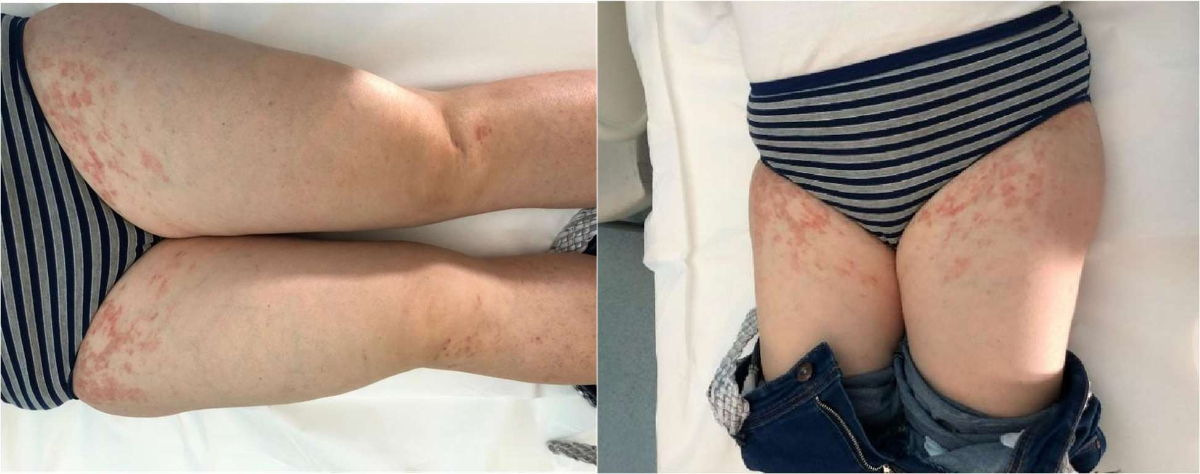
Figure 1Maculo-erythematous lesions on thighs of patient 1.
DOI: https://doi.org/https://doi.org/10.57187/3993
Pigmented purpuric dermatosis is a benign form of dermal capillaritis prevalent in the general population, presenting a generally chronic relapsing-remitting course and clinically characterised by the occurrence of macules, patches or petechiae ranging in colour from red-purple (due to erythrocyte extravasation and haemosiderin deposition) to red-brown/golden-brown with haemosiderin resorption. The skin eruptions, either totally asymptomatic or associated with mild to severe itching, typically affect the legs, with the possibility of occasionally involving the trunk and arms. Eight subtypes or variants have been recognised, differing in peculiar clinical features, and some of them have unique histopathological patterns: Progressive Pigmentary Dermatosis (Schamberg Disease), Purpura Annularis Telangiectodes (Majocchi’s Disease), Lichen Aureus, Pigmented Purpuric Lichenoid Dermatitis (Gougerot and Blum’s Disease), Eczematid-Like Purpura (Doucas and Kapetanakis Disease), Disseminated Pruriginous Angiodermatitis (Itching Purpura), Unilateral Linear Capillaritis (Linear Pigmented Purpura) and Granulomatous Pigmented Purpura [1].
Progressive Purpuric Pigmentary Dermatosis, also known as Schamberg Disease, is the most common form of pigmented purpuric dermatosis. Eczematid-like Purpura of Doucas and Kapetanakis is one of the eight variants of pigmented purpuric dermatosis. This form is considered an inflammatory variant of Schamberg Disease with more extensive involvement. It is characterised by itchy skin lesions with scaling; furthermore, association with allergic contact dermatitis to rubber and clothes has been reported [2, 3].
The aetiology of pigmented purpuric dermatosis is, in most cases, idiopathic and not associated with coagulopathies or thrombocytopenia. Possible predisposing factors are venous stasis, exercise, local infection and capillary fragility. Sometimes an iatrogenic cause is recognised, as well as an association with various systemic diseases such as diabetes mellitus, hepatopathy, dyslipidaemia, thyroid disease, neoplasms and autoimmune diseases. The pathogenesis is immune-based, both cell-mediated and humoral, as confirmed on direct immunofluorescence by the recent findings of vascular immunoglobulin deposits [4, 5].
Common variable immunodeficiency (CVID) is a heterogeneous disease characterised by hypogammaglobulinaemia, recurrent upper and lower airway and digestive tract infections, and defective antibody responsiveness. The disease can be associated with interstitial lung disease, granulomatous disease resembling sarcoidosis, autoimmune manifestations (20–30%), inflammatory bowel disease and malignancies (such as gastric cancer and lymphomas) [6, 7]. The clinical association between a dermatological condition and an immunological disorder finds its foundation in the well-established view of the skin as an immune organ. This organ is topologically heterogeneous and structured with sophisticated threat-sensing mechanisms, which activate specific defensive responses to restore homeostasis. Additionally, highly specialised immune cell populations are anatomically compartmentalised across various cutaneous microniches (including the interfollicular epidermis; pilosebaceous unit; papillary, reticular and hypo-dermis; along with vascular and neuronal niches) and communicate with adjacent parenchyma to maintain barrier function. Specifically, the epidermis is constantly monitored by Langerhans cells, CD8+ resident memory T cells and innate lymphoid cells, whose recruitment and survival are tightly regulated by the local release of cytokines and chemokines from the epithelium.
Deeper within, the pilosebaceous unit in the papillary and reticular dermis serves as a crucial, dynamic immune hub. This unit synergises with commensal microbes through unique cytokine and chemokine release patterns, recruiting and retaining both epidermal and dermal immune cells, including dermal antigen-presenting cells like CD103 dendritic cells and dermal dendritic cells. In the deeper dermis, vascular endothelium is surveilled by innate immune cells, such as mast cells and macrophages, which are particularly enriched in the perivascular space. During skin inflammation, these perivascular immune cells release cytokines and chemokines that activate the endothelium, thus enabling the entry of neutrophils, monocytes and lymphocytes from the systemic circulation. Furthermore, the perivascular space functions as an epicentre for the activation of effector T cells by clustering with dermal dendritic cells [8].
Among the skin manifestations associated with common variable immunodeficiency, chronic autoimmune-based skin conditions such as alopecia totalis, vitiligo and psoriasis are well described alongside the more predictable infectious complications of various origins [9]. In such a setting, clinical association between pigmented purpuric dermatosis and common variable immunodeficiency is not established in the literature. We report below two cases of such an association, suggesting new aetiopathogenic perspectives.
We report the cases of two patients with common variable immunodeficiency, women aged 50 and 59, clinically managed over the years for this condition at our institution (table 1).
Table 1Clinical and laboratory characteristics of the two patients.
| Patient 1 (age: 50 yr) | Patient 2 (age: 59 yr) | |
| Personal medical history | Negative for atopy and chronic autoimmune diseases | Negative for atopy and chronic autoimmune diseases |
| Family history | Negative for PPD and CVID | Negative for PPD and CVID |
| CVID-PPD temporal span | 3 years | 10 years |
| Genetic analysis (performed by NGS) | Heterozygous variant c.239A>C in the SOCS1 gene (likely pathogenic) | No pathogenic variants |
| Freiburg classification | Group II | Subgroup Ia |
| EUROclass classification | B⁺ MB⁺ smB⁻ Trnorm | B⁺ MB⁺ smB⁻ Trnorm |
| CVID complications | Bronchiectasis, hepatic regenerative nodular hyperplasia, portal circulation abnormalities, hepatosplenomegaly, ITP | Splenomegaly, chronic enteropathy with diffuse nodular lymphoid hyperplasia, ITP |
| PPD sites | Pretibial and thighs | Pretibial and highs |
| PPD clinical features | Eczematidic-like variant, mildly itchy, relapsing-remitting, responsive to topical corticosteroids leaving dyschromic exits | Eczematidic-like variant, asymptomatic, more noticeably fixed presentation, poorly responsive to topical corticosteroids |
| Dermatosis flare-up subsequent to Ig infusion | Observed | Not observed |
| Naranjo Adverse Drug Reaction Probability Scale | Final score: 6 | Final score: 7 |
PPD: pigmented purpuric dermatosis; CVID: common variable immunodeficiency
A 50-year-old woman with a personal pathological history negative for atopy and chronic autoimmune diseases, positive for inappropriate sinus tachycardia, was diagnosed with common variable immunodeficiency in 2017. Family history was negative for pigmented purpuric dermatosis and common variable immunodeficiency (no family members are affected by antibody deficiencies). Molecular genetic analysis for common variable immunodeficiency, performed by Next Generation Sequencing (NGS) on DNA extracted from peripheral whole blood, revealed the presence of the heterozygous variant c.239A>C in the Suppressor of Cytokine Signaling 1 gene, resulting in the amino acid substitution p.Tyr80Ser. This variant, not previously reported in the literature, can be classified as likely pathogenic (C4) according to the American College of Medical Genetics (ACMG) guidelines [10].
The patient was deficient in all three classes of immunoglobulins. Since then, she has been undergoing intravenous immunoglobulin (Ig) replacement therapy, overall tolerating this treatment and experiencing only on one occasion an adverse reaction with symptoms such as shaking chills, lower back and chest pain. The disease has complicated over the years with bronchiectasis, hepatic regenerative nodular hyperplasia to cirrhogenic evolution accompanied by portal circulation abnormalities and hepatosplenomegaly. She also presented with likely immune thrombocytopenia (ITP); although autoimmunity markers were negative, other reversible causes of isolated thrombocytopenia were nevertheless ruled out. The clinical course of ITP over the years has typically been relapsing-remitting, with some severe episodes (platelet count <30,000/mcL) requiring transfusions and oral corticosteroid therapy but never complicated by life-threatening bleeding, except for occasional epistaxis. The patient underwent flow cytometric analysis for the evaluation of B-cell subsets, which revealed the following findings (table 2): B lymphocytes (CD19⁺): 6.4%; switched memory B cells (CD19⁺ IgD⁻ CD27⁺): 0.7%; CD21low B cells (CD19⁺ CD21low CD38⁻): 34.7%. Based on the switched memory B cell population of 0.7% (cut-off ≥0.4%), the patient was classified as Group II according to the Freiburg classification [11]. More thoroughly, according to the EUROclass classification [12], the patient was classified as B⁺ MB⁺ smB⁻ Trnorm [Total B cells (CD19⁺): 6.4% (≥1%) → B⁺; Memory B cells (CD27⁺): 10.7% (>2%) → MB⁺; Switched memory B cells: 0.7% (≤2%) → smB⁻; transitional B cells: 4.3% (<9%) → smB⁻Trnorm].
Table 2Flow cytometric analysis for the study of B lymphocyte subpopulations in patient 1.
| B lymphocyte subpopulation | Phenotype | Value | Range |
| B lymphocytes | CD19+ | 6.4 | 4.9–18.4 |
| Naive B lymphocytes | CD19+ IgD+ CD27- | 83.5 | 42.6–82.3 |
| IgM-memory B lymphocytes | CD19+ IgD+ CD27+ | 10.7 | 7.4–32.5 |
| Switched memory B lymphocytes and plasmablasts | CD19+ IgD- CD27+ | 0.7 | 6.5–29.1 |
| IgG+ B lymphocytes | CD19+ IgG+ | 2.9 | 3.6–13.4 |
| CD21+ low subpopulation | CD19+ CD21+ low CD38- | 34.7 | 0.9–7.6 |
| Transitional B lyphocytes | CD19+ IgM++ CD38++ | 4.3 | 0.6–3.4 |
| Plasmablasts | CD19+ IgM-/(+) CD38+++ | 0.1 | 0.4–3.6 |
In 2020, the appearance of chronic, mildly itchy, nummular maculo-erythematous lesions with initial pretibial localisation, then spreading to the thighs, was observed (figure 1). Their clinical course was recurrent-remitting, with the “purpuric phase” lasting about 15 days and on several occasions immediately following Ig infusion therapy. To estimate the likelihood of a causal relationship between the skin manifestations and the infusion therapy, we applied the Naranjo algorithm [13], which yielded a final score of 6 in our patient, consistent with a probable association. Therefore, a dermatological evaluation was requested, which raised the suspicion of pigmented purpuric dermatosis, confirmed, after a skin punch biopsy, by histopathological examination in its eczematid-like variant. However, the personal history was negative for both drug intake and systemic diseases usually correlated with pigmented purpuric dermatosis. Treatment of the lesions with topical corticosteroids led to clinical remission, but left residual dyschromic lesions.
Figure 1Maculo-erythematous lesions on thighs of patient 1.
A 59-year-old woman with a personal pathological history negative for atopy and chronic autoimmune diseases, positive for congenital hip dysplasia with multi-prosthetic interventions, was diagnosed with CVID in 2006. Family history was negative for pigmented purpuric dermatosis and CVID (no family members are affected by antibody deficiencies). Molecular genetic analysis for CVID, performed by Next Generation Sequencing (NGS) on DNA extracted from peripheral whole blood, did not reveal any variants that could be classified as pathogenic and/or likely pathogenic for the condition under investigation.
The deficit involved serum levels of IgA, IgG and IgM for which she started intravenous Ig replacement therapy. The disease complicated over the years with splenomegaly, chronic enteropathy with diffuse nodular lymphoid hyperplasia. The patient also presented with plausible ITP, with negative autoimmunity markers, but absence of other reversible causes of isolated thrombocytopenia. The clinical course of ITP was reported to be typically relapsing-remitting, in the absence of severe episodes (platelet count >30,000/mcL) thus never treated with oral corticosteroids and never involving life-threatening bleeding. The patient underwent flow cytometric analysis for the evaluation of B-cell subsets, which revealed the following findings (table 3): B lymphocytes (CD19⁺): 15.1%; switched memory B cells (CD19⁺ IgD⁻ CD27⁺): 0.3%; CD21low B cells (CD19⁺ CD21low CD38⁻): 69.9%. Based on the finding of switched memory B cells at 0.3% (below the 0.4% cut-off) and CD21low B cells at 69.9% (exceeding the 20% threshold), the patient was classified as Freiburg subgroup Ia [11]. More thoroughly, according to the EUROclass classification [12], the patient was classified as B⁺ MB⁺ smB⁻ Trnorm [Total B cells (CD19⁺): 15.1% (≥1%) → B⁺; Memory B cells (CD27⁺): 9.2% (>2%) → MB⁺; Switched memory B cells: 0.3% (≤2%) → smB⁻; transitional B cells: 0.1% (<9%) → smB⁻Trnorm].
Table 3Flow cytometric analysis for the study of B lymphocyte subpopulations in patient 2.
| B lymphocyte subpopulation | Phenotype | Value | Range |
| B lymphocytes | CD19+ | 15.1 | 4.9–18.4 |
| Naive B lymphocytes | CD19+ IgD+ CD27- | 89.5 | 42.6–82.3 |
| IgM-memory B lymphocytes | CD19+ IgD+ CD27+ | 9.2 | 7.4–32.5 |
| Switched memory B lymphocytes and plasmablasts | CD19+ IgD- CD27+ | 0.3 | 6.5–29.1 |
| IgG+ B lymphocytes | CD19+ IgG+ | 0.3 | 3.6–13.4 |
| CD21+ low subpopulation | CD19+ CD21+ low CD38- | 69.9 | 0.9–7.6 |
| Transitional B lymphocytes | CD19+ IgM++ CD38++ | 0.1 | 0.6–3.4 |
| Plasmablasts | CD19+ IgM-/(+) CD38+++ | 0 | 0.4–3.6 |
In 2016, the appearance of erythematous-nummular macules with a chronic course and more nuanced relapsing-remitting character, localised to the thighs and pretibial area, was observed. To estimate the likelihood of a causal relationship between the skin manifestations and the infusion therapy, we applied the Naranjo algorithm [13], which yielded a final score of 7 in our patient, consistent with a probable association. Dermatological counselling posed suspicion for pigmented purpuric dermatosis, with subsequent histopathological confirmation of eczematid-like variant after skin punch biopsy. Even in this case, however, the personal history was negative for both drug intake and systemic diseases possibly related to pigmented purpuric dermatosis. As the skin lesions were characterised by a natural course of less marked clinical remission, the patient did not significantly benefit from the application of topical corticosteroids.
The current literature identifies concurrent venous hypertension as the primary trigger for pigmented purpuric dermatosis in association with dermal capillaritis [1].
The association between pigmented purpuric dermatosis and common variable immunodeficiency or other forms of primary immunodeficiency has never been documented in the international scientific literature. Instead, rare cases of occurrence of the concerned capillaritis in patients with secondary forms of immunodeficiency have been reported. Pantanowitz et al. first characterised pigmented purpuric dermatosis as a pharmacologically induced regression lesion of a cutaneous Kaposi’s sarcoma related to acquired immunodeficiency syndrome (AIDS) [14]. Interestingly, Garrido’s group described a purpuric pigmented lichenoid dermatitis in a patient with AIDS. The lesion was not objectified until after the initiation of antiretroviral therapy, leading the authors to hypothesise that it resulted from an immune imbalance and thus could be framed as a clinical manifestation of immune reconstitution inflammatory syndrome [15]. Similarly, Bonnet and colleagues reported a case of progressive pigmentary dermatosis having a recurrent course in a patient with a history of potus and moderate alcohol-related combined immunodeficiency. Characteristically, the capillaritis reexacerbated in close concomitance with chronic and sustained alcohol intake, then regressed within the first few days of abstinence. This “purpuric phase” was serologically associated with elevation of IgE levels, within a framework of more general humoral and cell-mediated immune activation (IgA elevation and activated T lymphocytes). In light of these findings, it was speculated that alcohol-induced immunoreconstitution (otherwise defined by the authors as hypersensitivity-like reaction) was also clinically extrinsic to the dermatosis in question [16].
In view of these insights from the literature and our patient investigations, an interesting aetiopathogenic watershed for pigmented purpuric dermatosis in common variable immunodeficiency appears to be emerging. Capillaritis itself, as a pivotal pathophysiological event in dermatosis, could result from a local autoimmune reaction, especially given the well-documented clinical profile of autoimmunity associated with both pigmented purpuric dermatosis and common variable immunodeficiency [4, 9]. Alternatively, and even more unexpectedly, also in light of the recent findings in dermal vascular Ig deposits [5], pigmented purpuric dermatosis could be framed as a clinical inflammatory expression of the immunoreconstitution resulting fromIg replacement therapy. This last consideration suggests,for the first time to our knowledge, a new potential trigger that would open up interesting new scenarios in the understanding of common variable immunodeficiency, at the same time expanding its already wide clinical heterogeneity.
Written informed consent for publication was obtained from the patients.
The authors did not receive any financial support for the preparation of this manuscript.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. No potential conflict of interest related to the content of this manuscript was disclosed.
1. Tolaymat L, Hall MR. Pigmented Purpuric Dermatosis. 2023 Apr 17. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan.
2. Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106(2):86–95. doi: https://doi.org/10.1159/000256830
3. Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented Purpuric Dermatosis: A Review of the Literature. Actas Dermosifiliogr (Engl Ed). 2020 Apr;111(3):196–204. doi: https://doi.org/10.1016/j.ad.2019.02.013
4. Spigariolo CB, Giacalone S, Nazzaro G. Pigmented Purpuric Dermatoses: A Complete Narrative Review. J Clin Med. 2021 May;10(11):2283. doi: https://doi.org/10.3390/jcm10112283
5. Ilagan FM, Wu YH. A retrospective study on the direct immunofluorescence findings in pigmented purpuric dermatosis. J Cutan Pathol. 2024 Jan;51(1):63–9. doi: https://doi.org/10.1111/cup.14507
6. Gangemi S, Allegra A, Musolino C. Lymphoproliferative disease and cancer among patients with common variable immunodeficiency. Leuk Res. 2015 Apr;39(4):389–96. doi: https://doi.org/10.1016/j.leukres.2015.02.002
7. Amato G, Vita F, Quattrocchi P, Minciullo PL, Pioggia G, Gangemi S. Involvement of miR-142 and miR-155 in Non-Infectious Complications of CVID. Molecules. 2020 Oct;25(20):4760. doi: https://doi.org/10.3390/molecules25204760
8. Naik S. One Size Does Not Fit All: Diversifying Immune Function in the Skin. J Immunol. 2022 Jan;208(2):227–34. doi: https://doi.org/10.4049/jimmunol.2100758
9. Remiker A, Bolling K, Verbsky J. Common Variable Immunodeficiency. Med Clin North Am. 2024 Jan;108(1):107–21. doi: https://doi.org/10.1016/j.mcna.2023.06.012
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405–24. doi: https://doi.org/10.1038/gim.2015.30
11. Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002 Mar;99(5):1544–51. doi: https://doi.org/10.1182/blood.V99.5.1544
12. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008 Jan;111(1):77–85. doi: https://doi.org/10.1182/blood-2007-06-091744
13. Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981 Aug;30(2):239–45. doi: https://doi.org/10.1038/clpt.1981.154
14. Pantanowitz L, Dezube BJ, Pinkus GS, Tahan SR. Histological characterization of regression in acquired immunodeficiency syndrome-related Kaposi’s sarcoma. J Cutan Pathol. 2004 Jan;31(1):26–34. doi: https://doi.org/10.1046/j.0303-6987.2004.0132.x
15. Garrido PM, Espinosa-Lara P, Aguado-Lobo M, Soares-Almeida L, Borges-Costa J. Dermoscopy of Pigmented Purpuric Lichenoid Dermatitis of Gougerot and Blum in an HIV-infected Patient. Dermatol Pract Concept. 2020 Oct;10(4):e2020075. doi: https://doi.org/10.5826/dpc.1004a75
16. Bonnet U, Selle C, Isbruch K, Isbruch K. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016 Oct;10(1):301. doi: https://doi.org/10.1186/s13256-016-1065-6