
Figure 1Patient selection flowchart.
DOI: https://doi.org/https://doi.org/10.57187/s.3633
Lipoprotein (a) [Lp(a)] is a serum lipoprotein with potential pro-atherogenic, pro-inflammatory and anti-fibrinolytic properties [1, 2]. It consists of an apolipoprotein B100 molecule and an apolipoprotein(a) [apo(a)] molecule covalently bound to one another [1, 3]. In contrast to LDL cholesterol (LDL-C) levels, Lp(a) plasma levels are mainly genetically determined and thus barely affected by diet or lifestyle modifications; they vary widely in the population. These variations are mainly caused by polymorphisms in the LPA gene [4].
Elevated Lp(a) levels have been identified as a causal cardiovascular risk factor [5]. Specifically, there is emerging evidence that Lp(a) contributes to the residual cardiovascular risk in patients with dyslipidaemia who have achieved their target LDL-C levels with pharmacological LDL-lowering therapies [6].
High Lp(a) levels have been causally associated with ischaemic stroke [5, 7]. Despite the known association between high Lp(a) levels and the risk for ischaemic stroke, only a few studies have addressed the association of Lp(a) on different stroke subtypes [8, 9, 10]. Since Lp(a) promotes atherosclerosis, an association with large artery atherosclerosis (LAA) can be expected. This is supported by the findings of a recent study by Arnold et al. which showed that Lp(a) was associated with LAA stroke aetiology (aOR 1.48, 95% CI 1.14–1.90) [11]. The interplay between Lp(a) and other cardiovascular risk factors for ischaemic stroke, such as arterial hypertension or dyslipidaemia (mainly driven by LDL-C), has received little attention. Only age – not dyslipidaemia, diabetes, or active and former smoking – was a strong effect modifier of this association [11].
In a well-described, prospectively collected cohort of patients with ischaemic stroke, we aimed to externally validate the association between Lp(a) and LAA stroke aetiology, as well as explore potential effect modifiers of the association.
The CoRisk (CoPeptin for Risk Stratification in Acute Patients) study [NCT00878813] was a prospective observational multicentre study. The protocol has been described elsewhere [12]. The responsible ethics committee approved the study (ID: KEK 001/09). We followed the STROBE guidelines for reporting observational studies.
In the current analyses, we included patients with acute ischaemic stroke admitted to the University Hospital of Bern between 2009 and 2011. Acute ischaemic stroke was defined as a new focal neurological deficit lasting longer than 24h without signs of acute intracranial bleeding on brain imaging. Only patients with available blood samples for lipid analysis and stroke subtype aetiology identified according to the TOAST (Trial of Org 10172 in Acute Stroke Treatment) classification were included [13]. Patients who did not give written informed consent and those with diagnoses other than ischaemic stroke after a diagnostic workup were excluded.
We assessed demographic data, pre-stroke modified Rankin Scale (mRS), National Institutes of Health Stroke Scale (NIHSS) score on admission, medical history (arterial hypertension, dyslipidaemia, diabetes mellitus, atrial fibrillation, coronary heart disease, previous cerebrovascular event, family history, and smoking status) and prior medication (platelet inhibitors, anticoagulants, antihypertensive agents, and statins).
Computed tomography (CT) with angiography or magnetic resonance imaging (MRI) with/without angiography was performed during hospitalisation. For stroke subtype classification according to the TOAST criteria, additional diagnostic tests were performed at the treating physician's discretion, including duplex ultrasound of the carotid artery, electrocardiogram, and transthoracic echocardiography.
In the CoRisk study, on which the current study was based, information on cardiovascular risk factors and their treatment was collected systematically during the initial patient contact using the Charlson comorbidity index [14], self-reported medical history, and medication upon admission. Smoking was classified as current or former (smoking cessation >2 years before study entry).
For the Lp(a) measurements, blood was drawn within 24 hours of symptom onset. After centrifugation for 20min at 3000g at room temperature, plasma was aliquoted, and samples were stored in the freezer until further analysis. Lp(a) concentration was assessed using the Roche Tina-quant Lp(a) Gen.2 assay. Plasma levels were recorded in nmol/l. The lower detection limit was 6nmol/l.
Our primary outcome was LAA stroke as the underlying stroke aetiology according to the TOAST classification. The secondary outcome was the incidence of recurrent stroke after 3 months.
The baseline characteristics of LAA stroke patients and patients with any other stroke aetiology (non-LAA) were compared using descriptive statistics. Categorical variables were compared using Fisher’s exact test, and continuous variables using the Mann-Whitney U test. Results were presented as numbers (with percentages) or medians (with interquartile ranges [IQR]), respectively. Univariable and multivariable logistic regression analyses were performed to assess the association of log10 Lp(a) with LAA stroke. Log10 transformation was performed due to the skewed distribution of Lp(a). For the multivariable models, we used (1) covariables that showed a significant association with LAA stroke aetiology (i.e., p <0.05) in the univariable analyses, and (2) covariables that have been reported in the literature to influence the risk of LAA (i.e., age, history of smoking, LDL-C [log transformed], statin pre-treatment) [15]. In the final model, we included age, sex, hypertension, diabetes, LDL-C (log transformed), smoking, statin treatment on admission, and platelet inhibitor treatment on admission. Only cases with complete covariable sets were considered. As sensitivity analyses, we performed backward selection based on the prespecified covariable sets, removing terms with p ≥ 0.10, and repeated the multivariable analyses after excluding patients with undetermined stroke aetiology (after full evaluation) and patients with more than one possible stroke aetiology.
In addition, we performed interaction analysis to identify possible effect modifiers of the observed effects. P-values <0.05 were considered significant; we report 95% confidence intervals. Statistical analysis was performed using STATA version 17 (STATA Corp LLC, College Station, Texas, USA). Do-files are available from the corresponding authors upon reasonable request.
Ethical approval: The ethics committee of Kanton Bern approved this study (ID: KEK 001/09).
Informed consent: Written informed consent was obtained from all subjects before the study.
Lp(a) measurements were available for 743 of 783 patients (94.9%). According to the TOAST classification, 105 (14%) patients had LAA stroke aetiology (figure 1).
Figure 1Patient selection flowchart.
Median Lp(a) levels were 16.9 nmol/l (IQR 6.1–60), with significantly higher levels in patients with LAA stroke (23.0 nmol/l [IQR 9.8–80.0] than in those with non-LAA stroke (16.3 nmol/l [IQR 5.8–57.0], p = 0.01). Patients with LAA stroke, compared with non-LAA stroke patients, had a higher BMI (26.4 kg/m2 vs 25.6 kg/m2, p = 0.01), were more often male (73% vs 61%, p = 0.02), were less likely to have atrial fibrillation (1% vs 21%, p ≤0.001), and were more likely to have arterial hypertension (81% vs 66%, p = 0.003), dyslipidaemia (68% vs 57%, p = 0.03), and diabetes mellitus (24% vs 14%, p = 0.02). On admission, patients with LAA stroke were more likely to be taking platelet inhibitors and antihypertensive drugs (table 1).
Table 1Baseline characteristics of LAA and non-LAA stroke patients.
| Total (n = 743) | LAA (n = 105) | Non-LAA (n = 638) | p-value | Missing patients (%) | |
| Demographic data | |||||
| Age (ys) – median [IQR] | 71 [61–80] | 72 [63–79] | 71 [60–80] | 0.60 | 0 (0) |
| Male sex – n (%) | 465 (63) | 77 (73) | 388 (61) | 0.02 | 0 (0) |
| Medical history – n (%) | |||||
| Dyslipidaemia | 426 (58) | 71 (68) | 355 (57) | 0.03 | 10 (1) |
| Previous cerebrovascular event | 153 (21) | 29 (28) | 124 (19) | 0.07 | 1 (<1) |
| Arterial hypertension | 508 (68) | 85 (81) | 423 (66) | 0.003 | 0 (0) |
| Current smoking | 133 (18) | 26 (25) | 107 (17) | 0.05 | 0 (0) |
| Former smoking | 105 (17) | 11 (14) | 94 (18) | 0.52 | 133 (18) |
| Ever smoking | 238 (32) | 37 (35) | 201 (32) | 0.50 | 0 (0) |
| Atrial fibrillation | 136 (18) | 1 (1) | 135 (21) | <0.001 | 13 (2) |
| Coronary heart disease | 132 (18) | 19 (18) | 113 (18) | 0.89 | 0 (0) |
| Diabetes mellitus | 115 (16) | 25 (24) | 90 (14) | 0.02 | 1 (<1) |
| Positive family history | 112 (20) | 12 (16) | 100 (21) | 0.44 | 182 (25) |
| Clinical data – median [IQR] | |||||
| BMI (kg/m2) | 25.7 [23.1–28.3] | 26.4 [24.4–29.1] | 25.6 [22.9–28.2] | 0.01 | 172 (23) |
| NIHSS on admission | 6 [3–13] | 5 [3–12] | 6 [3–13] | 0.63 | 2 (<1) |
| mRS before | 0 [0–0] | 0 [0–0] | 0 [0–0] | 0.69 | 6 (<1) |
| Medication on admission – n (%) | |||||
| Statins | 194 (26) | 35 (34) | 159 (25) | 0.07 | 3 (<1) |
| Platelet inhibitors | 278 (37) | 52 (50) | 226 (35) | 0.007 | 1 (<1) |
| Antihypertensive drugs | 440 (59) | 76 (73) | 364 (57) | 0.002 | 3 (<1) |
| Anticoagulation | 74 (10) | 0 (0) | 74 (12) | <0.001 | 2 (<1) |
| Laboratory values – median [IQR] | |||||
| Lp(a), nmol/l | 16.9 [6.1–60] | 23 [9.8–80] | 16.3 [5.8–57] | 0.01 | 0 (0) |
| Lp(a) >100 nmol/l- n (%) | 127 (17) | 23 (22) | 104 (16) | 0.16 | 0 (0) |
| LDL-C, mmol/l | 2.6 [2.0–3.3] | 2.6 [2.1–3.4] | 2.6 [2.0–3.3] | 0.33 | 20 (3) |
| Apolipoprotein B, g/l | 0.95 [0.78–1.13] | 0.94 [0.8–1.13] | 0.95 [0.77–1.13] | 0.74 | 27 (4) |
| Cholesterol, mmol/l | 4.7 [4.0–5.5] | 4.6 [4.1 –5.4] | 4.7 [4.0–5.5] | 0.75 | 0 (0) |
| Triglycerides, mmol/l | 1.4 [1.0–2.0] | 1.4 [1.1–2.0] | 1.4 [1.0–2.0] | 0.92 | 0 (0) |
| Non-HDL-C, mmol/l | 3.3 [2.7–4.2] | 3.3 [2.9–4.2] | 3.3 [2.7–4.1] | 0.58 | 1 (<1) |
| HbA1c, % | 5.8 [5.6–6.2] | 6.0 [5.7–6.3] | 5.8 [5.6–6.2] | 0.002 | 51 (7) |
| TOAST aetiology – n (%) | |||||
| Macroangiopathy | 105 (14) | 105 (100) | 0 (0) | – | 0 (0) |
| Cardioembolism | 286 (38) | 0 (0) | 286 (45) | – | 0 (0) |
| Microangiopathy | 43 (6) | 0 (0) | 43 (7) | – | 0 (0) |
| Other etiologies | 28 (4) | 0 (0) | 28 (4) | – | 0 (0) |
| Undetermined, evaluations complete | 116 (16) | 0 (0) | 116 (18) | – | 0 (0) |
| Undetermined, evaluations incomplete | 95 (13) | 0 (0) | 95 (15) | – | 0 (0) |
| More than one possible aetiology | 70 (9) | 0 (0) | 70 (11) | – | 0 (0) |
LAA: large artery atherosclerosis; IQR: interquartile range; BMI: body mass index; NIHSS: National Institutes of Health Stroke Scale; mRS: modified Rankin Stroke scale; Lp(a): lipoprotein(a); LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol; TOAST: Trial of Org 10172 in Acute Stroke Treatment.
In univariable analysis, Lp(a) was associated with LAA stroke aetiology (OR 1.54 [95% CI 1.11–2.15], p = 0.01; table 1). In multivariable regression, Lp(a) remained independently associated with LAA stroke after adjusting for cardiovascular risk factors (aOR 1.47 [95% CI 1.03–2.09], p = 0.03; table 2). These results were consistent with the sensitivity analysis conducted with alternative covariable selection using backward selection and after excluding individuals with undetermined aetiology (with incomplete evaluation) and patients with more than one possible underlying aetiology (tables S1, S2, S3 in the appendix).
Table 2Univariable and multivariable regression of the association of log10 Lp(a) with LAA stroke aetiology.
| Univariable models (LAA vs non-LAA) | OR | 95% CI | p-value |
| Log10 Lp(a) | 1.54 | 1.11–2.15 | 0.01 |
| Age | 1.01 | 0.99–1.02 | 0.27 |
| Female sex | 0.56 | 0.36–0.89 | 0.02 |
| Arterial hypertension | 2.16 | 1.29–3.61 | 0.003 |
| Diabetes | 1.90 | 1.15–3.14 | 0.01 |
| Ever smoking | 1.18 | 0.77–1.83 | 0.45 |
| Log10 LDL-C | 1.77 | 0.51–6.10 | 0.37 |
| Statins on admission | 1.52 | 0.98–2.37 | 0.06 |
| Antiplatelets on admission | 1.78 | 1.18–2.70 | 0.01 |
| Multivariable model (LAA vs non-LAA) | aOR | 95% CI | p-value |
| Log10 Lp(a) | 1.47 | 1.03–2.09 | 0.03 |
| Age | 1.00 | 0.98–1.02 | 0.96 |
| Female sex | 0.52 | 0.31–0.85 | 0.009 |
| Arterial hypertension | 1.99 | 1.13–3.50 | 0.02 |
| Diabetes | 1.99 | 1.16–3.39 | 0.01 |
| Ever smoking | 1.03 | 0.64–1.65 | 0.90 |
| Log10 LDL-C | 3.99 | 0.97–16.49 | 0.06 |
| Statins on admission | 1.02 | 0.60–1.74 | 0.94 |
| Antiplatelets on admission | 1.54 | 0.95–2.50 | 0.08 |
LAA: large artery atherosclerosis; (a)OR: (adjusted) odds ratio; CI: confidence interval; Lp(a): lipoprotein a; LDL-C: low-density lipoprotein cholesterol.
The interaction between Lp(a) and diabetes mellitus was statistically significant in univariable analysis (p-value for interaction = 0.045). However, Lp(a) was not associated with LAA stroke (aOR 0.85 [95% CI: 0.43–1.68]) among patients with diabetes mellitus but rather only among patients without diabetes mellitus (aOR 1.9[95% CI 1.29–2.80]). To further explore this interaction, we compared Lp(a) levels among patients with and without diabetes mellitus (14.6 [IQR 5.5–60] vs 17.4 [IQR 6.3–61], p = 0.45). No significant interaction was found between Lp(a) and age, gender, hypertension, smoking, statin intake on admission, or LDL-C levels on admission (figure 2, table S4 in the appendix).
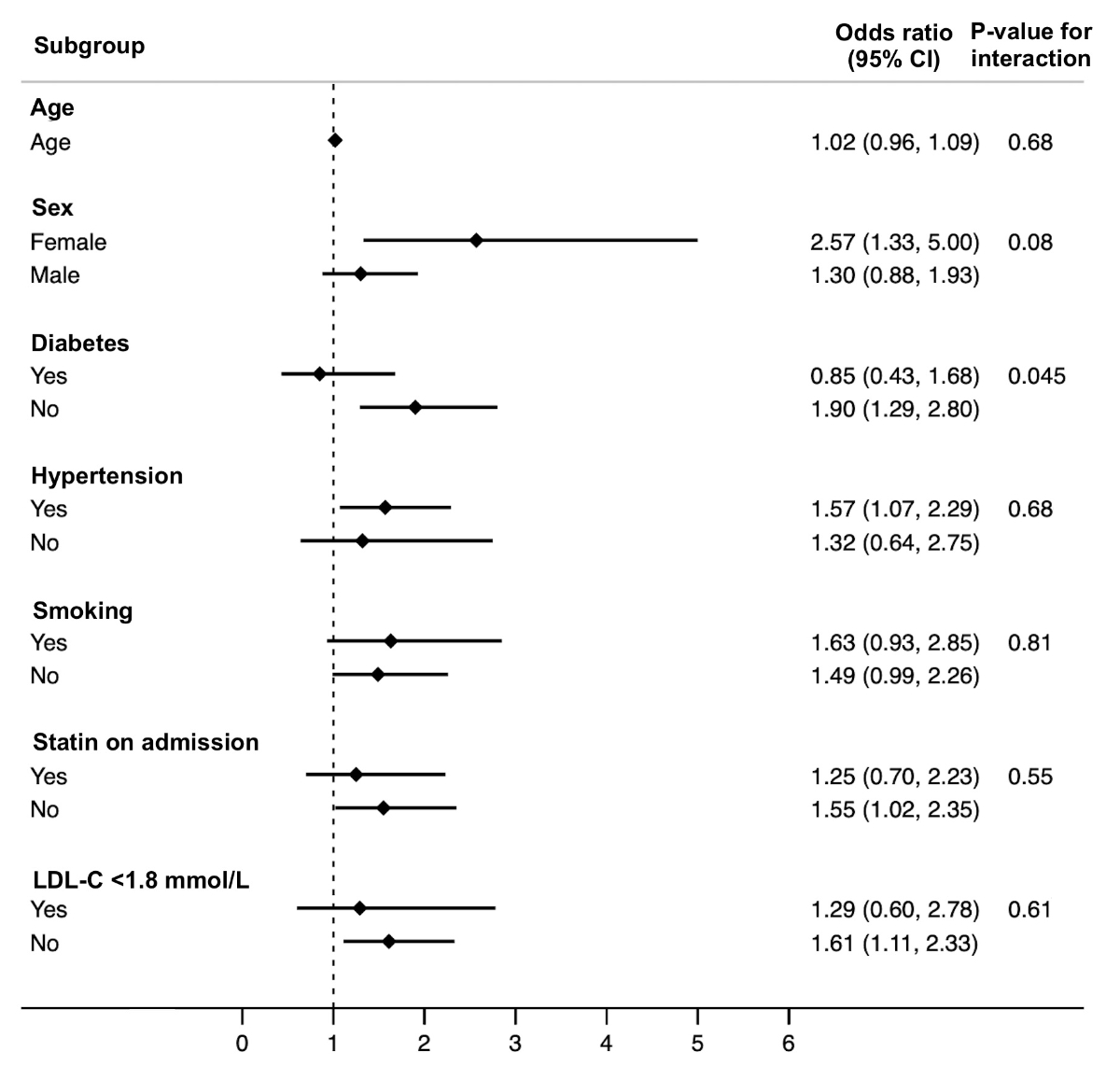
Figure 2Interaction analysis for the association of Lp(a) with large artery atherosclerosis stroke aetiology in different subgroups, odds ratios (95% CI).
Our main findings are: (1) among ischaemic stroke patients, those with LAA stroke aetiology had higher plasma Lp(a) levels than patients with non-LAA stroke; (2) after adjusting for traditional cardiovascular risk factors, Lp(a) remained associated with LAA stroke aetiology, validating higher Lp(a) blood levels in LAA stroke patients compared to other etiological subtypes; (3) among patients with diabetes mellitus, no significant association between Lp(a) and LAA was found.
Our finding of higher plasma Lp(a) among patients with stroke of LAA aetiology is consistent with the results of the multicentre, prospective BIOSIGNAL study. The latter yielded Lp(a) levels in patients with LAA stroke and non-LAA stroke that were similar to those observed in our cohort. In addition, two cohort studies with smaller sample sizes reported similar findings about the association of Lp(a) and LAA stroke [16, 17]. An Asian cohort study found that Lp(a) was associated with LAA stroke and intra- or extracranial stenosis [8].
A significant interaction between Lp(a) and diabetes mellitus was present. Among patients with diabetes, Lp(a) was not associated with LAA stroke. The lack of association between Lp(a) and LAA among patients with diabetes can be explained by (1) diabetes itself being a strong independent risk factor for LAA stroke, diluting the association between Lp(a) and LAA, and (2) by numerically lower Lp(a) levels in patients with diabetes, although the difference was small. The relationship between Lp(a) and diabetes mellitus is complex.[18] Several studies have reported an inverse correlation between Lp(a) levels and the risk of type 2 diabetes [19, 20, 21]. The pathophysiological mechanism of the inverse association between Lp(a) levels and diabetes risk is still unclear. A causal relationship has been suspected but has not yet been demonstrated by genetic association studies [21, 22]. Elevated Lp(a) levels in people with diabetes are an independent risk factor for cardiovascular disease in general, beyond LAA stroke [23].
In contrast to the BIOSIGNAL investigators, we did not observe an interaction between Lp(a) and age. In BIOSIGNAL, patients younger than 60 years showed the strongest association between Lp(a) and LAA (aOR for patients <60 years: 3.64 [95 % CI 1.76–7.53], p <0.001] [11]. The modest number of patients <60 years with LAA stroke may explain the lack of a statistically significant interaction.
Other interactions between Lp(a) and concomitant risk factors could not be found in our data. Effect modification between Lp(a) and statin use, as well with LDL-C – both as a continuous and dichotomous variable – was non-significant. However, the sample size of our cohort prevented us from assessing of whether low LDL or lowering LDL pharmacologically can mitigate the increased risk of LAA associated with high Lp(a) levels.
No approved pharmacological agents specifically lower Lp(a). The PCSK-9 inhibitors alirocumab and evolocumab reduce Lp(a) levels by 25–30% in patients with Lp(a) levels above 50 mg/dl. Yet, the contribution of PCSK-9-induced Lp(a) lowering to the reduction in cardiovascular events is modest, as shown by a recent Mendelian randomisation study [24]. Pelacarsen, a pharmacological agent designed to reduce Lp(a) levels specifically, is being investigated in a phase 3 clinical trial – Lp(a)-HORIZON (NCT04023552) – among patients with established but stable cardiovascular disease and Lp(a) >70 mg/dl. Pelacarsen impairs apo(a) synthesis in hepatocytes. In previous phase 1 and 2 studies, Pelacarsen showed promising results in lowering Lp(a) [25].
Our study’s strengths include the use of a large prospective data set with imaging-proven ischaemic stroke and the high completeness of the data on the cardiovascular risk factors.
We also acknowledge limitations. (1) The sample size prevents the drawing of conclusions about the interactions among other cardiovascular risk factors, especially age and LDL-C. Moreover the incidence of recurrent stroke was low (10/743 patients; 1.3%), making further regression analyses in this domain inappropriate. (2) 25–33% of patients were treated with statins, antihypertensive agents or platelet inhibitors potentially biasing the association between Lp(a) and LAA and the effect modification with concomitant cardiovascular risk factors. (3) There is an inherent risk of misclassification bias in classifying causes of stroke, as more than one possible stroke aetiology may be present. Classification systems that provide additional information on the probability of the underlying pathomechanism (e.g. the Causative Classification of Stroke) had not been established at the time this study was planned. (4) CoRisk recruited patients in Switzerland, and they were predominantly of Caucasian ethnicity, limiting the generalisability of the results.
We externally validated the independent association of higher Lp(a) levels with LAA stroke aetiology in a predominantly Caucasian cohort. This contributes to the emerging evidence of Lp(a) as a risk factor for LAA stroke and might be important for randomised control trials investigating the effect of Lp(a) lowering agents.
The original data will be made available by the corresponding authors upon reasonable request.
We would like to thank all CoRisk contributors.
Author contributions statement: SR and GMDM planned the work. SR and TD performed the analyses. SR drafted the manuscript. All authors interpreted the results and substantially contributed to the final manuscript. Guarantors: SR and GMDM.
The CoRisk study was funded by an unrestricted research grant from Thermo Fisher Scientific, Thermo Scientific Biomarkers; by the Clinical Trial Unit, University of Bern through the De-Quervain research grant for young clinical investigators; the Foundation of the Inselspital Bern; by the Foundation Pro Scientia et Arte, Bern and by the Swisslife Jubiläumsstiftung for Medical Research.
This analysis was supported by the Swiss National Science Foundation. Roche provided the assays for Lp(a) measurements. The funder did not influence the design of the study, data collection, analysis, or interpretation of the data.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. M. Arnold received research grants from the Swiss National Science Foundation and the Swiss Heart Foundation, honoraria for lectures from Astra Zeneca, Bayer, Novartis and Sanofi and honoraria for scientific advisory boards from Amgen, Bayer, Boehringer Ingelheim, BMS, Daiichi Sankyo, Novartis and Novo Nordisk. G. M. De Marchis declares a research grant from Novartis for a study unrelated to the present study. Payments have been made to the research fund of the University Hospital Basel. T. D. Dittrich was or is supported by the Swiss Heart Foundation, the science funds of the University Hospital Basel and the Research fund for excellent young researchers of the University of Basel. He holds shares from Johnson & Johnson, Roche, Lilly, Bristol-Myers Squibb, Merck and Novo Nordisk. U. Fischer reported research support of the Swiss National Science Foundation and the Swiss Heart Foundation. PI of the ELAN trial, Co-PI of the DISTAL, TECNO, SWIFT DIRECT and SWITCH trial. Research grants from Medtronic (BEYOND SWIFT, SWIFT DIRECT) and from Stryker, Rapid medical, Penumbra and Phenox (DISTAL). Consultancies for Medtronic, Stryker, and CSL Behring (fees paid to institution). Participation in an advisory board for Alexion/Portola, Boehringer Ingelheim, Biogen and Acthera (fees paid to institution). Member of a clinical event committee (CEC) of the COATING study (Phenox) and member of the data and safety monitoring committee (DSMB) of the TITAN, LATE_MT and IN EXTREMIS trials. Vice-presidency of the Swiss Neurological Society. S. Jung has been receiving support from the Swiss National Science Foundation and Swiss Heart Foundation. M. Katan received funding from the Swiss National Science Foundation (PZ00P3_142422, 32003B_182267) the Swiss Heart Foundation, the Göhner Foundation as well as the Baasch Medicus Foundation; She has acted on advisory boards for Astra Zeneca, Medtronic and Bayer. N. Peters received honoraria for lectures/presentations from Vifor Pharma, OM Pharma and Novo Nordisk and has served on advisory boards for Novo Nordisk, AstraZeneca, OM Pharma, Vifor Pharma and Medtronic. The other authors report no conflicts of interest related to this work.
1. Gencer B, Kronenberg F, Stroes ES, Mach F. Lipoprotein(a): the revenant. Eur Heart J. 2017 May;38(20):1553–60. 10.1093/eurheartj/ehx033
2. Nordestgaard BG, Chapman MJ, Ray K, Borén J, Andreotti F, Watts GF, et al.; European Atherosclerosis Society Consensus Panel. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010 Dec;31(23):2844–53. 10.1093/eurheartj/ehq386
3. Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017 Feb;69(6):692–711. 10.1016/j.jacc.2016.11.042
4. Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al.; PROCARDIS Consortium. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009 Dec;361(26):2518–28. 10.1056/NEJMoa0902604
5. Langsted A, Nordestgaard BG, Kamstrup PR. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J Am Coll Cardiol. 2019 Jul;74(1):54–66. 10.1016/j.jacc.2019.03.524
6. Hoogeveen RC, Ballantyne CM. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin Chem. 2021 Jan;67(1):143–53. 10.1093/clinchem/hvaa252
7. Nave AH, Lange KS, Leonards CO, Siegerink B, Doehner W, Landmesser U, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015 Oct;242(2):496–503. 10.1016/j.atherosclerosis.2015.08.021
8. Kim BS, Jung HS, Bang OY, Chung CS, Lee KH, Kim GM. Elevated serum lipoprotein(a) as a potential predictor for combined intracranial and extracranial artery stenosis in patients with ischemic stroke. Atherosclerosis. 2010 Oct;212(2):682–8. 10.1016/j.atherosclerosis.2010.07.007
9. Pan Y, Li H, Wang Y, Meng X, Wang Y. Causal Effect of Lp(a) [Lipoprotein(a)] Level on Ischemic Stroke and Alzheimer Disease: A Mendelian Randomization Study. Stroke. 2019 Dec;50(12):3532–9. 10.1161/STROKEAHA.119.026872
10. Arora P, Kalra R, Callas PW, Alexander KS, Zakai NA, Wadley V, et al. Lipoprotein(a) and Risk of Ischemic Stroke in the REGARDS Study. Arterioscler Thromb Vasc Biol. 2019 Apr;39(4):810–8. 10.1161/ATVBAHA.118.311857
11. Arnold M, Schweizer J, Nakas CT, Schütz V, Westphal LP, Inauen C, et al. Lipoprotein(a) is associated with large artery atherosclerosis stroke aetiology and stroke recurrence among patients below the age of 60 years: results from the BIOSIGNAL study. Eur Heart J. 2021 Jun;42(22):2186–96. 10.1093/eurheartj/ehab081
12. De Marchis GM, Katan M, Weck A, Brekenfeld C, Mattle HP, Buhl D, et al. Copeptin and risk stratification in patients with ischemic stroke and transient ischemic attack: the CoRisk study. International journal of stroke : official journal of the International Stroke Society. 2013;8(3):214-8. 10.1111/j.1747-4949.2011.00762.x
13. Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993 Jan;24(1):35–41. 10.1161/01.STR.24.1.35
14. Goldstein LB, Samsa GP, Matchar DB, Horner RD. Charlson Index comorbidity adjustment for ischemic stroke outcome studies. Stroke. 2004 Aug;35(8):1941–5. 10.1161/01.STR.0000135225.80898.1c
15. Banerjee C, Chimowitz MI. Stroke Caused by Atherosclerosis of the Major Intracranial Arteries. Circ Res. 2017 Feb;120(3):502–13. 10.1161/CIRCRESAHA.116.308441
16. Cerrato P, Imperiale D, Fornengo P, Bruno G, Cassader M, Maffeis P, et al. Higher lipoprotein (a) levels in atherothrombotic than lacunar ischemic cerebrovascular disease. Neurology. 2002 Feb;58(4):653–5. 10.1212/WNL.58.4.653
17. Petersen NH, Schmied AB, Zeller JA, Plendl H, Deuschl G, Zunker P. Lp(a) lipoprotein and plasminogen activity in patients with different etiology of ischemic stroke. Cerebrovasc Dis. 2007;23(2-3):188–93. 10.1159/000097640
18. Lamina C, Ward NC. Lipoprotein (a) and diabetes mellitus. Atherosclerosis. 2022 May;349:63–71. 10.1016/j.atherosclerosis.2022.04.016
19. Mora S, Kamstrup PR, Rifai N, Nordestgaard BG, Buring JE, Ridker PM. Lipoprotein(a) and risk of type 2 diabetes. Clin Chem. 2010 Aug;56(8):1252–60. 10.1373/clinchem.2010.146779
20. Lan NS, Chan DC, Pang J, Fegan PG, Yeap BB, Rankin JM, et al. Lipoprotein(a) in Patients With Type 2 Diabetes and Premature Coronary Artery Disease in the Coronary Care Unit. Heart Lung Circ. 2021 May;30(5):734–40. 10.1016/j.hlc.2020.09.932
21. Kamstrup PR, Nordestgaard BG. Lipoprotein(a) concentrations, isoform size, and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2013 Nov;1(3):220–7. 10.1016/S2213-8587(13)70064-0
22. Tolbus A, Mortensen MB, Nielsen SF, Kamstrup PR, Bojesen SE, Nordestgaard BG. Kringle IV Type 2, Not Low Lipoprotein(a), as a Cause of Diabetes: A Novel Genetic Approach Using SNPs Associated Selectively with Lipoprotein(a) Concentrations or with Kringle IV Type 2 Repeats. Clin Chem. 2017 Dec;63(12):1866–76. 10.1373/clinchem.2017.277103
23. Waldeyer C, Makarova N, Zeller T, Schnabel RB, Brunner FJ, Jørgensen T, et al. Lipoprotein(a) and the risk of cardiovascular disease in the European population: results from the BiomarCaRE consortium. Eur Heart J. 2017 Aug;38(32):2490–8. 10.1093/eurheartj/ehx166
24. De Marchis GM, Dittrich TD, Malik R, Zietz AV, Kriemler LF, Ference BA, et al. Genetic proxies for PCSK9 inhibition associate with lipoprotein(a): effects on coronary artery disease and ischemic stroke. Atherosclerosis. 2022 Nov;361:41–6. 10.1016/j.atherosclerosis.2022.09.007
25. Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, et al.; AKCEA-APO(a)-LRx Study Investigators. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med. 2020 Jan;382(3):244–55. 10.1056/NEJMoa1905239
Table S1Sensitivity analysis: Multivariable regression of the association of log10 Lp(a) with LAA stroke aetiology with alternative covariable selection using backward selection.
| LAA vs non-LAA | aOR | 95% CI | p-value |
| Log10 Lp(a) | 1.04 | 1.00–1.09 | 0.04 |
| Female sex | 0.93 | 0.89–0.98 | 0.01 |
| Arterial hypertension | 1.07 | 1.01–1.13 | 0.02 |
| Diabetes | 1.10 | 1.02–1.18 | 0.01 |
| Log10 LDL-C | 1.14 | 0.99–1.33 | 0.08 |
| Antiplatelets on admission | 1.06 | 1.00–1.11 | 0.05 |
aOR: adjusted odds ratio; CI: confidence interval; Lp(a): lipoprotein(a): LDL-C: LDL cholesterol.
Table S2Sensitivity analysis: Multivariable regression of the association of log10 Lp(a) with LAA stroke aetiology with excluded individuals with undetermined aetiology (with incomplete evaluation) and patients with more than one possible aetiology. N = 561 (only cases with complete sets of covariables of interest were considered).
| LAA vs non-LAA | aOR | 95% CI | p-value |
| Log10 Lp(a) | 1.59 | 1.09–2.31 | 0.02 |
| Age | 1.01 | 0.99–1.03 | 0.43 |
| Female sex | 0.46 | 0.27–0.76 | 0.003 |
| Arterial hypertension | 1.92 | 1.07–3.44 | 0.03 |
| Diabetes | 2.37 | 1.34–4.19 | 0.003 |
| Ever smoking | 1.09 | 0.66–1.78 | 0.74 |
| LDL-C | 1.24 | 0.96–1.60 | 0.09 |
| Statins on admission | 1.07 | 0.61–1.87 | 0.81 |
| Antiplatelets on admission | 1.59 | 0.96–2.61 | 0.07 |
(a)OR: (adjusted) odds ratio; CI: confidence interval; Lp(a): lipoprotein a.
Table S3Multivariable regression of the association of log10 Lp(a) with LAA stroke aetiology, LDL-C (absolute).
| LAA vs non-LAA | aOR | 95% CI | P-value |
| Log10 Lp(a) | 1.46 | 1.03–2.09 | 0.04 |
| Age | 1.00 | 0.98–1.02 | 0.98 |
| Female sex | 0.52 | 0.31–0.85 | 0.009 |
| Arterial hypertension | 2.00 | 1.14–3.52 | 0.02 |
| Diabetes | 2.00 | 1.17–3.43 | 0.01 |
| Ever smoking | 1.03 | 0.64–1.65 | 0.91 |
| LDL-C | 1.25 | 0.99–1.58 | 0.06 |
| Statins on admission | 1.04 | 0.61–1.78 | 0.89 |
| Antiplatelets on admission | 1.54 | 0.95–2.50 | 0.08 |
aOR: adjusted odds ratio; CI: confidence interval; Lp(a): lipoprotein(a); LDL-C: LDL cholesterol.
Table S4Interaction analysis for the association of Lp(a) with large artery atherosclerosis stroke aetiology in different subgroups, with adjusted odds ratios (95% CI). All models were adjusted for following covariables: age, sex, hypertension, diabetes, LDL-C (log transformed), smoking, statin treatment on admission, and platelet inhibitor treatment on admission.
| LAA vs non-LAA | aOR* | 95% CI* | p-value (for interaction) |
| Age x log10 Lp(a) | 0.99 | 0.97–1.02 | 0.67 |
| Female sex x log10 Lp(a) | 1.00 | 1.00–1.01 | 0.24 |
| Diabetes x log10 Lp(a) | 0.52 | 0.23–1.18 | 0.12 |
| Arterial hypertension x log10 Lp(a) | 1.31 | 0.57–3.04 | 0.53 |
| Ever smoking x log10 Lp(a) | 1.26 | 0.60–2.63 | 0.54 |
| Statins on admission x log10 Lp(a) | 0.75 | 0.36–1.58 | 0.46 |
| Antiplatelets on admission x log10 Lp(a) | 0.89 | 0.44–1.79 | 0.75 |
| Log10 LDL-C x log10 Lp(a) | 0.88 | 0.62–1.24 | 0.46 |
aOR: adjusted odds ratio; CI: confidence interval; Lp(a): lipoprotein(a); LDL-C: LDL cholesterol. * for the interaction term