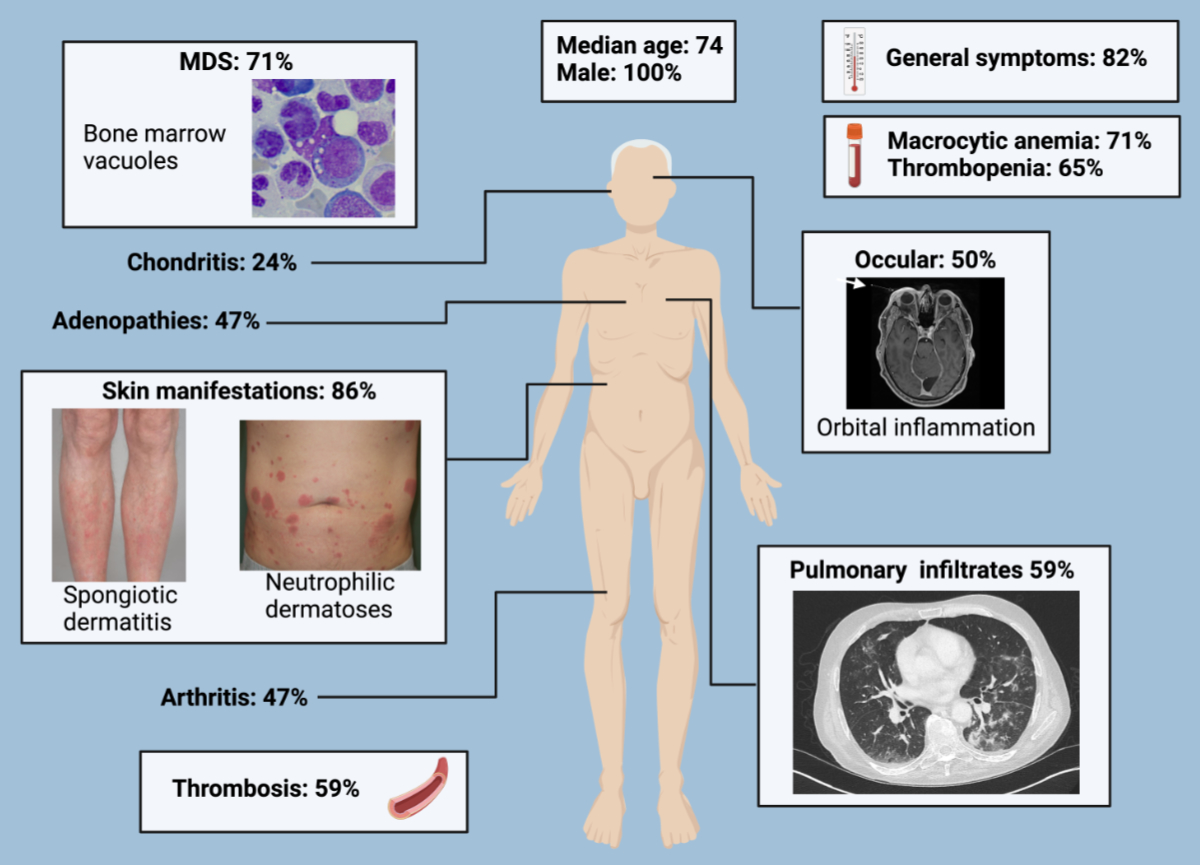
Figure 1Illustrations of the signs and symptoms of VEXAS syndrome. The images are from patients within our cohort and illustrate the wide range of clinical manifestations of VEXAS syndrome. MDS: myelodysplastic syndrome.
DOI: https://doi.org/https://doi.org/10.57187/s.3879
Janus kinase
ubiquitin-like modifier activating enzyme 1
Vacuoles, E1 Enzyme, X-linked, Auto-inflammatory, Somatic
Identified in 2020, VEXAS syndrome (Vacuoles, E1 Enzyme, X-linked, Auto-inflammatory, Somatic) is a unique monogenic auto-inflammatory disease that emerges predominantly in the later stages of life. It is caused by a somatic mutation in the ubiquitin-like modifier activating enzyme 1 (UBA1) gene, predominantly impacting myeloid precursor cells. Current scientific understanding suggests that UBA1 mutation reduces ubiquitylation, leading to heightened oxidative stress and the accumulation of unfolded proteins, resulting in inflammation [1, 2]. The clinical features of VEXAS syndrome include general symptoms (fever, weight loss and sweats), musculoskeletal complaints, pulmonary infiltrates, skin manifestations (e.g. neutrophilic dermatosis), eye involvement and thromboembolism [3]. Interestingly, VEXAS syndrome has specific features that distinguish it from other immune-mediated inflammatory diseases: it is highly associated with myelodysplastic syndromes and is more resistant to immunosuppressive drugs [3].
Currently, our understanding of the management of VEXAS syndrome remains nascent. The therapeutic approach varies according to the presence or absence of myelodysplastic syndrome. When myelodysplastic syndrome is present, targeting clonal haematopoiesis with a hypomethylating agent, such as azacytidine, is suggested as first-line therapy. Conversely, when myelodysplastic syndrome is absent, the treatment aims to mitigate inflammation. Here, immunosuppressants such as ruxolitinib or tocilizumab have shown promise [4, 5]. Haematopoietic stem cell transplantation emerges as a potential curative treatment not only for those with myelodysplastic syndrome but also for some cases without myelodysplastic syndrome. Those patients usually suffer severe autoinflammatory manifestations and are refractory to immunosuppressants [5, 6].
Given the emerging nature of VEXAS syndrome and the diversity of therapeutic approaches, there is an urgent need to consolidate data and experience. This study was designed as a national retrospective cohort study involving nine major hospitals across Switzerland, spanning multiple regions. Our primary objective is to analyse the phenotypic aspects of VEXAS syndrome and investigate the various treatments used in Switzerland.
We performed a retrospective study across nine major hospitals in Switzerland from July 2022 to July 2023, including institutions in Bern, Zurich, Geneva, Fribourg, Sion, Lausanne, Lucerne, Neuchâtel and Basel. These hospitals represent both university centres and regional medical facilities, ensuring a broad geographic distribution and diverse representation of the Swiss population. Physicians from relevant specialities, including internal medicine, rheumatology, haematology, and immunology, were contacted to identify potential patients. The patient could be included if he or she was diagnosed with a mutation in the UBA1 gene, independent of the sequencing method. The treating physician completed a case report form for each patient in a Microsoft Excel spreadsheet (version 16.73). The case report form encompassed the patient’s demographics, epidemiological data, detailed clinical presentation, laboratory results – including UBA1 mutation analysis – bone marrow examination, treatment modalities and clinical response. Two authors (LW and LC) compiled the case report forms, with a team of three authors (LW, LC and DC) conducting subsequent analyses. This study adhered to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines [7]. Quantitative variables are presented as the median (interquartile range [IQR]), and categorical variables are presented as the number (percentage). The clinical response was defined as the induction of a state without relapse and no need to transition to an alternative immunosuppressor. The laboratory results were recorded at the time of diagnosis, which was defined as the discovery of the mutation.
This study was approved by the Ethics Committee of the Canton of Vaud (CER-VD 2022-01365) and performed according to the Declaration of Helsinki, as revised in 2013, and written informed consent was obtained from all participating patients.
Of the 23 patients identified as suffering from VEXAS syndrome, 17 were included in our study: two refused to participate, and four were excluded due to incomplete data. All included patients were male. The median age at symptom onset and diagnosis were 67.5 (58–75) and 70.3 (59–77) years, respectively. The youngest recorded age for disease onset and diagnosis were 49 and 52 years, respectively. By the end of the study period, 29% (5/17) of the patients had died (table 1).
Table 1Epidemiology and manifestations: characteristics at diagnosis.
| Age (years), median (IQR) | 74 (59–77) | ||
| Male sex, number (%) | 17/17 (100) | ||
| Death, number (%) | 5/17 (29) | ||
| General symptoms, number (%) | 14/17 (82) | ||
| Fever, number (%) | 11/17 (65) | ||
| Weight loss, number (%) | 7/17 (41) | ||
| Sudation, number (%) | 7/17 (41) | ||
| Skin manifestations, number (%) | 15/17 (86) | ||
| Neutrophilic dermatosis, number (%) | 5/17 (29) | ||
| Dermatitis spongiotic, number (%) | 3/17 (18) | ||
| Panniculitis, number (%) | 2/17 (12) | ||
| Atopic dermatitis, number (%) | 1/17 (6) | ||
| Pulmonary manifestations, number (%) | 10/17 (59) | ||
| Cryptogenic organising pneumonia (COP), number (%) | 4/17 (23) | ||
| Nodules, number (%) | 2/17 (12) | ||
| Usual interstitial pneumonia (UIP), number (%) | 2/17 (12) | ||
| Nonspecific interstitial pneumonia (NSIP), number (%) | 1/17 (6) | ||
| Pleural effusion, number (%) | 1/17 (6) | ||
| Chondritis, number (%) | 4/17 (24) | ||
| Auricular, number (%) | 2/17 (12) | ||
| Nasal, number (%) | 2/17 (12) | ||
| Costal, number (%) | 1/17 (6) | ||
| Tracheal, number (%) | 1/17 (6) | ||
| Conductive hearing loss, number (%) | 1/17(6) | ||
| Adenopathy, number (%) | 8/17 (47) | ||
| Mediastinal, number (%) | 5/17 (29) | ||
| Paraoeosphageal, number (%) | 1/17 (6) | ||
| Inguinal, number (%) | 2/17 (12) | ||
| Axillary, number (%) | 2/17 (12) | ||
| Cervical, number (%) | 4/17 (24) | ||
| Polyarthralgia, number (%) | 8/17 (47) | ||
| Polyarthritis, number (%) | 6/17 (35) | ||
| Peripheral, number (%) | 8/17 (47) | ||
| Small articulations, number (%) | 7/17 (41) | ||
| Large articulations, number (%) | 8/17 (47) | ||
| Axial, number (%) | 1/17 (6) | ||
| Ocular manifestations, number (%) | 10/17 (50) | ||
| Orbital inflammation, number (%) | 4/17 (24) | ||
| Scleritis, number (%) | 2/17 (12) | ||
| Episcleritis, number (%) | 3/17 (18) | ||
| Anterior uveitis, number (%) | 2/17 (12) | ||
| Ocular veinous thrombosis, number (%) | 1/17 (6) | ||
| Anterior ischaemic optic neuropathy, number (%) | 1/17 (6) | ||
| Digestive, number (%) | 0/17 (0) | ||
| Central nervous system, number (%) | 1/17 (6) | ||
| Peripheral nervous system, number (%) | 2/17 (12) | ||
| Orchitis, number (%) | 1/17 (6) | ||
| Heart, number (%) | 0/17 (0) | ||
| Vasculitis, number (%) | 7/17 (41) | ||
| Leukocytoclastic vasculitis, number (%) | 5/17 (29) | ||
| Aortitis, number (%) | 1/17 (6) | ||
| Aneurysm of renal arteries, number (%) | 1/17 (6) | ||
| Acute renal insufficiency, number (%) | 1/17 (6) | ||
| Venous thromboembolism, number (%) | 10/17 (59) | ||
| Deep vein thrombosis, number (%) | 6/17 (35) | ||
| Pulmonary embolism, number (%) | 3/17 (18) | ||
| Haematological manifestations, number (%) | 15/17 (86) | ||
| Myelodysplastic syndrome, number (%) | 12/17 (71) | ||
| Lymphoproliferative disease, number (%) | 1/17 (6) | ||
| Monoclonal gammopathy of undetermined significance (MGUS), number (%) | 3/17 (18) | ||
IQR: interquartile range.
Most patients exhibited general symptoms: fever (11/17, 64%), night sweats or weight loss (14/17, 82%). Skin manifestations were the most commonly reported (15/17, 86%), including neutrophilic dermatosis (6/17, 35%), leukocytoclastic vasculitis (5/17, 29%), spongiotic dermatitis (3/17, 18%) and unspecified panniculitis (2/17, 12%). Chondritis was observed in four patients (4/17, 24%), primarily affecting the ear, nose, costal cartilage and upper airways.
Musculoskeletal involvement was present in nearly half of the patients, presenting as arthralgias (8/17, 47%) and arthritis (6/17, 35%). The affected joints included the small joints (7/17, 41%), such as the metacarpophalangeal (MCP), proximal interphalangeal (PIP) and distal interphalangeal (DIP), as well as large joints (6/17, 35%), such as the knees, wrists, ankles and elbows. Moreover, sacroiliitis was present in one patient.
Ophthalmologic manifestations were present in more than half of the patients (10/17, 59%) and manifested as orbital inflammation (4/17, 24%), scleritis (2/17, 12%), episcleritis (3/17, 18%), ocular venous thrombosis (1/17, 6%), anterior uveitis (2/17, 12%) and anterior ischaemic optic neuropathy (1/17, 6%). Venous thromboembolism was observed in ten patients (10/17, 59%), with deep vein thrombosis (6/17, 35%) and pulmonary embolism (3/17, 18%) being the most common forms. Confounding factors such as anticoagulant use or specific triggers were not investigated. Vasculitis was found in seven patients (7/17, 41%), including leukocytoclastic vasculitis (4/17, 24%), aortitis (2/17, 12%) and renal artery aneurysms in one case. Lung involvement was present in ten patients (10/17, 59%), including cryptogenic organising pneumonia (4/17, 23%), usual interstitial pneumonia (2/17, 12%), micro and macro nodules (2/17, 12%) and nonspecific interstitial pneumonia (1/17, 6%). One patient presented pleuritis with lymphohistiocytic reactions. Lymphadenopathies were observed in eight patients (8/17, 41%), predominantly in the mediastinal, cervical, axillary and inguinal chains, with biopsies showing follicular and interfollicular hyperplasia and haemophagocytic lymphohistiocytosis in one patient. Orchitis was reported in one patient, central nervous system involvement (stroke) in another patient, and peripheral nervous system involvement was observed in two patients (2/17, 12%), described as distal symmetric sensory polyneuropathy. One patienthad progressive chronic renal insufficiency of unknown origin (figure 1).
Figure 1Illustrations of the signs and symptoms of VEXAS syndrome. The images are from patients within our cohort and illustrate the wide range of clinical manifestations of VEXAS syndrome. MDS: myelodysplastic syndrome.
At the time of diagnosis, macrocytic anaemia was present in 12 patients (12/17, 71%), with all patients eventually developing macrocytosis. The mean corpuscular volume (MCV) was 101 fl (range: 100–108), and the mean haemoglobin level was 84 g/l (range: 80–100). Platelet level was found at 169 × 109/l (range: 97–261), with thrombopenia observed in seven patients (7/17, 41%). Notably, all patients with thrombopenia also exhibited anaemia.
The mean total white blood cell count was 3.9 × 109/l (range: 3.2–4.9), with lymphocyte and neutrophile counts of 0.65 × 109/l (range: 0.38–0.88) and 2.8 × 109/l (range: 2.2–3.6), respectively. Eosinophilia was noted in only one patient (1.44 × 109/l). Inflammatory markers showed elevated C-reactive protein (CRP) of 103 mg/l (range: 49–130) and an erythrocyte sedimentation rate (ESR) of 95 mm/h (range: 78–103). The mean creatinine was 75 mmol/l (range: 68–108). The coagulation profile included a mean INR of 1.1 (range: 1.05–1.2) and activated partial thromboplastin time (aPTT) of 30 s (range: 26–33). The antinuclear antibody titers (ANA) were equal to or greater than 1/160 in five patients (5/17, 29%), with titers of 1/160 in three patients and 1/640 in two patients. Complement components C3 and C4 were within normal limits in all seven patients tested (table 2).
Table 2Additional work up: characteristics at diagnosis.
| Laboratory parameter | ||
| Macrocytic anaemia, number (%) | 12/17 (71) | |
| Thrombopenia, number (%) | 11/17 (65) | |
| Haemoglobin (g/l), median (IQR) | 84 (80–100) | |
| MCV (fl), median (IQR) | 101 (100–108) | |
| Leucocytes (109/l), median (IQR) | 3.9 (3.2–4.9) | |
| Lymphocytes (109/l), median (IQR) | 0.66 (0.38–0.88) | |
| Neutrophils (109/l), median (IQR) | 2.9 (2.2–3.6) | |
| Eosinophils (109/l), median (IQR) | 0.01 (0–0.1) | |
| Platelets (109/l), median (IQR) | 169 (97–261) | |
| CRP (mg/l), median (IQR) | 103 (49–130) | |
| ESR (mm/h), median (IQR) | 95 (78–103) | |
| Creatinine (μmol/l), median (IQR) | 78 (68–108) | |
| INR (number), median (IQR) | 1.1 (1.05–1.2) | |
| aPTT (s), median (IQR) | 30 (26–33) | |
| Anti-nuclear antibodies (≥1/160), number (%) | 5/17 (29) | |
| Identified antinuclear antibodies, number (%) | 0/17 (0) | |
| Bone marrow | ||
| Vacuoles, number (%) | 16/17 (94) | |
| Dysplasia, number (%) | 12/17 (71) | |
| Trilinear dysplasia, number (%) | 5/17 (29) | |
| Bilinear dysplasia, number (%) | 5/17 (29) | |
| Presence of blasts, number (%) | 0/17 (0) | |
| Fibrosis, number (%) | 3/17 (18) | |
| Genetic mutations | ||
| c.122T>C (p.Met41Thr), number (%) | 11/17 (65) | |
| c.121A>G (p.Met41Val), number (%) | 2/17 (12) | |
| c.121A>C (p.Met41Leu), number (%) | 2/17 (12) | |
| c.118-1G>C, number (%) | 1/17 (6) | |
| c.118-2A>C, number (%) | 1/17 (6) | |
aPTT: activated partial thromboplastin time; CRP: c-reactive protein; ESR: erythrocyte sedimentation rate; INR : international normalised ratio; IQR: interquartile range; MCV: mean corpuscular volume.
Haematological manifestations were present in 13 patients (13/17, 75%), with 12 showing myelodysplastic syndrome (12/17, 71%). Transfusion dependence was reported in two patients (2/17, 12%). One case of small cell lymphocytic lymphoma and three cases of monoclonal gammopathy of undetermined significance were reported. Bone marrow analysis revealed vacuoles in 16 patients (16/17, 94%) and dysplasia in 12 patients, with all cases of myelodysplastic syndrome showing multilineage dysplasia. Bone marrow fibrosis was observed in three patients (3/17, 18%). Somatic mutations in UBA1 were identified in all patients. Canonical mutations were the most common: Met41Thr (65%, 11/17), Met41Val (12%, 2/17) and Met41Leu (12%, 2/17). Splicing site mutations were less common: c.118–1G>C (6%, 1/17) and c.118–2A>C (6%, 1/17). The variant allele frequency (VAF) was available for four patients and averaged 57% (range: 47–83).
All patients required glucocorticoids and various lines of immunosuppressants. Janus kinase inhibitors induced clinical remission in five of the treated patients (5/6, 83%). Specifically, ruxolitinib achieved clinical remission in all treated patients (4/4, 100%) and was well-tolerated. Upadacitinib also resulted in a clinical remission in the only treated patient (1/1, 100%). Tofacitinib, used in two cases, did not induce a clinical remission as a standalone treatment (0/1, 0%) but, when combined with cyclosporine, led to a clinical remission in another patient (1/1, 100%) (table 3).
Table 3Treatments.
| Medication | Clinical response | Intolerance | ||
| Janus kinase inhibitors | Ruxolitinib, number (%) | 4/4 (100) | 0/4 (0) | |
| Upadacitinib, number (%) | 1/1 (100) | 0/1 (0) | ||
| Tofacitinib, number (%) | 0/1 (0) | 0/1 (0) | ||
| Tofacitinib with cyclosporine, number (%) | 1/1 (100) | 0/1 (0) | ||
| IL-6R blockers | Tocilizumab, number (%) | 3/8 (37) | 2/8 (25): Cytopenia and anaphylaxis | |
| Tocilizumab with methotrexate, number (%) | 0/2 (0) | 1/2 (50): Neutropenia | ||
| IL-1 blockers | Anakinra, number (%) | 0/5 (0) | 5/5 (100): Reactions at the injection site | |
| Canakinumab, number (%) | 0/2 (0) | 0/2 (0) | ||
| TNF-alpha blockers | Adalimumab, number (%) | 1/3 (33) | 0/3 (0) | |
| Infliximab, number (%) | 0/3 (0) | 0/3 (0) | ||
| Infliximab with methotrexate, number (%) | 0/1 (0) | 1/1 (100): Pancytopenia | ||
| Other treatments | Cyclosporine, number (%) | 2/2 (100) | 0/2 (0) | |
| Rituximab, number (%) | 0/2 (0) | 0/2 (0) | ||
| Rituximab with mycophenolate mofetil, number (%) | ||||
| Cyclophosphamide, number (%) | 0/2 (0) | 0/2 (0) | ||
| Methotrexate, number (%) | 0/6 (0) | 1/6 (17): Pancytopenia | ||
| Hydroxychloroquine, number (%) | 0/2 (0) | 0/2 (0) | ||
| Mycophenolate mofetil, number (%) | 0/2 (0) | 0/2 (0) | ||
| Colchicine, number (%) | 0/5 (0) | 0/5 (0) | ||
| Azathioprine, number (%) | 0/3 (0) | 0/3 (0) | ||
| Intravenous immunoglobulin, number (%) | 0/1 (0) | 0/1 (0) | ||
| Abatacept, number (%) | 0/1 (0) | 0/1 (0) | ||
| Dapsone, number (%) | 0/1 (0) | 0/1 (0) | ||
| Treatments for myelodysplastic syndrome | Azacytidine, number (%) | 2/2 (100) | 0/2 (0) | |
| Azacytidine with anakinra, number (%) | 1/1 (100) | 1/1 (100): Reaction at the injection site | ||
| Azacytidine with canakinumab, number (%) | 1/1 (100) | 0/1 (0) | ||
| Azacytidine with tocilizumab, number (%) | 1/1 (100) | 0/1 (0) | ||
| Lenalidomide, number (%) | 0/1 (0) | 0/1 (0) | ||
| Haematopoietic stem cell transplantation, number (%) | 2/2 (0) | 0/2 (0) | ||
IL-6R: interleukin-6 receptor; IL-1: interleukin-1; TNF: tumour necrosis factor.
Tocilizumab, an interleukin-6 receptor (IL-6R) blocker, induced a clinical remission in three treated patients (3/8, 37%), with adverse events reported in two treated patients (2/8, 25%), including cytopenia and anaphylaxis. Combining tocilizumab with methotrexate failed to induce a clinical remission in two patients (0/2, 0%) and was discontinued in one patient due to neutropenia (1/2, 50%).
Regarding tumour necrosis factor (TNF)-alpha blockers, adalimumab led to a clinical remission in one patient, but infliximab did not result in a clinical remission in any treated patient (0/3, 0%). Combining infliximab with methotrexate did not achieve a clinical remission and caused pancytopenia. Cyclosporine alone led to a clinical remission in both treated patients. All patients treated with anakinra exhibited reactions at the injection site, necessitating treatment interruption. Six patients were treated, but no clinical remission was observed, even among those treated for more than a month. While it did not lead to cutaneous intolerance, canakinumab did not lead to a clinical remission in two patients. Even in combination with mycophenolate mofetil, rituximab failed to induce a clinical remission. Other treatments that failed to induce a clinical remission included cyclophosphamide, colchicine, hydroxychloroquine, methotrexate, mycophenolate mofetil, azathioprine, dapsone, abatacept and intravenous immunoglobulin.
In patients with myelodysplastic syndrome, treatment with azacytidine achieved a clinical remission in all cases (5/5, 100%) and was well-tolerated. Lenalidomide was used in one patient and did not lead to a clinical remission (0/1, 0%). Haematopoietic stem cell transplantation led to a clinical remission in two patients (2/2, 100%), both of whom are currently in remission after one year and one month, respectively.
All five deaths in the study were attributed to infectious complications. One patient with the Met41Val mutation (1/2, 50%) died at the age of 75 years, five years after diagnosis. Four patients with the Met41Thr or Met41Leu mutations had died by the time of this study (4/15, 27%).
We present the first Swiss cohort of patients diagnosed with VEXAS syndrome. Epidemiologically, their ages at presentation and diagnosis align with those reported in other case series [2, 3, 8]. Our cohort consisted exclusively of males, a characteristic similar to most reported series [2, 8]. Notably, the prevalence of VEXAS is approximately 23 in 100,000 for males aged >50 years, compared to 4 in 100,000 for females [9]. The lower prevalence in females is mainly due to its X-linked nature, with cases in females typically attributed to constitutive monosomy [10]. While the sample size of our cohort is limited due to the rarity of VEXAS syndrome, the inclusion of cases from nine hospitals across Switzerland strengthens the generalisability of our findings within the Swiss population and underscores the national scope of our study. This multi-centre approach provides valuable insights into the clinical characteristics and treatment responses in a real-world setting, highlighting the importance of collaboration in rare disease research.
In our cohort, patients presented with general symptoms (82%) and skin manifestations (88%), similar to findings in other cohorts. However, despite its limited size and important risk of random variability, our cohort showed a tendency toward higher rates of venous thromboembolism (59%), ocular manifestations (59%), pulmonary infiltrates (59%) and articular manifestations (47%), which were more common compared to previous studies [2, 3, 8]. Consistent with other cohorts, the arthritis observed was non-erosive, and cases of chondritis did not progress to saddle nose deformity. Moreover, the involvement of the central nervous system, kidneys, heart, and digestive tract was rare in our cohort, aligning with other reports [3, 11].
Orbital inflammation was observed more frequently in our cohort than in others. Notably, our study is the first to describe a case of anterior ischaemic optical neuropathy as a manifestation of VEXAS syndrome. Ocular manifestation in VEXAS syndrome can affect any structure within the eye and orbit, with approximately 12% of reported cases experiencing orbital or periorbital inflammation [12–14]. This type of inflammation is typically associated with granulomatosis with polyangiitis and immunoglobulin G4 (IgG4)-related disease but is rare in relapsing polychondritis and other autoinflammatory disease (except for tumour necrosis factor receptor-associated periodic fever syndrome [TRAPS]) [15–18]. In cases of polychondritis or autoinflammatory diseases, the presence of orbital inflammation may suggest VEXAS syndrome. However, the specific mechanisms leading to the development of orbital inflammation in VEXAS syndrome remain to be elucidated.
Like the French cohort, lung involvement in our cohort was often characterised by consolidations compatible with cryptogenic organising pneumonia, nodules or interstitial involvement (usual interstitial pneumonia or nonspecific interstitial pneumonia). However, pleural effusion was present in only one patient, contrasting with the French cohort, where 53% of the patients with pulmonary involvement had pleural effusions [19]. These effusions were predominantly small in volume. Given our study’s focus on the complete clinical picture of VEXAS syndrome rather than solely on its pulmonary manifestations, small effusions, which are common in the elderly, may have been overlooked.
The predilection site of chondritis in VEXAS syndrome is controversial. While two studies reported that VEXAS syndrome never affects upper airways and costal cartilage [18, 19], others found these sites to be affected, albeit less frequently [17, 20]. In our cohort, 50% of the patients with chondritis had costochondritis (one patient) or upper-airway chondritis (one patient). We conclude that the presence of costochondritis or upper-airway chondritis should not rule out VEXAS syndrome as a differential diagnosis of relapsing polychondritis.
Interestingly, we report one patient with sensorineural hearing loss without chondritis. Initially absent in the description by Beck et al., sensorineural hearing loss was later described in patients with VEXAS-related polychondritis [17, 18]. Its occurrence in relapsing polychondritis is well documented, although its development mechanism is largely unknown [21]. Our findings suggest that sensorineural hearing loss in VEXAS syndrome can occur independently of chondritis. Therefore, further studies are needed to investigate the relationship between VEXAS syndrome, chondritis and sensorineural hearing loss.
One patient in our cohort presented with genital involvement and renal artery aneurysms, initially leading to a diagnosis of polyarteritis nodosa. Post-mortem bone marrow analysis revealed a UBA1 mutation, confirming VEXAS syndrome. While orchiepididymitis was not reported in larger cohorts [2, 3], it was noted in two case series, with a prevalence of 33% in one study [8, 22]. This finding suggests that genital involvement could be a classical manifestation of VEXAS syndrome and should be further investigated. Additionally, two other patients in our cohort showed manifestations compatible with giant cell arteritis, consistent with a previous description of patients with VEXAS syndrome [23].
The classical haematologic manifestations of VEXAS syndrome are macrocytic anaemia and thrombopenia [3, 18]. However, in our cohort, only 71% of patients exhibited macrocytic anaemia at diagnosis, and 65% exhibited thrombopenia. Therefore, their absence should not preclude a diagnosis of VEXAS syndrome if the clinical presentation is suggestive. Notably, the proportion of patients with macrocytosis varies across cohorts, ranging from 91 to 96%, with the lowest proportions reported in more recent articles, perhaps indicating a more acutely presenting cohort [2, 9]. Studies have shown that plasma cell dyscrasia is more common in those with VEXAS syndrome than in the general population, particularly in the form of monoclonal gammopathy of undetermined significance [24, 25]. In our cohort, 18% of the patients had monoclonal gammopathy of undetermined significance, but none had multiple myeloma. Most patients with myelodysplastic syndrome exhibited vacuoles and multilineage dysplasia without blasts. Remarkably, only one case of myelodysplastic syndrome progressing to acute myeloid leukaemia has been reported in VEXAS syndrome [26]. Our cohort exhibited the three primary mutations previously identified in VEXAS syndrome: p.Met41Thr, p.Met41Leu, and p.Met41Val, with p.Met41Thr being the most common. Notably, patients with the Met41Val mutation appeared to have a lower survival rate, as previously documented [3]. Additionally, the lymphohistiocytic reactions in the bone marrow, adenopathy and pleural fluid observed in three patients align with VEXAS syndrome’s association with macrophage activation syndrome or similar features, likely correlating with the high inflammatory state and monocyte dysregulation characteristic of the disease [27–29].
All patients in our cohort treated with ruxolitinib, upadacitinib, cyclosporine, azacytidine and haematopoietic stem cell transplantation achieved a clinical remission. These findings are consistent with previous retrospective studies on the efficacy of Janus kinase inhibitors, particularly ruxolitinib, in VEXAS syndrome [30, 31]. Azacytidine was effective in managing cases of concomitant myelodysplastic syndrome [32]. The successful use of cyclosporine, including in combination with tofacitinib, has already been reported in previous case reports [33, 34]. Therefore, cyclosporine could be considered a treatment option in case of limited access to biotherapies. We report the successful use of haematopoietic stem cell transplantation in two patients, which currently remains the only curative therapy [6].
Regarding outcomes, all patient deaths were attributed to infections, highlighting the importance of immunosuppression due to either the disease or the treatment. One patient received intravenous immunoglobulin for five months without improvement or reduced infection frequency.
Our study had several limitations. Firstly, as a retrospective cohort study with a relatively small cohort, the generalisability of our findings may be limited. Secondly, the case report forms were completed by treating physicians without a centralised and standardised review process by a dedicated investigator, potentially leading to variability in data reporting and interpretation. Thirdly, our definition of a clinical remission was based on the absence of relapse (recurrence or symptom worsening) and the absence of a need for additional or escalated immunosuppressive therapy, which are both based on subjective parameters and not clearly defined.
In conclusion, our cohort highlights the importance of considering VEXAS syndrome in the differential diagnosis of patients presenting with multiple symptoms that do not fit the typical profile of vasculitis or connective tissue disease. Therefore, the screening for UBA1 mutations should not be limited to male patients with macrocytosis. While the optimal treatment approach warrants further investigation, existing data suggest the efficacy of Janus kinase inhibitors as a first therapeutic option. In addition, haematopoietic stem cell transplantation is currently the only curative treatment available for VEXAS syndrome.
We used the ChatGPT 3.5 language model and Whisperit.ai to check the spelling and grammar in this medical article.
This study received no funding.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. RB received support not related to this article for attending meetings or travel from AbbVie (Rheumaupadate 2024 in Mainz) and from Pfizer (EULAR Congress, Vienna). AE received fees for lectures, presentations, speakers beureaus, manuscript writing or educational events from BeiGene, and support for attending meetings and/or travel from BeiGene, Jazz Pharmaceuticals and Takeda, all not related to this work. No other potential conflict of interest related to the content of this manuscript was disclosed.
1. Jachiet V, Ricard L, Hirsch P, Malard F, Pascal L, Beyne-Rauzy O, et al.; MINHEMON: French Network of dysimmune disorders associated with hemopathies. Reduced peripheral blood dendritic cell and monocyte subsets in MDS patients with systemic inflammatory or dysimmune diseases. Clin Exp Med. 2023 Jul;23(3):803–13. doi: https://doi.org/10.1007/s10238-022-00866-5
2. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020 Dec;383(27):2628–38. doi: https://doi.org/10.1056/NEJMoa2026834
3. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al.; French VEXAS group; GFEV, GFM, CEREMAIA, MINHEMON. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022 Mar;186(3):564–74. doi: https://doi.org/10.1111/bjd.20805
4. Boyadzhieva Z, Ruffer N, Kötter I, Krusche M. How to treat VEXAS syndrome: a systematic review on effectiveness and safety of current treatment strategies. Rheumatology (Oxford). 2023 Nov;62(11):3518–25.
5. Sujobert P, Heiblig M, Jamilloux Y. VEXAS: where do we stand 2 years later? Curr Opin Hematol. 2023 Mar;30(2):64–9. doi: https://doi.org/10.1097/MOH.0000000000000750
6. Diarra A, Duployez N, Fournier E, Preudhomme C, Coiteux V, Magro L, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. 2022 Feb;6(3):998–1003.
7. von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007 Oct;370(9596):1453–7. doi: https://doi.org/10.1016/S0140-6736(07)61602-X
8. van der Made CI, Potjewijd J, Hoogstins A, Willems HP, Kwakernaak AJ, de Sevaux RG, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunol. 2022 Jan;149(1):432–439.e4. doi: https://doi.org/10.1016/j.jaci.2021.05.014
9. Beck DB, Bodian DL, Shah V, Mirshahi UL, Kim J, Ding Y, et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA. 2023 Jan;329(4):318–24. doi: https://doi.org/10.1001/jama.2022.24836
10. Stubbins RJ, McGinnis E, Johal B, Chen LY, Wilson L, Cardona DO, et al. VEXAS syndrome in a female patient with constitutional 45,X (Turner syndrome). Haematologica. 2022 Apr;107(4):1011–3. doi: https://doi.org/10.3324/haematol.2021.280238
11. Ferrada MA, Savic S, Cardona DO, Collins JC, Alessi H, Gutierrez-Rodrigues F, et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood. 2022 Sep;140(13):1496–506. doi: https://doi.org/10.1182/blood.2022016985
12. Beecher MB, Tong JY, Halliday LA, Hissaria P, Selva D. Recurrent orbital inflammation associated with VEXAS syndrome. Orbit. 2022;0(0):1–4.
13. Koster MJ, Samec MJ, Warrington KJ. VEXAS Syndrome-A Review of Pathophysiology, Presentation, and Prognosis. J Clin Rheumatol. 2023 Sep;29(6):298–306. doi: https://doi.org/10.1097/RHU.0000000000001905
14. Vitale A, Caggiano V, Martin-Nares E, Frassi M, Dagna L, Hissaria P, et al. Orbital/ocular inflammatory involvement in VEXAS syndrome: data from the international AIDA network VEXAS registry. Semin Arthritis Rheum. 2024 Jun;66:152430.
15. Sota J, Vitale A, Fabiani C, Frediani B, Rigante D, Tosi GM, et al. The eye involvement in monogenic autoinflammatory diseases: literature review and update. Clin Exp Rheumatol. 2018 Jan-Feb;36 Suppl 110(1):44-53.
16. Sfiniadaki E, Tsiara I, Theodossiadis P, Chatziralli I. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther. 2019 Jun;8(2):227–34. doi: https://doi.org/10.1007/s40123-019-0176-8
17. Khitri MY, Guedon AF, Georgin-Lavialle S, Terrier B, Saadoun D, Seguier J, et al.; French VEXAS group and MINHEMON. Comparison between idiopathic and VEXAS-relapsing polychondritis: analysis of a French case series of 95 patients. RMD Open. 2022 Jul;8(2):e002255. doi: https://doi.org/10.1136/rmdopen-2022-002255
18. Ferrada MA, Sikora KA, Luo Y, Wells KV, Patel B, Groarke EM, et al. Somatic Mutations in UBA1 Define a Distinct Subset of Relapsing Polychondritis Patients With VEXAS. Arthritis Rheumatol. 2021 Oct;73(10):1886–95. doi: https://doi.org/10.1002/art.41743
19. Borie R, Debray MP, Guedon AF, Mekinian A, Terriou L, Lacombe V, et al.; French VEXAS Group. Pleuropulmonary Manifestations of Vacuoles, E1 Enzyme, X-Linked, Autoinflammatory, Somatic (VEXAS) Syndrome. Chest. 2023 Mar;163(3):575–85. doi: https://doi.org/10.1016/j.chest.2022.10.011
20. Tsuchida N, Kunishita Y, Uchiyama Y, Kirino Y, Enaka M, Yamaguchi Y, et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis. 2021 Aug;80(8):1057–61. doi: https://doi.org/10.1136/annrheumdis-2021-220089
21. Akdoğan Ö, Exilus S, Ward BK, McArthur JC, Della Santina CC, Carey JP. Sudden Sensorineural Hearing and Vestibular Loss in a Case of Relapsing Polychondritis. Ann Otol Rhinol Laryngol. 2021 Dec;130(12):1412–6. doi: https://doi.org/10.1177/00034894211005979
22. Muratore F, Marvisi C, Castrignanò P, Nicoli D, Farnetti E, Bonanno O, et al. VEXAS syndrome: a case series from a single-center cohort of Italian patients with vasculitis. Arthritis Rheumatol. 2021; n/a(n/a). Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/art.41992
23. Watanabe R, Kiji M, Hashimoto M. Vasculitis associated with VEXAS syndrome: A literature review. Front Med (Lausanne). 2022 Aug;9:983939. Available from: https://www.frontiersin.org/articles/10.3389/fmed.2022.983939
24. Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021 Aug;5(16):3203–15. doi: https://doi.org/10.1182/bloodadvances.2021004976
25. Gutierrez-Rodrigues F, Kusne Y, Fernandez J, Lasho T, Shalhoub R, Ma X, et al. Spectrum of clonal hematopoiesis in VEXAS syndrome. Blood. 2023 Jul;142(3):244–59. doi: https://doi.org/10.1182/blood.2022018774
26. Battipaglia G, Vincenzi A, Falconi G, Fiore A, D’Agostino F, Iannotta R, et al. New scenarios in Vacuoles, E1 enzyme, X linked, Autoinflammatory, Somatic (VEXAS) syndrome: evolution from myelodysplastic syndrome to acute myeloid leukemia. Curr Res Transl Med. 2023;71(2):103386. doi: https://doi.org/10.1016/j.retram.2023.103386
27. Terrier B, Posseme C, Temple M, Corneau A, Carbone F, Chenevier-Gobeaux C, et al. Le syndrome VEXAS se caractérise par une activation des voies de l’inflammasome dans le sang et les tissus et par une dérégulation du compartiment monocytaire. Rev Med Interne. 2022 Dec;43:A384.
28. Staels F, Betrains A, Woei-A-Jin FJ, Boeckx N, Beckers M, Bervoets A, et al. Case Report: VEXAS Syndrome: From Mild Symptoms to Life-Threatening Macrophage Activation Syndrome. Front Immunol. 2021 Apr;12:678927. doi: https://doi.org/10.3389/fimmu.2021.678927
29. Yoon JG, Lee S, Kim S, Kim MJ, Chang YH, Park JK, et al. The First Korean Case of VEXAS Syndrome Caused by a UBA1 Somatic Variant. Ann Lab Med. 2023 Mar;43(2):217–20. doi: https://doi.org/10.3343/alm.2023.43.2.217
30. Heiblig M, Ferrada MA, Gerfaud-Valentin M, Barba T, Mékinian A, Koster M, et al. Clinical Efficacy of JAK Inhibitors in Patients with Vexas Syndrome: A Multicenter Retrospective Study. Blood. 2021 Nov;138 Supplement 1:2608. doi: https://doi.org/10.1182/blood-2021-150394
31. Heiblig M, Ferrada MA, Koster MJ, Barba T, Gerfaud-Valentin M, Mékinian A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multicenter study. Blood. 2022 Aug;140(8):927–31. doi: https://doi.org/10.1182/blood.2022016642
32. Comont T, Heiblig M, Rivière E, Terriou L, Rossignol J, Bouscary D, et al.; French VEXAS study group, Groupe Francophone des Myélodysplasies (GFM) and MedecineINterne, HEmato et ONco (MINHEMON) group. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. 2022 Feb;196(4):969–74. doi: https://doi.org/10.1111/bjh.17893
33. Bourbon E, Heiblig M, Gerfaud Valentin M, Barba T, Durel CA, Lega JC, et al. Therapeutic options in VEXAS syndrome: insights from a retrospective series. Blood. 2021 Jul;137(26):3682–4. doi: https://doi.org/10.1182/blood.2020010177
34. Campochiaro C, Tomelleri A, Cavalli G, De Luca G, Grassini G, Cangi MG, et al. Successful use of cyclosporin A and interleukin-1 blocker combination therapy in VEXAS syndrome: a single-center case series. Arthritis Rheumatol. 2022 Jul;74(7):1302–3. doi: https://doi.org/10.1002/art.42101