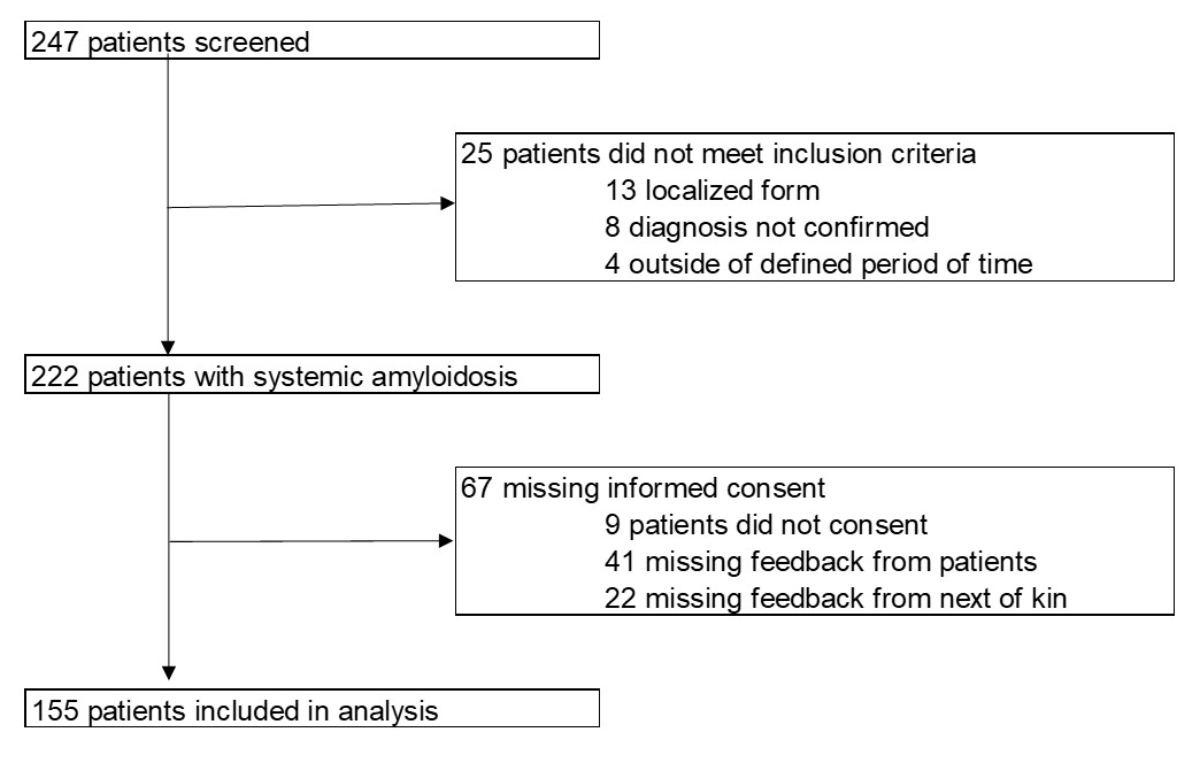
Figure 1Patients included in the analysis.
DOI: https://doi.org/https://doi.org/10.57187/s.3485
The term “amyloidosis” refers to a heterogeneous group of diseases with typical histopathological features defined as tissue deposition of insoluble proteins and consequent organ dysfunction [1, 2]. To date, more than thirty different proteins are known to be amyloidogenic [3]. The most common types include immunoglobulin light-chain amyloidosis due to an underlying haematological disorder, transthyretin amyloidosis in which wild-type and hereditary variant forms are distinguished, and amyloid A amyloidosis due to chronic inflammatory processes. Although organ tropism and hence clinical manifestations may differ between the various protein subtypes, there are nevertheless some common clinical features such as heart failure, nephropathy and peripheral neuropathy [2]. Most of these symptoms are not specific to amyloidosis, but can also occur with common diseases such as diabetes mellitus or hypertension. Therefore, the diagnosis of systemic amyloidosis is challenging, and treatment initiation often delayed [4]. Diagnostic work-up of patients with suspected amyloidosis must allow unambiguous characterisation of the amyloid subtype and should provide information about the spectrum and extent of organ involvement.
With rare diseases such as these, which present with a variety of symptoms, the path to correct diagnosis and the choice of optimal treatment is a challenge. Systemic amyloidosis is therefore a prototypical disease entity that requires an interdisciplinary diagnostic and and therapeutic approach [5].
Until recently, there have been little epidemiological data on systemic amyloidosis in Switzerland. Therefore, in 2013, the Collaborative Amyloidosis Network Zurich was founded at University Hospital Zurich (USZ) as the first network of its kind in Switzerland. It brings together specialists in haematology/oncology, cardiology, nephrology, pathology, neurology, medical genetics, rheumatology and gastroenterology, in order to improve and standardise the diagnostic work-up and treatment of these often critically ill patients with rare diseases.
It has long been recognised that medical registries do not just provide basic epidemiological data, but contribute to better understanding of mechanisms of disease within specific healthcare systems, ultimately resulting in better medical outcomes, especially in rare diseases for which randomised controlled trials are scarce [6]. Therefore, one of the central pillars of the network is the Swiss Amyloidosis Registry, a patient registry that collects data for all types of systemic amyloidosis. The objective of the present study was to describe the epidemiological pattern of systemic amyloidosis in Switzerland and to study patient outcomes in the different amyloidosis subtypes.
Patients have been included since 2005. Patient data were collected retrospectively for the period 2005 to 2014 and prospectively from 2015. The cut-off date for the current data analysis is February 2020. Screening of eligible patients was performed locally; data monitoring was performed centrally at University Hospital Zurich.
Patients 18 years or older diagnosed with any subtype of systemic amyloidosis were eligible for inclusion if they had been or were being treated in one of the four referring centres (USZ; Graubunden cantonal hospital [KSGR]; the oncological institute of southern Switzerland [IOSI]; St Gallen cantonal hospital [KSSG]). Patients with any form of amyloidosis were included in the analysis, except those with localised amyloidosis. Written informed consent was required from either the patient or, if deceased, his/her next of kin prior to inclusion in the registry. The local ethics committee approved the study (KEK-ZH-NR 2014-0490 / PB_2016_01744), but did not allow for inclusion of patients without written informed consent, if they were lost to follow-up, and/or could not be followed due to changes of address or death.
Diagnosis of systemic amyloidosis required direct or indirect proof of organ involvement, as well as one or more of the following features: (a) a positive biopsy with typical birefringence of a Congo red-stained specimen under polarised light, and/or (b) the presence of a known amyloidogenic mutation, and/or (c) non-biopsy-proven diagnosis of cardiac transthyretin amyloidosis by scintigraphy [7, 8].
Amyloid subtyping was performed locally by immunohistochemistry, generally by using commercially available antibodies. After the Collaborative Amyloidosis Network was founded in 2013, the tissue samples of inconclusive cases were sent for external review, mostly by the German reference pathology centre (Prof. Röcken, Kiel, Germany). If the diagnosis remained inconclusive, the Amyloidosis Research and Treatment Center Pavia, Italy, was contacted for evaluation using mass spectrometry (MS)-based proteomics analysis [9, 10]. The Institute of Medical Genetics at the University of Zurich performed routine genetic testing.
Primary data were extracted locally from the electronic and non-electronic patient charts and entered into electronic case report forms in SecuTrial®. SecuTrial® is a web-based application allowing access by authorised personnel at all participating centres. A unique patient ID number was computer-generated upon data entry. The information linking patient name to Study ID (key) was printed and stored locally separate from all other data. Accuracy and completeness of the data were analysed centrally at University Hospital Zurich.
Baseline data were captured at the time of diagnosis ( ± 3 months). Baseline data consisted of approximately 300 parameters including personal medical history, symptoms leading to diagnosis, demographic data and other data (see electronic case report forms available as supplementary file for download at https://doi.org/10.57187/s.3485). Organ involvement was defined as biopsy-proven amyloid deposition in the respective organ (or tissue) and/or typical organ alterations as defined by the working group for light-chain and transthyretin amyloidosis [7, 11]. Follow-up data were assessed half-yearly for the first two years then annually. The follow-up data included, among others, pharmacological and non-pharmacological treatment, changes in organ function and laboratory values (see electronic case report forms).
Statistical analyses were performed with IBM SPSS 28.010 (IBM, Armonk, New York, USA) and R statistics version 4.1.3. Survival analysis was performed by the Kaplan-Meier method and compared using the log rank test. The median observation time was estimated by the inverse Kaplan-Meier method [12].
Between 1 January 2005 and 29 February 2020, 247 patients were screened, and 155 patients with confirmed systemic amyloidosis were included in the present analysis (figure 1): 69 and 86 in the retrospective and prospective cohorts, respectively.
Figure 1Patients included in the analysis.
Overall, the most common amyloidosis type was light-chain amyloidosis (49.7%, n = 77), followed by transthyretin amyloidosis (40%, n = 62) (25.8%, n = 40 wild-type transthyretin amyloidosis and 8.4%, n = 13 hereditary transthyretin amyloidosis); in 5.8%, n = 9, transthyretin gene (TTR) mutation analysis was not performed. Amyloid A amyloidosis accounted for 5.2%, n = 8. In some of the patients (4.5%, n = 7), subtyping was not productive. Concomitant transthyretin and light-chain amyloidosis was present in one patient (0.6%).
Mean age at diagnosis was 64.7 years (median 68.2 years, range 18.6–85) with a predominance of males (76%, n = 118; light-chain amyloidosis n = 55, transthyretin amyloidosis n = 54, amyloid A amyloidosis n = 4, unclear n = 5). Patients were included from the four centres (USZ, KSGR, IOSI, KSSG). Most patients originated from Switzerland (63%, n = 98), followed by Italy (11%, n = 17) and Portugal (4%, n = 6).
Table 1Baseline characteristics.
| Light-chain amyloidosis | Transthyretin amyloidosis, wild-type | Transthyretin amyloidosis, variant | Transthyretin amyloidosis without genetic testing | Amyloid A amyloidosis | ||
| Age (yrs) | Mean ± SEM | 63.3 ± 1.3 | 73.1 ± 1.4 | 43.2 ± 4.5 | 77.1 ± 1.6 | 46.2 ± 6.2 |
| Range | 38.3–84.9 | 51.4–84.9 | 18.6–73.2 | 68.2–84.3 | 24.7–76.4 | |
| Data available for no/total cases | 77/77 | 40/40 | 12/13 | 9/9 | 8/8 | |
| Male sex | n (%) | 54 (70%) | 38 (95%) | 7 (53%) | 9 (100%) | 4 (50%) |
| BMI (kg/m2) | Mean ± SEM | 25.4 ± 0.5 | 27.1 ± 0.6 | 24.2 ± 1.5 | 28.4 ± 2.9 | 25 ± 2.3 |
| Range | 17.8–36 | 18.3–37 | 18.7–36 | 22–39 | 21.8–36.6 | |
| Data available for no/total cases | 69/77 | 40/40 | 11/13 | 5/9 | 4/8 | |
| Number of organs involved, no (%) | 0 | 0 | 0 | 1 (7.7%) * | 0 | 0 |
| 1 | 22 (28.6%) | 16 (40%) | 2 (15.4%) | 5 (55.5%) | 6 (75%) | |
| 2 | 30 (38.9%) | 21 (52.5%) | 5 (38.5%) | 3 (33.3%) | 1 (12.5%) | |
| 3 | 13 (16.9%) | 3 (7.5%) | 2 (15.4%) | 1 (11.1%) | 1 (12.5%) | |
| ≥4 | 12 (15.6%) | 0 | 2 (15.4%) | 0 | 0 | |
| Unknown | 0 | 0 | 1 (7.7%) | 0 | 0 | |
| Involved organs, no (%) | Heart | 55 (71.4%) | 38 (95%) | 7 (53.8%) | 9 (100%) | 1 (12.5%) |
| Kidney | 46 (59.7%) | 0 | 1 (7.7%) | 1 (11.1%) | 7 (87.5%) | |
| GI | 22 (28.6%) | 2 (5%) | 1 (7.7%) | 0 | 1 (12.5%) | |
| Liver | 6 (7.8%) | 2 (5%) | 0 | 0 | 0 | |
| Nerve | 18 (23.4%) | 5 (12.5%) | 11 (84.6%) | 0 | 1 (12.5%) | |
| Soft tissue | 15 (19.5%) | 2 (5%) | 1 (7.7%) | 0 | 1 (12.5%) | |
| Carpal tunnel syndrome | 8 (10.4%) | 23 (57.5%) | 6 (46.2%) | 4 (44.4%) | 0 | |
| NT-pro BNP (ng/l) | Mean ± SEM | 4433 ± 990 | 2913 ± 587 | 1153 ± 512 | 3851 ± 1288 | 10657 |
| Range | 81–44085 | 46–18143 | 24–3710 | 527–6748 | (one value) | |
| Data available for no/total cases | 66/77 | 38/40 | 7/13 | 4/9 | 1/8 | |
| Troponin T (ng/l) | Mean ± SEM | 41 ± 5.1 | 218 ± 162 | 41 ± 28 | 36 ± 10 | NA |
| Range | 0–166 | 11–4578 | 5–152 | 26–46 | ||
| Data available for no/total cases | 51/77 | 28/40 | 5/13 | 2/9 | 0/8 | |
| Creatinine (µmol/l) | Mean ± SEM | 109 ± 6 | 118 ± 19 | 98 ± 16 | 101 ± 14 | 132 ± 32 |
| Range | 52–282 | 52–832 | 54–173 | 51–140 | 79–188 | |
| eGFR (ml/min) | Mean ± SEM | 66 ± 3 | 67 ± 3 | 82 ± 13 | 65 ± 9 | 50 ± 14 |
| Range | 22–111 | 5–104 | 40–140 | 43–101 | 22–70 | |
| Data available for no/total cases (creatinine and eGFR) | 72/77 | 39/40 | 8/13 | 6/9 | 3/8 | |
| eGFR ≤30 or dialysis, no (%) | 8 (10%) | 2 (5%) | 0 | 0 | 2 (25%) | |
BMI: body mass index; eGFR: estimated glomerular filtration rate; SEM: standard error of the mean.
*asymptomatic carrier
Diagnosis was made by tissue biopsy in 87.8% (n = 136) of patients and by a non-biopsy approach by scintigraphy in 10.3%, n = 16 (transthyretin amyloidosis patients) [8]; in 3 patients, data were missing. The most common biopsy sites were the heart (n = 49), kidney (n = 39), gastrointestinal tract (n = 38), abdominal fat tissue aspirate (n = 46), skin (n = 4) and lung (n = 3). One site was biopsied in 52.3% (n = 71; transthyretin amyloidosis n = 32, light-chain amyloidosis n = 39) of patients, two sites in 25.8% (n = 40) patients and three sites in 9% (n = 14) patients. In 5.2% (n = 8) patients, amyloid was found in the bone marrow biopsy. Notably not all bone marrow biopsies were routinely stained with Congo red.
Table 2Organ involvement of the most common types of amyloidosis.
| Type | n | Number of organs, median | Heart | Kidney | GI | Liver | PNS/ANS | CTS | Lung |
| Light-chain amyloidosis | 77 | 1.5 (0–3) | +++ | ++ | + | + | + | + | – |
| Transthyretin amyloidosis, wild-type | 40 | 1.5 (0–3) | +++ | – | – | – | + | ++ | – |
| Transthyretin amyloidosis, variant | 13 | 1 (0–2) | ++ | + | + | – | +++ | ++ | – |
| Amyloid A amyloidosis | 8 | 1.3 (1–3) | + | +++ | + | – | + | – | – |
CTS: carpal tunnel syndrome; GI: gastrointestinal tract; PNS/ANS: peripheral nervous system / autonomic nervous system;
–: 0–5%; +: 6–35%; ++ 36–70%; +++: 71–100%.
In 54.8% of transthyretin amyloidosis patients (n = 34), an endomyocardial biopsy was done to establish the diagnosis. In light-chain amyloidosis patients, the most common site of biopsy was the kidney (39%, n = 30), followed by the gastrointestinal tract (19.5%, n = 15) and the heart (16.9%, n = 13). In 9.1% of light-chain amyloidosis patients (n = 7), diagnosis was established with abdominal fat aspirate only.
Organ involvement was defined by either proven amyloid in organ biopsy, elevated biomarkers or typical symptoms such as weight loss, nausea, diarrhoea or constipation, polyneuropathy, orthostatic dysregulation, erectile dysfunction in absence of other causes.
Most patients (61.9%, n = 96) presented with multiorgan involvement with typical tropism determined by the amyloidosis type. However, single organ involvement was seen in all types of amyloidosis (n = 56, 36.1%; wild-type transthyretin amyloidosis n = 16/40, 40%; hereditary transthyretin amyloidosis n = 2/13, 15.3%; light-chain amyloidosis n = 22/77, 28.6% and amyloid A amyloidosis n = 6/8, 75%).
The number of patients with transthyretin and light-chain amyloidosis in our registry increased over time. From 2011, the numbers of diagnosed transthyretin amyloidosis patients increased rapidly (figure 2).
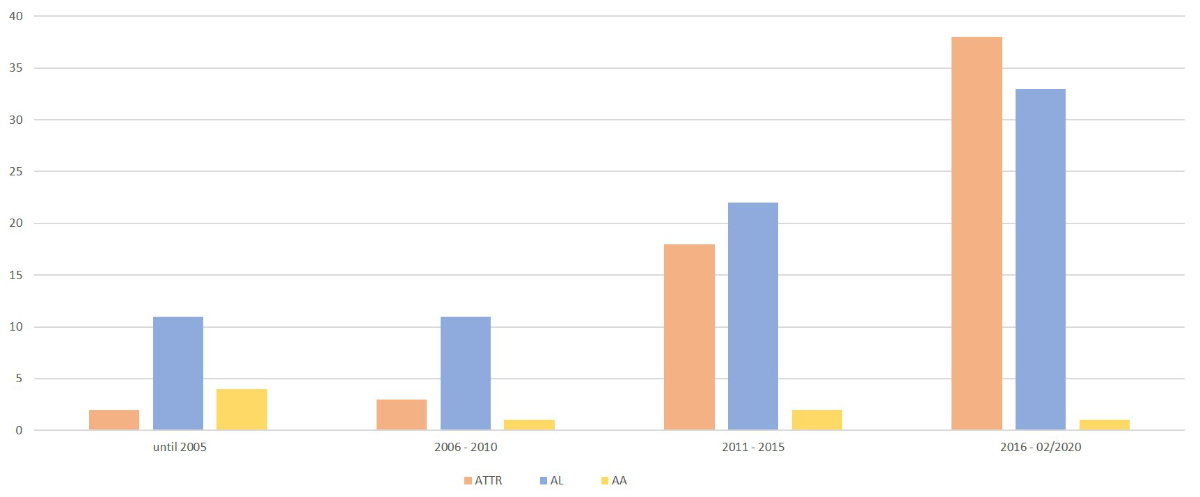
Figure 2Number of amyloidosis patients included in the registry over time. ATTR: transthyretin amyloidosis; AL: light-chain amyloidosis; AA: amyloid A amyloidosis.
Of the 77 patients with light-chain amyloidosis, free light chains were found in 57.1% (n = 44), IgG in 28.6% (n = 22), IgM in 9.1% (n = 7) and IgA in 5.2% (n = 4). Light-chain lambda was present in most of the patients (70.1%; n = 54). Translocation t(11;14) was found in 27.3% (n = 21). In 53.2% (n = 41) of the patients, FISH analysis was not performed (30 in the retrospective cohort, 11 in the prospective cohort).
Patients who were included in our registry were staged according to the revised Mayo staging system [11]. Stage 1, 2, 3 and 4 were present in 10.3%, n = 8, 22.1%, n = 17, 13%, n = 10 and 16.9%, n = 13, respectively. 37.7%, n = 29 patients could not be staged due to missing data (22 in the retrospective cohort, 7 in the prospective cohort).
Plasma cell infiltration grade was ≥10% in 62.3% (n = 48) and <10% in 24.7% (n = 19). In 13% (n = 10, 9 in retrospective cohort), the infiltration grade was not known.
Of all transthyretin amyloidosis patients, 87.1% (n = 54) had cardiac involvement. Overall, 53.2% (n = 33) presented with Gillmore stage 1, 16.1% (n = 10) with stage 2 and 9.7% (n = 6) with stage 3. In 21% (n = 13; n = 5 in the retrospective cohort), the Gillmore stage could not be determined [13].
In 22.6% (n = 14) patients, concomitant monoclonal gammopathy of undetermined significance (MGUS) was found; in 11.3%, n = 7 patients (n = 5 from the retrospective cohort), gammopathy screening was not done. 56.5% (n = 35) of the patients suffered from carpal tunnel syndrome, 14.5% (n = 9) from lumbar spinal stenosis.
In the retrospective cohort, the main trigger for genetic testing was neurological symptoms or a positive family history. In the prospective cohort, testing was recommended in all newly admitted transthyretin amyloidosis patients, if cost coverage by the Swiss health insurance fund was guaranteed. Of the 62 patients with transthyretin amyloidosis, TTR mutation analysis was performed in 74% (n = 46). Variant transthyretin amyloidosis with an amyloidogenic mutation in TTR was detected in 13 patients (28%).
In patients with variant transthyretin amyloidosis, the following mutations were found: p.(Val50Met) (n = 5, Portuguese ancestry, 4 different families), p.(Ile127Met) (n = 3, Bosnian ancestry, 1 family), p.(Phe53Val) (n = 1, Macedonian ancestry), p.(Thr79Lys) (n = 1, Egyptian ancestry), p.(Thr80Ala) (n = 1, Swiss ancestry), p.(Val142Ile) (n = 2, Swiss ancestry, individuals unrelated).
p.(Val50Met), p.(Ile127Met), p.(Phe53Val) patients presented with a predominantly neurological phenotype, whereas p.(Thr79Lys), p.(Thr80Ala) and p.(Val142Ile) patients showed a predominantly cardiac phenotype.
Of the 8 amyloid A amyloidosis patients, familial Mediterranean fever was the most common underlying disease (63%; n = 5), one patient had rheumatoid arthritis, one had Crohn’s disease, one had an unknown disease.
The median observation time for surviving patients calculated by the reverse Kaplan-Meier method was 3.29 years (95% confidence interval [CI] 2.33–4.87); 4.87 years (95% CI 3.14–7.22) in light-chain amyloidosis patients and 1.85 years (95% CI 1.48–3.66) in transthyretin amyloidosis patients. 32% of the patients (n = 50) died during the observation period. The average age at death was 68.7 years (median 70.2 years, range 34.5–87.3 years).
The survival analysis for light-chain and transthyretin amyloidosis patients is shown in figure 3. 1-, 3- and 5-year survival rates were 87.0% (95% CI 79.4–95.3%), 68.5% (95% CI 57.4–81.7%) and 66.0% (95% CI 54.6–79.9%) respectively for light-chain amyloidosis patients and 91.2% (95% CI 83.2–99.8%), 77.0% (95% CI 63.4–93.7%) and 50.6% (95% CI 31.8–80.3%) respectively for transthyretin amyloidosis patients. There was no significant difference between the two groups (p = 0.81) (figure 3).
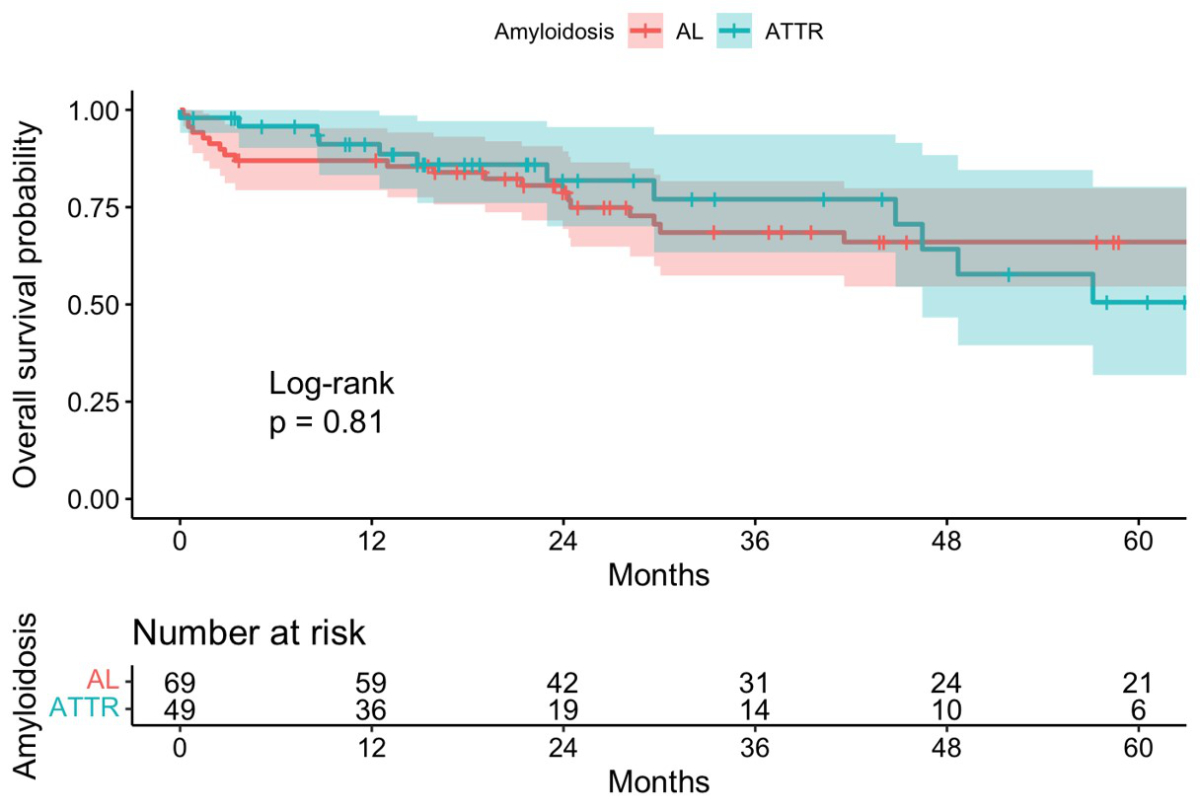
Figure 3Overall survival in light-chain and transthyretin amyloidosis. AL: light-chain amyloidosis; ATTR: transthyretin amyloidosis.
We report here for the first time data from the Swiss Amyloidosis Registry on systemic amyloidosis. Our patients show a distribution of different amyloidosis subtypes consistent with data from the literature [14]. Light-chain amyloidosis and wild-type transthyretin amyloidosis account for most of the cases. The relatively high number (8%) of variant amyloidosis in our patient cohort likely reflects our status as one of the two Swiss referral centres for RNA interference therapy such as Patisiran and Inotersen and as a tertiary referral centre with organ transplantation activity [15]. Our patient registry does not yet cover the whole of Switzerland, which should be the goal for a small country such as ours.
Most patients presented with cardiac or renal involvement. Clinically significant organ failure is almost always associated with advanced disease and unfavourable outcome [11, 13]. Therefore, efforts must be made to enable early diagnosis by raising awareness of these rare diseases and investigating suspected cases in a timely manner.
In transthyretin amyloidosis, the diagnosis was established mostly through tissue biopsy. A non-biopsy approach for transthyretin amyloidosis was considered in patients from 2016 onwards, as data revealed an excellent specificity and sensitivity of the semi-quantitative measurement of the cardiac tracer uptake in bone scintigraphy in patients in whom a monoclonal gammopathy has been ruled out [8, 13, 16].
Our data show an almost exponential increase in transthyretin amyloidosis within the Collaborative Amyloidosis Network since 2015, as experienced by other centres [17]. This is most likely due to increased awareness and better diagnostic pathways [18]. Patients with heart failure and preserved ejection fraction (HFpEF) within our network undergo systematic evaluation with echocardiography, MRI and, in case of absence of MGUS, bone scintigraphy to detect or rule out transthyretin amyloidosis [19]. We suspect that there will be a further shift towards wild-type transthyretin amyloidosis as the approval of new treatments increases awareness of the disease.
More than half of the patients with light-chain amyloidosis in our registry have a clonal plasma cell infiltration of ≥10% in bone marrow samples, as compared to 20–40% in patient cohorts reported in the literature [20, 21]. These patients have a comparatively high plasma cell burden and worse outcome compared to patients with light-chain amyloidosis and <10% bone marrow plasma cells [20]. We can only speculate on the reasons for this relatively high percentage of patients in our registry, but late diagnosis with expansion of the plasma cell clone might be an explanation. Diagnosis of light-chain amyloidosis is often delayed by more than one year from the onset of first symptoms and advanced disease is associated with unfavourable outcome [4]. In order to address the challenge of early disease detection, we included sensitive biomarkers of cardiac amyloidosis (NT-proBNP) and renal amyloidosis (albuminuria) in the regular follow-up of patients with MGUS and abnormal free light chain (FLC) ratio [22–24].
Of note, amyloid A amyloidosis patients were comparatively rare and mainly found in the retrospective patient cohort. This phenomenon has been described by others, and is due to earlier diagnosis of predisposing conditions for amyloid A amyloidosis as well as widespread access to more effective treatments for inflammatory and infectious diseases [25, 26].
In our cohort, overall survival of patients with light-chain amyloidosis is better than described in the literature. This is probably due to a selection bias as the ethics committee only permitted retrospective inclusion of deceased patients in the registry with informed consent of a next of kin. In the transthyretin amyloidosis cohort, the median overall survival of around 5 years reflects published data from other centres [27, 28]. Most of our transthyretin amyloidosis patients were diagnosed during the prospective data collection period; we assume that these data are more accurate.
Since the availability of the tetramer stabilising drug Tafamidis and the RNA interference therapies Patisiran and Inotersen in Switzerland, the number of referrals of wild-type and variant transthyretin amyloidosis patients in our network has increased [15]. After a drug is approved by Swissmedic and subsequently included in the Federal Office of Public Health (FOPH) list of specialties, patients must be referred to a designated centre to apply for reimbursement [15]. With the new treatment options, awareness of the disease is increasing.
In our registry, the cohort consists of a retrospective and a prospective part, with a larger proportion of missing data in the retrospective cohort. This is understandable and yet we believe it highlights the importance and impact of such registries in terms of patient outcome. Missing data might lead to incomplete staging and suboptimal treatment, given that risk adaptation is a cornerstone of the treatment of patients with systemic amyloidosis. Of our light-chain amyloidosis patient data, 75.6% of the missing staging data came from the retrospective cohort. The same applies for data on iFISH (73.2%) and plasma cell infiltration in the bone marrow (90%). These parameters must be considered when choosing the best treatment options for patient. However, our data do not allow us to reach a conclusion of worse outcome as the general number of patients in the registry is still low.
When founding the Collaborative Amyloidosis Network, great efforts were undertaken to standardise our diagnostic pathways and treatment recommendations. The data from our registry not only shows a more complete dataset, but also shows the implementation of change of practice, such as comprehensive testing for TTR mutations in transthyretin amyloidosis patients as recommended by ISA guidelines. However, and in contrast to other European countries, Swiss health insurers assess the necessity for genetic testing on a case-by-case basis. This might explain why genetic testing could only be performed in 74% of the patients. Collecting information on the mutational landscape in Switzerland is important because hereditary amyloidosis is very heterogeneous with regionally different clinical manifestations and mutation patterns. Therapy options may differ for patients with and without TTR mutations [29, 30].
These practice changes within the network further resulted in a national initiative, where experts from all over Switzerland drew up Guidelines for the treatment of light-chain and transthyretin amyloidosis, also published in Swiss Medical Weekly [19, 31].
Our registry has some limitations, which are inherent to all multicentre registries. As stressed before, the retrospectively collected data are prone to be less homogeneous and less complete than the data collected prospectively from 2015 and even though we have standardised follow-up recommendations for the members of the Collaborative Amyloidosis Network, some centres might prefer local guidelines, leading to a partially incomplete dataset. Also, the absence of a central laboratory where all samples on amyloidosis would be analysed and the absence of a centralised imaging team might lead to some variability in the reporting of the results. Our cohort is still quite small, which is in part attributable to the rather strict inclusion criteria that allowed for the inclusion of only confirmed systemic amyloidosis cases, and the restriction imposed by the local ethics committee to not include patients without informed consent if they were lost to follow-up, and/or could not be tracked due to changes in address or due to death. Further, funding for medical registries is a challenge as the operating period is usually several years and direct endpoints/ outcome are not always clear.
However, given the prospective character of the registry including a standardised follow-up and data collection procedure from 2015 onwards, we expect higher-quality data from this database in the future.
During the set-up of the registry, a more comprehensive work-up of our patients suffering mainly from light-chain and transthyretin amyloidosis was implemented. Survival rates were remarkably high and similar between light-chain and transthyretin amyloidosis, a finding which was noted in similar historic registries of international centres. However, further studies are needed to depict morbidity and mortality as the amyloidosis landscape is changing rapidly.
Manuela Averaimo (Istituto Cardiocentro Ticino, Ente Ospedaliero Cantonale, Lugano, Switzerland)
Luc Biedermann (Departement of Gastroenterology, University Hospital Zurich, Switzerland)
Marco Bühler (Department of Pathology, University Hospital Zurich, Zurich, Switzerland)
Oliver Distler (Departement of Rheumatology, University Hospital Zurich, Zurich, Switzerland)
Nadia Djerbi (Departement of Medical Oncology and Hematology, University Hospital Zurich, Zurich, Switzerland)
Heiko Pohl (Departement of Neurology, University Hospital Zurich, Zurich, Switzerland)
Juli Leo-Stickelberger (Departement of Medical Oncology and Hematology, University Hospital Zurich, Zurich, Switzerland)
Erika Lerch (Clinic of Oncology, Oncology Institute of Southern Switzerland, Ente Ospedaliero Cantonale, Bellinzona, Switzerland)
Daniela Leupold (Departement of Neurology, Cantonal Hospital St. Gallen, St. Gallen, Switzerland)
Robert Manka (University Heart Center, University Hospital Zurich, Zurich, Switzerland)
Stephanie McKeown (Departement of Medical Oncology and Hematology, University Hospital Zurich, Zurich, Switzerland)
Violeta Mihaylova (Departement of Neurology, Cantonal Hospital Lucerne, Lucerne, Switzerland)
Holger Moch (Department of Pathology, University Hospital Zurich, Zurich, Switzerland)
Nilufar Mohebbi (Departement of Nephrology, University Hospital Zurich, Zurich, Switzerland)
Aju Pazhenkottil (University Heart Center, University Hospital Zurich, Zurich and Cardiac Imaging, Department of Nuclear Medicine, University Hospital and University of Zurich, Zurich, Switzerland)
Anita Rauch (Institute of Medical Genetics, University of Zurich, Schlieren-Zurich, Switzerland)
Lenka Schilg-Hafer (Departement of Neurology, Cantonal Hospital St. Gallen, St. Gallen, Switzerland)
Paolo Servida (Clinic of Oncology, Oncology Institute of Southern Switzerland, Ente Ospedaliero Cantonale, Bellinzona, Switzerland)
Felix Tanner (University Heart Center, University Hospital Zurich, Zurich, Switzerland)
Friederike Vetter (Departement of Medical Oncology and Hematology, University Hospital Zurich, Zurich, Switzerland)
This research received no specific grant from any funding agency.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. AA is the editor in chief of Swiss Medical Weekly. He did play no role in the editorial assessment of the paper and was blinded to the reviewers. In addition, he serves on the Board of Directors and is a shareholder of Babylon AG, a company that develops immunotherapeutic against amyloids. LB reports fees for consulting/advisory board from AbbVie, BMS, MSD, Vifor, Falk, Escape, Calypso, Ferring, Pfizer, Takeda, Janssen, Sanofi, Ewopharma. AJR received financial support from Alnylam and Pfizer, related to this article and financial support from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Medtronic, MSD, Unipharm, Novartis, Pierre Fabre, Roche, Vifor and Zoll unrelated to this work. BG reports non-financial support and funding for accredited continuing medical education program from Axonlab, and Thermo Fisher Scientific; personal fees and funding for accredited continuing medical education program from Alnylam, Pfizer and Sanofi; funding for accredited continuing medical education program from Bayer, Bristol Myers Squibb, Daiichi-Sankyo, Takeda, Octapharma, SOBI, Janssen, Novo Nordisk, Mitsubishi Pfizer, Tanabe Pharma, outside the submitted work. FH reports support for attending meetings and/or travel from Roche, Janssen Pharmaceuticals, Takeda, Amgen. HHJ reports fees from consulting/ advisory boards from Alnylam and SOBI related to this article. FR has not received personal payments by pharmaceutical companies or device manufacturers in the last 3 years (remuneration for the time spent in activities, such as participation as steering committee member of clinical trials and member of the Pfizer Research Award selection committee in Switzerland, were made directly to the University of Zurich). The Department of Cardiology (University Hospital of Zurich/University of Zurich) reports research-, educational- and/or travel grants from Abbott, Amgen, Astra Zeneca, Bayer, Berlin Heart, B. Braun, Biosense Webster, Biosensors Europe AG, Biotronik, BMS, Boehringer Ingelheim, Boston Scientific, Bracco, Cardinal Health Switzerland, Corteria, Daiichi, Diatools AG, Edwards Lifesciences, Guidant Europe NV (BS), Hamilton Health Sciences, Kaneka Corporation, Kantar, Labormedizinisches Zentrum, Medtronic, MSD, Unipharm Medical Company, Novartis, Novo Nordisk, Orion, Pfizer, Quintiles Switzerland Sarl, Roche Diagnostics, Sahajanand IN, Sanofi, Sarstedt AG, Servier, SIS Medical, SSS International Clinical Research, Terumo Deutschland, Trama Solutions, V- Wave, Vascular Medical, Vifor, Wissens Plus, ZOLL. The research and educational grants do not impact on Prof. Ruschitzka`s personal remuneration. RS received financial support from Alnylam, Pfizer, SOBI, Janssen related to this article and financial support from Takeda, BMS, Amgen not related to this article. None of the other authors have reported a conflict of interest in relation to this work.
1. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003 Aug;349(6):583–96. 10.1056/NEJMra023144
2. Nuvolone M, Merlini G. Systemic amyloidosis: novel therapies and role of biomarkers. Nephrol Dial Transplant. 2017 May;32(5):770–80.
3. Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016 Dec;23(4):209–13. 10.1080/13506129.2016.1257986
4. Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light Chain Amyloidosis: Patient Experience Survey from the Amyloidosis Research Consortium. Adv. Ther; 2015.
5. Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current Concepts of Cardiac Amyloidosis: Diagnosis, Clinical Management, and the Need for Collaboration. Heart Fail Clin. 2017 Apr;13(2):409–16. 10.1016/j.hfc.2016.12.003
6. Pop B, Fetica B, Blaga ML, Trifa AP, Achimas-Cadariu P, Vlad CI, et al. The role of medical registries, potential applications and limitations. Med Pharm Rep. 2019 Jan;92(1):7–14. 10.15386/cjmed-1015
7. Gertz MA, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10thInternational Symposium on Amyloid and Amyloidosis. in American Journal of Hematology, 2005.
8. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016 Jun;133(24):2404–12. 10.1161/CIRCULATIONAHA.116.021612
9. Schönland SO, Hegenbart U, Bochtler T, Mangatter A, Hansberg M, Ho AD, et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood. 2012 Jan;119(2):488–93. 10.1182/blood-2011-06-358507
10. Brambilla F, Lavatelli F, Di Silvestre D, Valentini V, Rossi R, Palladini G, et al. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood. 2012 Feb;119(8):1844–7. 10.1182/blood-2011-07-365510
11. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012 Mar;30(9):989–95. 10.1200/JCO.2011.38.5724
12. Gillespie BW, Chen Q, Reichert H, Franzblau A, Hedgeman E, Lepkowski J, et al. Estimating population distributions when some data are below a limit of detection by using a reverse Kaplan-Meier estimator. Epidemiology. 2010 Jul;21(4 Suppl 4):S64–70. 10.1097/EDE.0b013e3181ce9f08
13. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018 Aug;39(30):2799–806. 10.1093/eurheartj/ehx589
14. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016 Jun;387(10038):2641–54. 10.1016/S0140-6736(15)01274-X
15. Spezialitätenliste (SL) - Präparate. Online. Available: https://www.spezialitaetenliste.ch/ShowPreparations.aspx. Accessed: 13-Feb-2023.
16. Y. Rauf, et al. Real World Experience of Tc-DPD scintigraphy as a diagnostic imaging tool in amyloidosis. 2022.
17. M. Zampieri et al., Changes in the perceived epidemiology of amyloidosis: 20 year-experience from a Tertiary Referral Centre in Tuscany. Int. J. Cardiol., vol. 335, 2021.
18. Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and Survival Trends in Amyloidosis, 1987-2019. N Engl J Med. 2020 Apr;382(16):1567–8. 10.1056/NEJMc1917321
19. Condoluci A, Théaudin M, Schwotzer R, Pazhenkottil AP, Arosio P, Averaimo M, et al. Management of transthyretin amyloidosis. Swiss Med Wkly. 2021 Oct;151(4142):w30053. 10.4414/SMW.2021.w30053
20. Kourelis TV, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, Hayman SR, et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013 Dec;31(34):4319–24. 10.1200/JCO.2013.50.8499
21. Dittus C, Uwumugambi N, Sun F, Sloan JM, Sanchorawala V. The Effect of Bone Marrow Plasma Cell Burden on Survival in Patients with Light Chain Amyloidosis Undergoing High-Dose Melphalan and Autologous Stem Cell Transplantation. Biol Blood Marrow Transplant. 2016 Sep;22(9):1729–32. 10.1016/j.bbmt.2016.05.027
22. Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003 May;107(19):2440–5. 10.1161/01.CIR.0000068314.02595.B2
23. Wechalekar AD, Gillmore JD, Wassef N, Lachmann HJ, Whelan C, Hawkins PN. Abnormal N-terminal fragment of brain natriuretic peptide in patients with light chain amyloidosis without cardiac involvement at presentation is a risk factor for development of cardiac amyloidosis. Haematologica. 2011 Jul;96(7):1079–80. 10.3324/haematol.2011.040493
24. Milani P, Merlini G, Palladini G. Light chain amyloidosis. Mediterr J Hematol Infect Dis. 2018 Mar;10(1):e2018022. 10.4084/mjhid.2018.022
25. Lane T, Pinney JH, Gilbertson JA, Hutt DF, Rowczenio DM, Mahmood S, et al. Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral centre. Amyloid. 2017 Sep;24(3):162–6. 10.1080/13506129.2017.1342235
26. Immonen K, Finne P, Grönhagen-Riska C, Pettersson T, Klaukka T, Kautiainen H, et al. A marked decline in the incidence of renal replacement therapy for amyloidosis associated with inflammatory rheumatic diseases - data from nationwide registries in Finland. Amyloid. 2011 Mar;18(1):25–8. 10.3109/13506129.2010.549252
27. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019 Jul;140(1):16–26. 10.1161/CIRCULATIONAHA.118.038169
28. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al.; ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018 Sep;379(11):1007–16. 10.1056/NEJMoa1805689
29. D. Adams and S. Ole, Patisiran, an investigational RNAi therapeutic for patients with hereditary transthyretin-mediated (hATTR) amyloidosis: Results from the phase 3 APOLLO study. Rev. Neurol. (Paris)., 2018.
30. Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul;379(1):22–31. 10.1056/NEJMoa1716793
31. Schwotzer R, Flammer AJ, Gerull S, Pabst T, Arosio P, Averaimo M, et al. Expert recommendation from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med Wkly. 2020 Dec;150(4950):w20364. 10.4414/smw.2020.20364