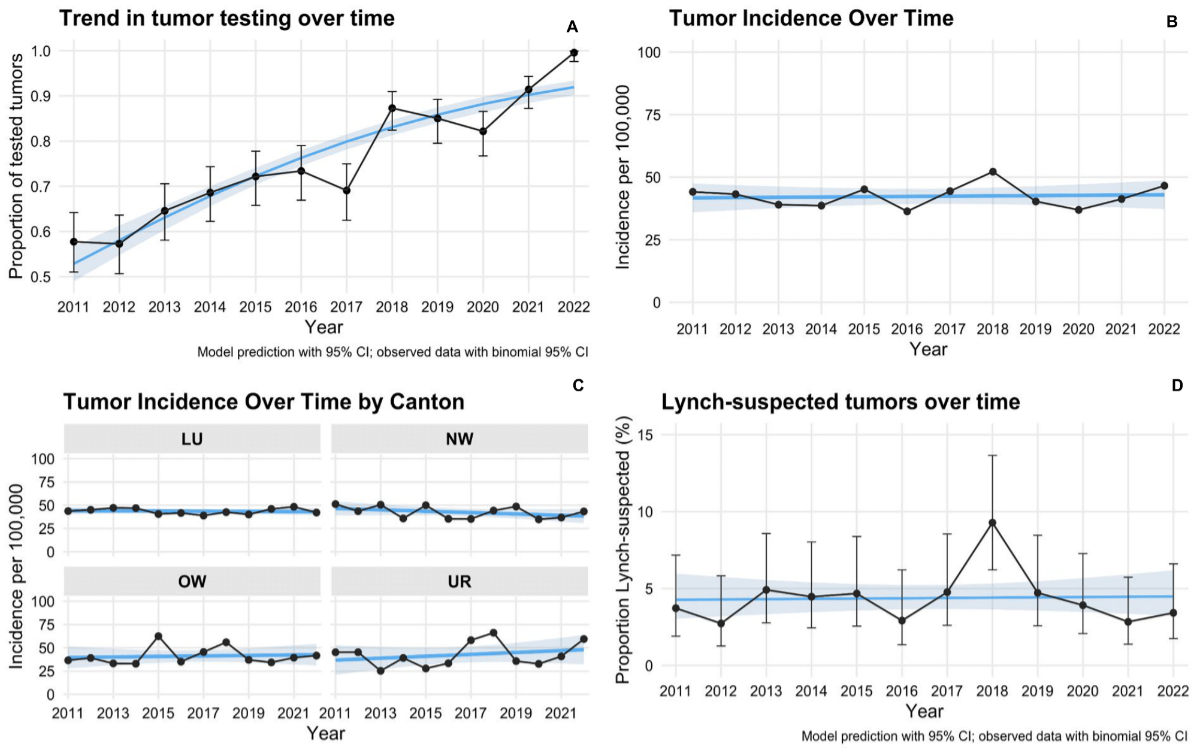
Figure 2Time trend analysis (2011–2022). A: Proportion of
tumours tested for mismatch repair proteins over time, increasing from 0.57 in
2011 to 0.99 in 2022. B: Tumour
incidence per 100,000 over time. C:
Tumour incidence per 100,000 by canton over time. D: Proportion of Lynch-suspected
tumours (%) over time. CI: confidence interval; LU: Lucerne; NW:
Nidwalden; OW: Obwalden; UR: Uri.
Tumour incidence remained stable over the
study period. In the Poisson regression model adjusted for population size, no
significant association between year and incidence was observed (β = −0.001,
SE = 0.006, p = 0.886), corresponding to an incidence rate ratio of 0.999 per
calendar year. A likelihood ratio test comparing a full model including canton
to a reduced model excluding canton showed no improvement in model fit (χ²(3) =
0.740, p = 0.860), indicating no substantial regional variation in incidence
rates (figures 2B, 2C).
The proportion of Lynch-suspected tumours
among all tumours remained stable over time. The binomial regression model
yielded a year coefficient of β = 0.005 (SE = 0.027, p = 0.862), corresponding
to an odds ratio of 1.005 (95% CI: 0.953–1.060) per calendar year. While the
proportion in 2018 exceeded the model-predicted range (9%), this deviation was
considered likely due to random variation (figure 2D).
In contrast, the proportion of tumours
tested showed a significant upward trend. The binomial regression model
estimated a year coefficient of β = 0.211 (SE = 0.015, p <0.001),
corresponding to an odds ratio of 1.235 (95% CI: 1.200–1.271) per calendar
year. This reflects a substantial annual increase of approximately 24% in the
odds of tumour testing throughout the study period (figure 2A).
Comparison between the MRPd and MRPp cohorts
Significant differences were found between
MRPd and MRPp tumours across several factors, as shown in table 2. The median
age of diagnosis was higher for MRPd tumours (76 years) compared to MRPp tumours
(70 years). MRPd tumours were more likely to be high-grade (grade 3), were more
common in females and were predominantly located in the right colon.
Table 2Comparison between the MRPd and
MRPp cohorts and between Lynch-suspected and sporadic cohorts. All values
represent the number of cases (n); unless otherwise specified, the percentages
in brackets (%) are based on the total number of available cases per category.
Missing data is indicated at the bottom of the table.
| Descriptive data |
MRPd vs MRPp tumours |
Lynch-suspected vs sporadic tumours |
| MRPp tumours |
MRPd tumours |
Lynch-suspected tumours |
Sporadic tumours |
| Tumour count* |
1736 |
281 |
117 |
105 |
| Age of incidence |
Median (range) |
70 (24–101) |
76 (27–94) |
67 (27–92) |
78 (54–94) |
| <50 years** |
169 (10%) |
19 (7%) |
18 (15%) |
0 (0%) |
| Sex |
Male |
1069 (62%) |
105 (37%) |
55 (47%) |
32 (30%) |
| Female |
667 (38%) |
176 (63%) |
62 (53%) |
73 (70%) |
| Canton of residence |
Lucerne |
1322 (76%) |
240 (85%) |
101 (86%) |
93 (89%) |
| Nidwalden |
146 (8.4%) |
19 (7%) |
8 (7%) |
5 (5%) |
| Obwalden |
134 (7.7%) |
9 (3%) |
5 (4%) |
3 (3%) |
| Uri |
134 (7.7%) |
13 (5%) |
3 (3%) |
4 (4%) |
| Tumour location*** |
Right colon |
566 (33%) |
236 (84%) |
92 (79%) |
94 (90%) |
| Left colon |
711 (41%) |
34 (12%) |
16 (14%) |
11 (10%) |
| Rectum |
448 (26%) |
10 (4%) |
9 (8%) |
0 (0%) |
| Tumour grading**** |
Grade 1 (WHO low-grade) |
98 (6%) |
9 (3%) |
2 (2%) |
4 (4%) |
| Grade 2 (WHO low-grade) |
1125 (67%) |
109 (39%) |
46 (40%) |
42 (40%) |
| Grade 3 (WHO high-grade) |
461 (27%) |
161 (58%) |
68 (58%) |
58 (56%) |
Comparison between the Lynch-suspected cohort and
the sporadic cohort
The Lynch-suspected and sporadic cohorts
differed in terms of median age (67 vs 78 years, respectively) and minimum age
(27 vs 54 years, respectively). Notably, all 18 cases with an incidence age
younger than 50 years were exclusively observed in the Lynch-suspected cohort
(table 2).
Although right-sided tumours predominated
in both cohorts, all 9 rectal carcinomas were found exclusively in the
Lynch-suspected cohort.
A statistically significant difference in sex
distribution was observed (p = 0.017), with a higher proportion of females in
the sporadic cohort compared to the Lynch-suspected cohort.
No significant differences were detected in
grading or regarding the distribution of cases across cantons, suggesting a
lack of geographical clustering or territorial hotspots.
Discussion
This retrospective study provided a
comprehensive overview of testing behaviours for mismatch repair protein in all
colorectal cancer across Central Switzerland, identifying gaps and
non-adherence to published guidelines of the Swiss Academy for Quality in
Medicine (SAQM) in reflex testing for mismatch repair protein and, when
appropriate, molecular testing. By identifying inconsistencies and highlighting
positive trends, we aim to raise awareness among pathologists to improve the
identification of patients with Lynch syndrome and those who qualify for immune
checkpoint inhibitor (ICI) therapy.
In 2011 the SAQM guidelines recommended mismatch
repair protein testing by immunohistochemistry or microsatellite instability testing
by PCR according to the revised Bethesda Guidelines [20, 21]. This
resulted in testing rates of about 60–70% of the tumours, potentially missing
patients with MRPd tumours. Pathologists performing the analysis often lacked the
full clinical information needed to apply the revised Bethesda criteria, e.g. family
history, so relied on non-established criteria for the decision. For example, since
rectal tumours are known to show microsatellite instability in fewer than 10%
of the cases, these tumours were significantly less likely to undergo testing,
indicating that pathologists may be influenced by tumour location, whereas
patient age did not appear to impact testing decisions [22]. In the early days,
microsatellite status was performed mainly with the goal of ruling out Lynch
syndrome, but nowadays the status is crucial for selecting the appropriate
treatment. For example, a recent publication showed a nearly complete response
in locally advanced rectal carcinomas treated with ICI therapy [28]. In
2019, the SAQM guidelines were revised, stating that immunohistochemistry
analysis of the mismatch repair protein with a 4-antibody panel or assessment of
the microsatellite instability status by PCR should be performed for all
patients with a newly diagnosed colorectal cancer [22]. This
policy shift was strongly reflected in our results. Testing rates went up from
56% in 2011 to over 90% in 2021, achieving an impressive near-complete coverage
of >99% in 2022 (figure 2A). This substantial progress represents a major
achievement in improving colorectal cancer diagnostic practices and adherence
to guidelines across the region.
Despite these improvements, significant
gaps remained in testing practices, particularly during the early years of the period
analysed. Nearly a quarter of the tumours were not tested for mismatch repair
protein. Based on statistical estimates, 14% (n ≈ 88) of the tumours are
potentially MRPd, which were missed due to insufficient testing. Notably 44% of
those tumours were located in the rectum, compared to a rectal tumour
distribution of only 28% in the entire cohort. This suggests that n ≈ 5 Lynch-suspected
cases (6%) may have been missed due to selective bias, especially in the early
years of the study period. Age on the other hand played no role in the
decision. In 2017–2018, a 2-antibody approach was taken (PMS2 and MSH6) instead
of the usual 4-antibody panel. In 44 cases, incorrect testing for MSH2 and MLH1
instead of the recommended PMS2 and MSH6 was performed, impeding the
identification of isolated PMS2 or MSH6 deficiencies, leading to another 2% of cases
potentially harbouring microsatellite instability. In 12 cases, other mismatch
repair protein combinations were tested, due to already known mutations,
technical issues or insufficient tissue. Furthermore, MLH1 promotor
methylation analysis was only performed in 9 of the 52 Lynch-suspected tumours
with an MLH1 loss without a BRAF mutation. Six of them were found to be
methylated, ruling out Lynch syndrome. This in part is to be attributed to the
lack of clear established protocols at the institution. A standardised approach
could reduce the risk of misclassification and ensure consistent identification
of Lynch syndrome cases.
A major point of concern lies in incomplete
molecular testing following mismatch repair protein deficiency, especially in
the MLH1-negative cohort. Of the 216 tumours with lost MLH1 expression, only
157 were further tested for BRAF, meaning that of the remaining 59,
statistically seen, approximately 20 of these cases may be Lynch-suspected and
could have been missed due to insufficient testing.
On a positive note, a trend towards
improvement was observed in BRAF testing practices. While in 2011 only 67%
and in 2013 even 47% of cases underwent BRAF testing, the rate increased
to 82% in 2018, reaching 96% in 2022. This progressive improvement shows, once
again, a positive improvement in testing practices over time. Additionally, the
testing rates in patients under 50 years were higher for both mismatch repair
protein and molecular testing. Of the 188 patients <50 years, 87% were
tested for mismatch repair protein and 7 of 8 with a MLH1 loss underwent BRAF
testing. Recently Bläker et al. demonstrated that patients under the age of 50
could still have Lynch syndrome despite a BRAF mutation and recommended genetic
counselling independently of BRAF status [25].
Direct referral for genetic counselling could potentially save costs and time
on BRAF and methylation analysis for younger patients.
In addition to testing practices, our
analysis also revealed insights into demographic and clinicopathological
characteristics. While the total tumour count showed a 9% increase over the 12-year
study period (from 213 cases in 2011 to 232 in 2022), this trend did not
translate into a significant rise in tumour incidence when adjusted for
population growth (β = −0.001, SE = 0.006, p = 0.886). Also, no statistically
significant cantonal hotspot in incidence rates was identified (figure 2C). The
proportion of Lynch-suspected tumours among all tumours remained consistently
low and stable over time, indicating effective regional screening and no
apparent increase in Lynch syndrome-associated tumours in Central Switzerland.
MRPp tumours were more frequent in males,
primarily left-sided, of lower histological grade and affected younger people.
In contrast, MRPd tumours were more frequent in females, primarily right-sided,
of higher histological grade and found in older patients, aligning with several
previous publications (table 2) [29–31]. In Lynch-suspected
cases, compared to MRPd, both were more common in females, predominantly right-sided
and of higher grade. However, both groups differed in patient age, with Lynch-suspected
patients being younger. This also aligned as expected with several previous
publications [32, 33].
The mismatch repair protein-deficiency
incidence in our cohort (14%) aligns well with that of similar studies from
other Swiss regions. For example, a study conducted in Basel reported an
incidence rate of 15%, which also aligns with broader European data showing an
MMR-deficiency prevalence of approximately 15% in colorectal cancer cases [34]. The
global incidence of Lynch syndrome is estimated at about 3%, with some
geographical and demographic variations [2, 12]. However,
due to limitations in our dataset, particularly the lack of genetic testing
reports, we classified cases as suspicious for Lynch syndrome when MLH1 expression
was lost without a BRAF mutation, or when non-MLH1-related mismatch
repair protein deficiencies were present, representing 6% of the cohort. Naturally,
this rate seems elevated, as it still includes patients without BRAF
mutation but with MLH1 promotor methylation and Lynch-like patients, or
even false positives. A comparable Swiss study reported similar rates (6%),
supporting the robustness of our findings within the constraints of available
data [34].
While our study provides valuable insight into reflex testing practices, we
acknowledge that direct comparisons with studies using different inclusion
criteria (e.g. genetically confirmed Lynch syndrome cases) should be
interpreted with caution.
The retrospective design limited our
ability to gather complete data, particularly for cases diagnosed externally,
which introduced gaps in the cohort. Although most pathological reports came
from the Lucerne Cantonal Hospital, where we had access to most medical
records, a minority of cases diagnosed externally introduced gaps in the
cohort. Despite mandatory reporting of new cancer cases to the registry, some
entries were incomplete, particularly those involving molecular tests conducted
after the initial diagnosis (for example after metastasis). Changes in testing
practices such as using 2 antibodies vs 4 in some years, may also have
introduced some bias. Variable interpretation of immunohistochemistry might
have also introduced some bias. Initially a positive staining of >10% was sufficient
to classify a tumour as MRPp; however more recent publications suggest that any
staining pattern deviating from strong uniform positivity warrants further
testing, especially since abnormal patterns are more strongly associated with Lynch
syndrome [35, 36]. The
clinical records on genetic counselling were also mostly incomplete, making it
challenging to categorise the cases as Lynch or sporadic. In certain cases,
testing might not have taken place due to perceived low clinical relevance.
While current testing algorithms are
designed to detect most Lynch syndrome cases, some remain undetected, including
cases with a methylated MLH1 promoter where constitutive methylation
syndrome was not considered, a condition found in approximately 3% of patients
with Lynch syndrome [5].
These factors suggest that the true number of Lynch syndrome cases in our
cohort may be slightly higher than reported.
In conclusion, the tumour incidence and the
proportion of Lynch-suspected tumours remained stable over time, with no
evidence of cantonal hotspots. In parallel, colorectal cancer diagnostics in Central
Switzerland showed remarkable progress, culminating in near-complete compliance
with guidelines for mismatch repair protein and molecular testing by 2022. This
high adherence provides a solid foundation for better personalised surveillance
and treatment, ultimately improving the quality of care for colorectal cancer
patients in the region.
Data sharing statement
Open Science: Due to the retrospective nature
of the study, a protocol was not prepared.
Anonymised study data can be shared upon
request by contacting the corresponding author. Requests must include a
detailed study protocol outlining the intended use of the data.
Bettina
Marturet Fendt
Institute
of Pathology
Cantonal
Hospital Lucerne
Spitalstrasse
CH-6000 Lucerne
16
bettina.fendt[at]luks.ch
References
1. Valle L, Vilar E, Tavtigian SV, Stoffel EM. Genetic predisposition to colorectal cancer:
syndromes, genes, classification of genetic variants and implications for precision
medicine. J Pathol. 2019 Apr;247(5):574–88. doi: https://doi.org/10.1002/path.5229
2. Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL, et al. Cancer Susceptibility
Gene Mutations in Individuals With Colorectal Cancer. J Clin Oncol. 2017 Apr;35(10):1086–95.
doi: https://doi.org/10.1200/JCO.2016.71.0012
3. Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008 Jan;18(1):85–98.
doi: https://doi.org/10.1038/cr.2007.115
4. Castillejo A, Hernández-Illán E, Rodriguez-Soler M, Pérez-Carbonell L, Egoavil C,
Barberá VM, et al. Prevalence of MLH1 constitutional epimutations as a cause of Lynch
syndrome in unselected versus selected consecutive series of patients with colorectal
cancer. J Med Genet. 2015 Jul;52(7):498–502. doi: https://doi.org/10.1136/jmedgenet-2015-103076
5. Pinto D, Pinto C, Guerra J, Pinheiro M, Santos R, Vedeld HM, et al. Contribution of
MLH1 constitutional methylation for Lynch syndrome diagnosis in patients with tumor
MLH1 downregulation. Cancer Med. 2018 Feb;7(2):433–44. doi: https://doi.org/10.1002/cam4.1285
6. Tutlewska K, Lubinski J, Kurzawski G. Germline deletions in the EPCAM gene as a cause
of Lynch syndrome - literature review. Hered Cancer Clin Pract. 2013 Aug;11(1):9.
doi: https://doi.org/10.1186/1897-4287-11-9
7. Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer
Biol Med. 2016 Mar;13(1):120–35. doi: https://doi.org/10.20892/j.issn.2095-3941.2015.0103
8. Pawlik TM, Raut CP, Rodriguez-Bigas MA. Colorectal carcinogenesis: MSI-H versus MSI-L.
Dis Markers. 2004;20(4-5):199–206. doi: https://doi.org/10.1155/2004/368680
9. Elsayed FA, Kets CM, Ruano D, van den Akker B, Mensenkamp AR, Schrumpf M, et al. Germline
variants in POLE are associated with early onset mismatch repair deficient colorectal
cancer. Eur J Hum Genet. 2015 Aug;23(8):1080–4. doi: https://doi.org/10.1038/ejhg.2014.242
10. Martínez-Roca A, Giner-Calabuig M, Murcia O, Castillejo A, Soto JL, García-Heredia A,
et al. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers (Basel).
2022 Feb;14(5):1115. doi: https://doi.org/10.3390/cancers14051115
11. Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, Zapater P, Castillejo A, Barberá VM,
et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation.
Gastroenterology. 2013 May;144(5):926–932.e1. doi: https://doi.org/10.1053/j.gastro.2013.01.044
12. Win AK, Dowty JG, Reece JC, Lee G, Templeton AS, Plazzer JP, et al.; International
Mismatch Repair Consortium. Variation in the risk of colorectal cancer in families
with Lynch syndrome: a retrospective cohort study. Lancet Oncol. 2021 Jul;22(7):1014–22.
doi: https://doi.org/10.1016/S1470-2045(21)00189-3
13. Idos G, Valle L. Table 8 [Recommended Surveillance for Individuals with Lynch Syndrome]Seattle:
University of Washington; 2021.[Internet][ [cited 2025 Mar 18]], Available from https://www.ncbi.nlm.nih.gov/books/NBK1211/table/hnpcc.T.recommended_surveillance_for_ind/
14. Seppälä TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sánchez-Guillén L,
et al.; European Hereditary Tumour Group (EHTG) and European Society of Coloproctology
(ESCP). European guidelines from the EHTG and ESCP for Lynch syndrome: an updated
third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021 May;108(5):484–98.
doi: https://doi.org/10.1002/bjs.11902
15. Bhattacharya P, Leslie SW, McHugh TW. Lynch Syndrome (Hereditary Nonpolyposis Colorectal
Cancer). StatPearls [Internet]Treasure Island (FL): StatPearls Publishing; 2024.[
[cited 2024 Jun 26]], Available from http://www.ncbi.nlm.nih.gov/books/NBK431096/
16. Dunne PD, Arends MJ. Molecular pathological classification of colorectal cancer-an
update. Virchows Arch. 2024 Feb;484(2):273–85. doi: https://doi.org/10.1007/s00428-024-03746-3
17. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous
immune microenvironment of microsatellite instable colon cancer is balanced by multiple
counter-inhibitory checkpoints. Cancer Discov. 2015 Jan;5(1):43–51. doi: https://doi.org/10.1158/2159-8290.CD-14-0863
18. Chang L, Chang M, Chang HM, Chang F. Microsatellite Instability: A Predictive Biomarker
for Cancer Immunotherapy. Appl Immunohistochem Mol Morphol. 2018 Feb;26(2):e15–21.
doi: https://doi.org/10.1097/PAI.0000000000000575
19. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 Blockade
in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N Engl J Med. 2022 Jun;386(25):2363–76.
doi: https://doi.org/10.1056/NEJMoa2201445
20. Qualitaet-Qualitaetsrichtlinien-SGPath_DE_2011.pdf [Internet]. [cited 2024 Nov 12].
Available from: https://www.sgpath.ch/customer/files/231/Qualitaet-Qualitaetsrichtlinien-SGPath_DE_2011.pdf
21. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised
Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome)
and microsatellite instability. J Natl Cancer Inst. 2004 Feb;96(4):261–8. doi: https://doi.org/10.1093/jnci/djh034
22. Qualitaet-QRL-KolonRektum-SGPath-2019.pdf [Internet]. [cited 2024 Nov 12]. Available
from: https://www.sgpath.ch/customer/files/231/Qualitaet-QRL-KolonRektum-SGPath-2019.pdf
23. Organisation [Internet]. Luzerner Kantonsspital. [cited 2024 Aug 9]. Available from:
https://www.luks.ch/ihr-luks/organisation
24. Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF
mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status:
a literature review assessing utility of tumour features for MMR variant classification.
J Med Genet. 2012 Mar;49(3):151–7. doi: https://doi.org/10.1136/jmedgenet-2011-100714
25. Age‐dependent performance of BRAF mutation testing in Lynch syndrome diagnostics -
Bläker - 2020 - International Journal of Cancer - Wiley Online Library [Internet].
[cited 2024 Aug 14]. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ijc.33273
26. Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA, Eshleman JR. Detection of
microsatellite instability by fluorescence multiplex polymerase chain reaction. J
Mol Diagn. 2000 Feb;2(1):20–8. doi: https://doi.org/10.1016/S1525-1578(10)60611-3
27. Statistik B für. Bilanz der ständigen Wohnbevölkerung nach Kanton, 1991-2022 - 1991-2022
| Tabelle [Internet]. Bundesamt für Statistik. 2023 [cited 2024 Aug 9]. Available
from: https://www.bfs.admin.ch/asset/de/26565330
28. Trojan J, Stintzing S, Haase O, Koch C, Ziegler P, Demes M, et al. Complete Pathological
Response After Neoadjuvant Short-Course Immunotherapy with Ipilimumab and Nivolumab
in Locally Advanced MSI-H/dMMR Rectal Cancer. Oncologist. 2021 Dec;26(12):e2110–4.
doi: https://doi.org/10.1002/onco.13955
29. De Smedt L, Lemahieu J, Palmans S, Govaere O, Tousseyn T, Van Cutsem E, et al. Microsatellite
instable vs stable colon carcinomas: analysis of tumour heterogeneity, inflammation
and angiogenesis. Br J Cancer. 2015 Jul;113(3):500–9. doi: https://doi.org/10.1038/bjc.2015.213
30. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence.
Nat Rev Clin Oncol. 2010 Mar;7(3):153–62. doi: https://doi.org/10.1038/nrclinonc.2009.237
31. Ward R, Meagher A, Tomlinson I, O’Connor T, Norrie M, Wu R, et al. Microsatellite
instability and the clinicopathological features of sporadic colorectal cancer. Gut.
2001 Jun;48(6):821–9. doi: https://doi.org/10.1136/gut.48.6.821
32. Martin S, Katainen R, Taira A, Välimäki N, Ristimäki A, Seppälä T, et al. Lynch syndrome-associated
and sporadic microsatellite unstable colorectal cancers: different patterns of clonal
evolution yield highly similar tumours. Hum Mol Genet. 2024 Nov;33(21):1858–72. doi: https://doi.org/10.1093/hmg/ddae124
33. Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch
syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal
ramifications. Clin Genet. 2009 Jul;76(1):1–18. doi: https://doi.org/10.1111/j.1399-0004.2009.01230.x
34. Zumstein V, Vinzens F, Zettl A, Heinimann K, Koeberle D, Flüe M von, et al. Systematic
immunohistochemical screening for Lynch syndrome in colorectal cancer: a single centre
experience of 486 patients. Swiss Med Wkly. 2016 Apr 24;146(1718):w14315–w14315.
35. Rüschoff J, Schildhaus HU, Rüschoff JH, Jöhrens K, Bocker Edmonston T, Dietmaier W,
et al. Testing for deficient mismatch repair and microsatellite instability : A focused
update. Pathologie (Heidelb). 2023 Nov;44(S2 Suppl 2):61–70. doi: https://doi.org/10.1007/s00292-023-01208-2
36. Wang C, Zhang L, Vakiani E, Shia J. Detecting mismatch repair deficiency in solid
neoplasms: immunohistochemistry, microsatellite instability, or both? Mod Pathol.
2022 Nov;35(11):1515–28. doi: https://doi.org/10.1038/s41379-022-01109-4