Genomic characteristics of clinical non-toxigenic Vibrio cholerae isolates in Switzerland:
a cross-sectional study
DOI: https://doi.org/https://doi.org/10.57187/s.3437
Natalie Meyera,
Roger Stephana,
Nicole Cernelaa,
Jule Anna Horlbogb,
Michael Biggela
a Institute for Food Safety and Hygiene,
Vetsuisse Faculty, University of Zurich, Zurich, Switzerland
b National Reference Laboratory for
Enteropathogenic Bacteria and Listeria (NENT), Institute for Food Safety and Hygiene,
Vetsuisse Faculty, University of Zurich, Zurich, Switzerland
Summary
STUDY AIMS: Although non-toxigenic Vibrio cholerae lack the ctxAB genes encoding cholera toxin,
they can cause diarrhoeal disease and outbreaks in humans. In Switzerland, V.
cholerae is a notifiable pathogen and all clinical isolates are analysed at
the National Reference Laboratory for Enteropathogenic Bacteria and Listeria. Up
to 20 infections are reported annually. In this study, we investigated the population
structure and genetic characteristics of non-toxigenic V. cholerae isolates collected over five years.
METHODS: V. cholerae isolates were serotyped
and non-toxigenic isolates identified using a ctxA-specific PCR. Following
Illumina whole-genome sequencing, genome assemblies were screened for virulence
and antibiotic resistance genes. Phylogenetic analyses were performed in the context
of 965 publicly available V. cholerae genomes.
RESULTS: Out of 33 V. cholerae infections
reported between January 2017 and January 2022 in Switzerland, 31 were caused by
ctxA-negative isolates. These non-toxigenic isolates originated from gastrointestinal
(n = 29) or extraintestinal (n = 2) sites. They were phylogenetically diverse and
belonged to 29 distinct sequence types. Two isolates were allocated to the lineage
L3b, a ctxAB-negative but tcpA-positive clade previously associated
with regional outbreaks. The remaining 29 isolates were placed in lineage L4, which
is associated with environmental strains. Genes or mutations associated with reduced
susceptibility to the first-line antibiotics fluoroquinolones and tetracyclines
were identified in 11 and 3 isolates, respectively. One isolate was predicted to
be multidrug resistant.
CONCLUSIONS: V. cholerae infections in
Switzerland are rare and predominantly caused by lowly virulent ctxAB-negative
and tcpA-negative strains. As V. cholerae is not endemic in Switzerland,
cases are assumed to be acquired predominantly during travel. This assumption was
supported by the phylogenetic diversity of the analysed isolates.
Introduction
Vibrio
cholerae causes the severe diarrhoeal disease cholera
and has been responsible for seven major pandemics in the past two centuries. Although
improved sanitation and hygiene have reduced the threat of cholera [1], it is still
endemic in some countries and
causes 95,000 deaths per year globally [2]. The WHO has reported an increase in cholera
cases since 2021, mainly in Africa and the Eastern Mediterranean [3].
In routine diagnostics, V. cholerae isolates
are usually initially characterised by serotyping, with pandemic V. cholerae belonging to O-antigen types
O1 or O139. For epidemiological surveillance, serotype O1 isolates are further differentiated
into biotypes El Tor and classical. Whole-genome sequencing (WGS) enables more accurate
tracking of cholera outbreaks and transmission routes [4, 5]. Based on their genetic
phylogeny, V. cholerae strains were divided
into 9 major lineages (L1–L9) [6, 7]. Lineage L1 is assumed to have caused the
first six pandemics and comprises serotype O1 classical isolates. Lineage L2 is
responsible for the ongoing 7th pandemic and comprises serotype O1 El
Tor and O139 isolates. Lineages L3, L5, L6 and L8 (serotype O1 El Tor) cause sporadic
cholera cases in confined geographical regions [6, 7]. The L4 and L7 lineages comprise
environmental
isolates that rarely cause human disease [6]. Lineage L9 was recently described as
intermediate
between L1 and L4 [7].
The key virulence factors of pandemic V. cholerae strains are the cholera toxin
and the toxin-coregulated pilus (TCP). The cholera toxin is a heat-labile enterotoxin
and triggers the characteristic rice water stool in infected patients [8]. It is encoded
by the ctxA and ctxB genes, which are located on the mobile prophage
CTX. TCP plays a critical role in the colonisation of the host
intestine and additionally acts as a CTXφ phage receptor [9]. It is encoded by the
tcp gene cluster
located on Vibrio Pathogenicity Island I (VPI-1) [10]. V. cholerae
isolates often contain additional genomic regions enriched with virulence genes
such as Vibrio Pathogenicity Island II (VPI-2) and Vibrio Seventh Pandemic Island
I and II (VSP-1 and VSP-2).
While cholera outbreaks are mainly the result
of poor hygiene conditions involving transmission via the faecal-oral route, V.
cholerae is also a natural inhabitant of aquatic ecosystems. Although most environmental
V. cholerae isolates lack the ctxAB
genes, they can cause mild human infections upon exposure to water or the consumption
of seafood [11, 12]. Among ctxAB-negative (non-toxigenic)
V. cholerae, tcpA-positive strains are associated with an increased
risk of human infection and have occasionally been linked to local regional outbreaks,
including a recent seafood-borne diarrhoea outbreak in China [13–19].
Besides causing mild disease, non-toxigenic
V. cholerae are a concern for public health
as they may transfer antibiotic resistance to toxigenic strains [20] or acquire CTXφ
and transform into highly virulent
toxigenic strains [13, 21, 22]. To date, few studies have taken a
phylogenomic approach to investigating non-toxigenic V. cholerae. Although V. cholerae
is not endemic in Switzerland, human infections are reported each year [23]. In this
study, we genetically characterised
all non-toxigenic V. cholerae isolates
from human patients in Switzerland received between January 2017 and January 2022
at the National Reference Laboratory for Enteropathogenic Bacteria and Listeria
(NENT).
Materials and methods
Patients
Cholera is a notifiable disease in Switzerland
and all clinical V. cholerae isolates
from inpatients and outpatients must be sent to the National Reference Laboratory
for Enteropathogenic Bacteria and Listeria. Metadata available for this cross-sectional
study included the patients’ age, sex and place of residence. There were no data
on the patients’ travel history or symptoms. Institutional review board approval
or informed consent was not required as this analysis was conducted as part of the
tasks and duties of the NENT. A study protocol was not registered or published.
Bacterial isolates
Pure cultures were obtained on thiosulphate
citrate bile salts sucrose (TCBS) agar. Colonies growing in yellow, flat to slightly
convex colonies with a diameter of 2–3 mm on TCBS agar were considered V. cholerae candidates. Using ISO17025-accredited
methods, these were further tested for the ctxA
gene by PCR and O-antigen-serotyped with antisera (Denka Seiken Co.) against O1
El Tor Inaba, O1 El Tor Ogawa and O139.
Isolates with negative agglutination test
results were considered as non-O1 non-O139 V.
cholerae. The ctxA PCR reaction was
performed according to “CDC Chapter 7: Detection of Cholera Toxin” (pp 62–88) with
CTX2 and CTX3 as primers. The temperature programme was adapted as follows: initial
denaturation at 94 °C for 15 minutes, 30 cycles of denaturation at 94 °C for 30
seconds, annealing at 60 °C for 30 seconds and elongation at 72 °C for 30 seconds.
The final elongation was done at 72 °C for 7 minutes.
Whole-genome sequencing and genomic analyses
Genomic DNA was extracted using the DNeasy blood
and tissue kit (Qiagen). Sequencing libraries were prepared with the Nextera DNA
flex library preparation kit (Illumina) and sequenced on the Illumina MiniSeq platform
(2 × 150 bp). Paired-end Illumina reads were trimmed with fastp v0.22.0 [24] and assembled
using SPAdes v3.14.1 [25] implemented in the pipeline shovill 1.0.9 (https://github.com/tseemann/shovill).
For quality control, assemblies were passed to CheckM v1.2.2 [26] using the lineage_wf
workflow. For comparison,
we downloaded 965 publicly available assemblies of global V. cholerae isolates from the National Center for Biotechnology
Information (NCBI) (table S1, available for download as a separate file at
https://doi.org/10.57187/s.3437). The downloaded genomes reflect the collection
described by Wang et al. in 2020 [7] except for three assemblies that were flagged
as low quality by NCBI.
Core genome alignments were generated with parsnp
1.5.6 [27,28]. For analyses including global isolates
the “xtrafast” option was used. The generated alignments were used for construction
of phylogenetic trees using IQ-Tree v2.2.0.3 with the generalised time-reversible
(GTR) model and gamma distribution with 1000 bootstraps [29]. Trees were visualised
using iTOL V5 [30]
and annotated using Inkscape 1.2 [31]. SNP distances were determined from the core
genome alignment using snp-dists v0.8.2 (https://github.com/tseemann/snp-dists).
Lineages were defined based on phylogenetic clustering with isolates of known lineage
affiliations. Multi-locus sequence types (MLST) were determined using the PubMLST
suite and novel alleles and profiles submitted [32].
In silico O-antigen serotypes were determined
with VicPred [33] to complement the laboratory-based results.
Assemblies were annotated using Prokka v1.13 [34]. Virulence genes were detected using
ABRicate
V1.0.1 (https://github.com/tseemann/abricate) in combination with VFDB set B [35] (minimum coverage 70%; identity 70%). Using
VicPred [33], we examined for the presence of virulence-associated
islands (VPI-1, VPI-2, VSP-1 and VSP-2). A virulence island was considered present
when at least 80% of the genes were identified and partly present when 50– 80% of
the genes were identified. Genes associated with antibiotic resistance were identified
using AMRfinder 3.11.2 [36] and ABRicate in combination with the ResFinder
database (minimum coverage 50%, identity 90%) [37]. The presence of two mutations
associated with
fluoroquinolone resistance (gyrA S83I and parC S85L)[38] was manually investigated using CLC Main Workbench
22.0.2. Unless stated otherwise, default parameters were used for all analyses.
Results
Non-toxigenic V.
cholerae isolates from Switzerland belong to the L3b and L4 lineages
Between January 2017 and January 2022, a total
of 33 V. cholerae isolates were received
at the NENT, of which 31 tested PCR-negative for ctxA. The 31 non-toxigenic isolates originated from human faeces (n
= 28), urine (n = 1; isolate N18-0491), blood (n = 1; isolate N22-0171) and an unknown
clinical sample (n = 1). The annual number of reported infections varied from 6
to 19 between 2017 and 2019 but dropped to one infection in 2020 (table S1) when
international travel was restricted due to the COVID-19 pandemic. Although the patients’
travel history was not available, these data suggest that most cases were travel-acquired.
Agglutination tests identified one isolate (N18-1211) as O1 El Tor Inaba, two isolates
(N18-1982 and N18-1603) as O1 El Tor Ogawa and 28 isolates as non-O1/non-O139.
Whole-genome sequencing revealed substantial
diversity, with all non-O1 isolates (n = 28) differing by at least 7215 pairwise
SNPs in a core genome alignment. The two most closely related isolates (O1 isolates
N18-1211 and N18-1982) differed by 1271 SNPs, suggesting that all isolates are epidemiologically
unrelated. Multi-locus sequence typing assigned the isolates to 29 different sequence
types (STs), of which 16 were novel. Only two STs occurred more than once: ST579
(comprising two O1 isolates) and ST1378 (comprising both O5 isolates) (table S1).
To determine lineage affiliations, a phylogenetic
analysis was performed in the context of 965 additional V. cholerae isolates from global collections (figure 1). Of the 31 Swiss
isolates, all non-O1 isolates (n = 28) and one O1 El Tor isolate belonged to lineage
L4, a heterogeneous lineage lacking ctxAB and tcpA and associated
with environmental and lowly virulent clinical isolates [6]. The remaining two isolates
(N18-1211 and N18-1982,
both O1 ST579) grouped in the L3b lineage. L3b is a subclade of L3 recently associated
with a diarrhoeal epidemic of non-toxigenic strains in China [7] and otherwise comprised
isolates from Asia
or Latin America. The closest phylogenetic neighbours of N18-1211 and N18-1982 were
collected in Russia and Turkmenistan (see figure S1 in the appendix). In silico
serotyping with VicPred confirmed the O1 El Tor type of three isolates (figure 2).
The most frequent predicted O-antigen type was O8 (n = 6). Other predicted types
included O3, O4, O5, O7, O14, O37 and O49.
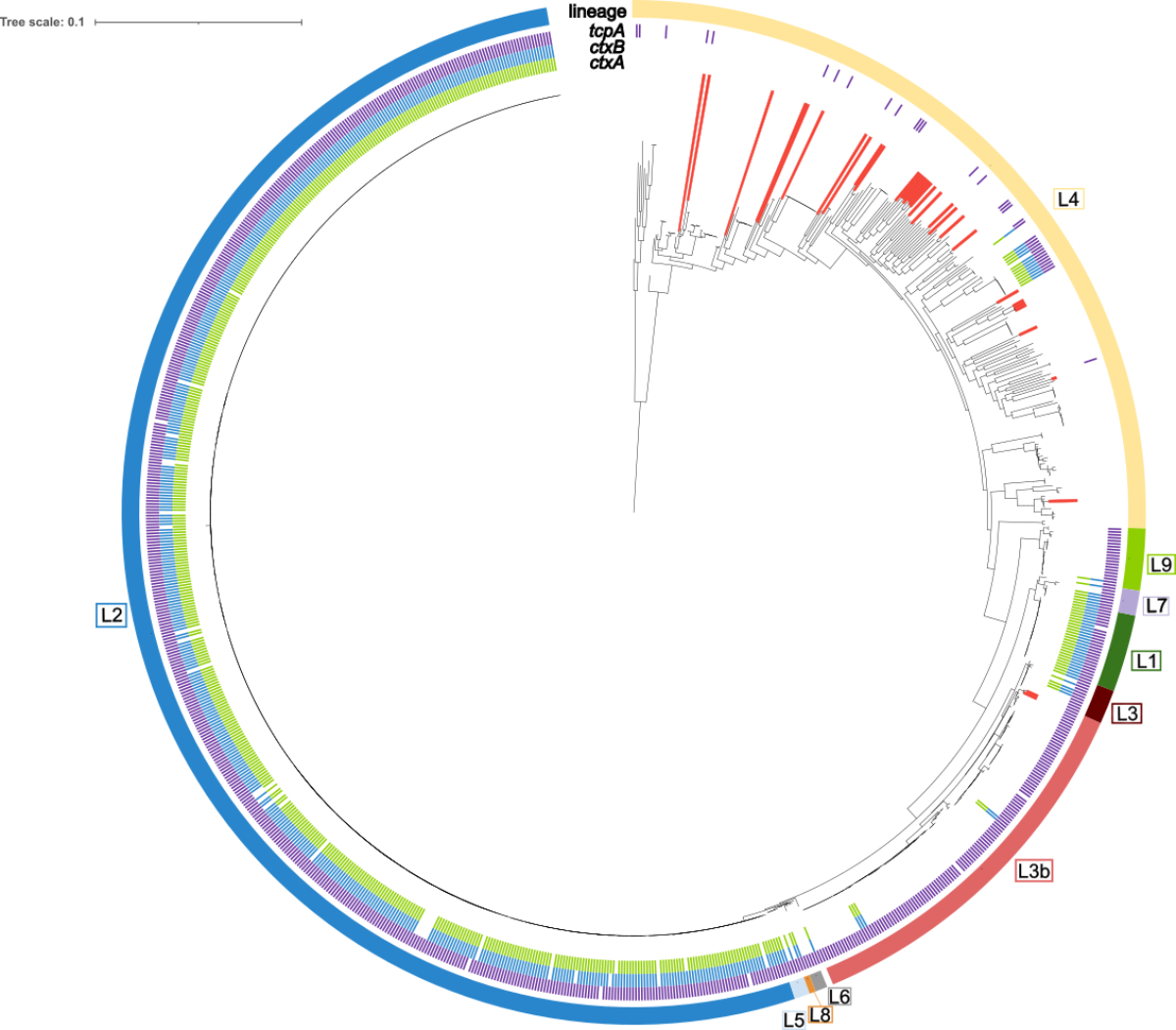
Figure 1Maximum-likelihood phylogenetic tree of 31
Swiss and 965 global V. cholerae
isolates. Swiss isolates are indicated by red lines. The presence of ctxA (green), ctxB (blue) and tcpA
(purple) is labelled (inner ring). The outer ring denotes lineage
affiliations (L1–L9). The tree is based on 32,851 polymorphic sites identified
in a multi-alignment-derived 0.45 Mbp core genome. The scale bar indicates the
number of substitutions per site in the core genome alignment. The tree was
visualised using iTOL.
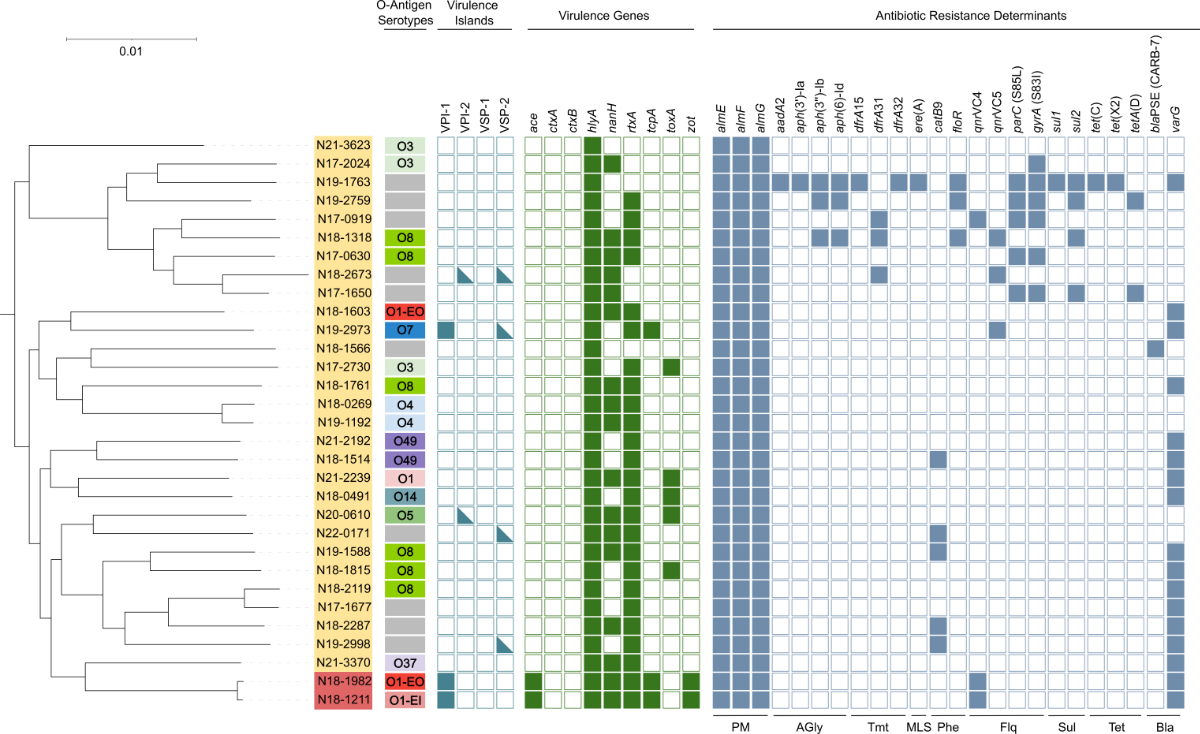
Figure 2Maximum-likelihood phylogenetic tree and
genetic characteristics of 31 Swiss non-toxigenic V. cholerae isolates. Lineage affiliations are indicated by the
yellow (L4) or red (L3b) background. In silico-predicted O-antigen serotypes
and the presence of virulence islands (filled square: complete presence;
half-filled: partial presence [>50% of the genes]), virulence genes and
antimicrobial resistance determinants are shown. The tree is based on 177,987
polymorphic sites identified in a multi-alignment-derived 2.8 Mbp core genome
and was visualised using iTOL. The scale bar indicates the number of
substitutions per site in the core genome alignment. AGly: aminoglycosides; Bla: beta-lactams;
Flq:
fluoroquinolones; MLS: macrolide-lincosamide-streptogramin; O1-EO: O1 EL Tor
Ogawa; O1-EI: O1 El Tor Inaba; Phe: phenicols; PM: polymyxines; Sul: sulphonamides;
Tet: tetracyclines; Tmt: trimethoprim.
No indications of a recent CTX prophage loss
All global and Swiss V. cholerae genomes were screened for the presence of virulence genes.
Importantly, all Swiss isolates belonged to branches consisting predominantly or
exclusively of ctxAB-negative isolates
(figure 1), suggesting that the absence of ctxAB
was unlikely to be due to a potential (partial) loss of the CTX prophage during
patient colonisation or subculturing in the laboratory. The tcpA gene (encoding a toxin-coregulated
pilus subunit) was identified in three isolates: in both L3b isolates (N18-1211
and N18-1982) and one L4 isolate (N19-2973). Further, the two L3b isolates were
the only isolates carrying the CTX prophage-associated genes zot (an enterotoxin affecting intestinal
tight junctions [39]) and ace (an enterotoxin causing fluid
secretion in rabbit ileal loops [40]) (figure 2), suggesting the presence of a putative
CTXφ precursor, as previously reported for L3b isolates [7]. The haemolysin gene hlyA was identified in all
31 isolates. The rtxA (cytotoxin)
and nanH (neuraminidase) genes were found in 25 and 17 of the Swiss isolates,
respectively. The toxA gene, which has
been associated with environmental strains [41], was identified in five L4 isolates.
The two
L3b isolates contained the complete VPI-1 island (comprising tcpA). Other
virulence pathogenicity islands or fragments thereof were identified in five L4
isolates, including the bloodstream isolate N22-0171, which carried fragments of
VSP-2.
Several isolates contain genes associated with tetracycline
and quinolone resistance
All Swiss isolates were screened for genes and
mutations associated with decreased antibiotic susceptibility with a focus on the
three first-line antibiotics doxycycline (a tetracycline), ciprofloxacin (a quinolone)
and azithromycin (a macrolide). The two screening approaches used (AMRfinder and
ResFinder) yielded identical results, except for one resistance gene (varG)
that was not included in the ResFinder database. Three isolates harboured tet
genes indicating potential tetracycline resistance (figure 2). Further, six isolates
contained qnrVC4 or qnrVC5, which are associated with ciprofloxacin
resistance when occurring in combination with target mutations in gyrA and parC [38], as observed in one of the six isolates (N17-0919).
An additional five isolates had mutations in quinolone-resistance determining regions
of parC or gyrA but did not carry qnrVC
genes. Isolate N19-1763 was identified as a potential multidrug-resistant strain,
harbouring genes or mutations associated with resistance against aminoglycosides,
beta-lactams, macrolides, phenicols, quinolones, sulphonamides, tetracycline and
trimethoprim.
Discussion
Although saline aquatic environments – the natural
habitat of V. cholerae – are absent in Switzerland, up to 20
infections caused by mostly ctxA-negative strains are reported annually.
Because infections with non-toxigenic strains are usually mild and patients may
not seek healthcare, the actual number of infections is likely higher. We assume
that most cases are associated with travel abroad. This is supported by our investigation
of 31 isolates, which demonstrated high genetic diversity suggesting distinct origins.
In addition, the number of reported infections dropped to only one infection in
2020 when international travel was restricted. Notably, although V. cholerae
occurs in the North Sea, a study of 836 V. cholerae infections in the
UK could link >99% of the (mostly non-toxigenic) cases with available metadata
to travel abroad [42, 43]. However contaminated imported seafood
cannot be excluded as a potential infection source. Prevalence studies in Switzerland
and neighbouring countries found non-toxigenic V. cholerae in 0.6% to 6.3%
of the examined seafood products [44–47].
In line with other studies on non-toxigenic
V. cholerae in Europe [48, 49], our isolates were phylogenetically diverse.
Half of the isolates belonged to previously unknown sequence types, suggesting that
the diversity of non-toxigenic strainsis largely unexplored. Further, these
results indicate diverse infection sources. This contrasts with a recent genomic
analysis of non-toxigenic V. cholerae from patients in China, which found
that most of the 104 investigated ctxAB-negative isolates belonged to few
phylogenetic SNP clusters and were epidemiologically linked [7, 13]. The source of
this outbreak could be traced
to aquatic food products. Whereas most of these outbreak-associated isolates were
tcpA-positive and belonged to lineages L3b and L9, most Swiss isolates investigated
here belonged to the environment-associated lineage L4. Among the Swiss isolates,
the tcpA gene was only detected in one L4 and two L3b isolates, which did not belong to the
Chinese outbreak clusters.
In recent decades, drug-resistant V. cholerae
have emerged,with resistance patterns fluctuating with changing epidemiology
and antibiotic use [20, 50–54]. Although antibiotics are not indicated
for mild cholera infections, antimicrobial resistance genes have also been acquired
by non-toxigenic strains [7, 55–57], possibly driven by inadequate treatment
of patients or the increasing use of antibiotics in aquaculture [58, 59]. In our study,
10% of the isolates carried
determinants associated with reduced susceptibility to both tetracycline and fluoroquinolone,
two important first-line antibiotics. Information on the patients’ treatment upon
disease diagnosis was unavailable, nor was their travel history, limiting the interpretation
of our findings.
In conclusion, our study provides insights into
the prevalence and characteristics of non-toxigenic V. cholerae in Switzerland.
Rising sea temperatures, intensified aquaculture production and global trade may
lead to an increasing prevalence of V. cholerae infections in the future
[60–62]. Continuous monitoring of the pathogen
and antimicrobial resistance rates is important for informing public health management.
Data availability
Assemblies and read data were deposited in the
NCBI repository under BioProject accession number PRJNA997795. Individual accession
numbers are listed in table S1 (available for download as a separate file at
https://doi.org/10.57187/s.3437).
Acknowledgments
We thank Andrea Diethelm from the National Reference
Laboratory for Enteropathogenic Bacteria and Listeria (NENT) for her assistance
in sample processing.
Dr. Michael
Biggel
Institute for
Food Safety and Hygiene
Vetsuisse Faculty
University of
Zurich
CH-8000 Zurich
michael.biggel[at]uzh.ch
References
1.Wolfe M, Kaur M, Yates T, Woodin M, Lantagne D. A Systematic Review and Meta-Analysis
of the Association between Water, Sanitation, and Hygiene Exposures and Cholera in
Case-Control Studies. Am J Trop Med Hyg. 2018 Aug;99(2):534–45. 10.4269/ajtmh.17-0897
2.Ali M, Nelson AR, Lopez AL, Sack DA. Updated global burden of cholera in endemic countries.
PLoS Negl Trop Dis. 2015 Jun;9(6):e0003832. 10.1371/journal.pntd.0003832
3.WHO. Disease Outbreak News; Cholera – Global situation [Internet]. 2022 [cited 2023
Jun 28]. Available from: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON426
4.Kuninobu KI, Takemura T, Takizawa Y, Hasebe F, Yamashiro T. Whole-genome analysis
of a Vibrio cholerae O1 biotype classical strain isolated in 1946 in Sasebo city, Nagasaki prefecture,
from a returnee from the northeast part of China. Trop Med Health. 2023 Feb;51(1):5.
10.1186/s41182-023-00500-4
5.Morita D, Morita M, Alam M, Mukhopadhyay AK, Johura FT, Sultana M, et al. Whole-Genome
Analysis of Clinical Vibrio cholerae O1 in Kolkata, India, and Dhaka, Bangladesh, Reveals Two Lineages of Circulating
Strains, Indicating Variation in Genomic Attributes. MBio. 2020 Nov;11(6):e01227-20.
10.1128/mBio.01227-20
6.Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several
waves of global transmission in the seventh cholera pandemic. Nature. 2011 Aug;477(7365):462–5.
10.1038/nature10392
7.Wang H, Yang C, Sun Z, Zheng W, Zhang W, Yu H, et al. Genomic epidemiology of Vibrio cholerae reveals the regional and global spread of two epidemic non-toxigenic lineages. PLoS
Negl Trop Dis. 2020 Feb;14(2):e0008046. 10.1371/journal.pntd.0008046
8.Kaper JB, Morris JG Jr, Levine MM. Cholera. Clin Microbiol Rev. 1995 Jan;8(1):48–86.
10.1128/CMR.8.1.48
9.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with
cholera toxin. Proc Natl Acad Sci USA. 1987 May;84(9):2833–7. 10.1073/pnas.84.9.2833
10.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad
Sci USA. 1998 Mar;95(6):3134–9. 10.1073/pnas.95.6.3134
11.Trubiano JA, Lee JY, Valcanis M, Gregory J, Sutton BA, Holmes NE. Non-O1, non-O139
Vibrio cholerae bacteraemia in an Australian population. Intern Med J. 2014 May;44(5):508–11. 10.1111/imj.12409
12.Cariri FA, Costa AP, Melo CC, Theophilo GN, Hofer E, de Melo Neto OP, et al. Characterization
of potentially virulent non-O1/non-O139 Vibrio cholerae strains isolated from human
patients. Clin Microbiol Infect. 2010 Jan;16(1):62–7. 10.1111/j.1469-0691.2009.02763.x
13.Hao T, Zheng W, Wu Y, Yu H, Qian X, Yang C, et al. Population genomics implies potential
public health risk of two non-toxigenic Vibrio cholerae lineages. Infect Genet Evol. 2023 Aug;112:105441. 10.1016/j.meegid.2023.105441
14.Pal A, Saha PK, Nair GB, Yamasaki S, Takeda T, Takeda Y, et al. Clonal analysis of
non-toxigenic Vibrio cholerae O1 associated with an outbreak of cholera. Indian J Med Res. 1999 Jun;109:208–11.
15.Zheng W, Yu H. Wang H qiu, Zhang W, Pan J cao [Molecular characteristics and antibiotic
resistances of Vibrio cholerae O1 isolates in Hangzhou in 2009]. Zhonghua Yu Fang
Yi Xue Za Zhi. 2011 Oct;45(10):895–8.
16.Onishchenko GG, Lomov IM, Moskvitina EA, Podosinnikova LS, Vodianitskaia SI, Prometnoĭ VI,
et al. [Cholera caused by Vibrio cholerae O1 ctxAB- tcpA+]. Zh Mikrobiol Epidemiol
Immunobiol. 2007;(1):23–9.
17.Nair GB, Safa A, Bhuiyan NA, Nusrin S, Murphy D, Nicol C, et al. Isolation of Vibrio cholerae O1 strains similar to pre-seventh pandemic El Tor strains during an outbreak of gastrointestinal
disease in an island resort in Fiji. J Med Microbiol. 2006 Nov;55(Pt 11):1559–62.
10.1099/jmm.0.46734-0
18.Monakhova EV, Pisanov RV, Mikhas’ NK. [The genome polymorphism of Vibrio cholerae
ctxAB(-) strains, containing the proximal part of the CTX element]. Zh Mikrobiol Epidemiol
Immunobiol. 2004;(1):23–9.
19. Monakhova EV. Phenotypic and Molecular Characteristics of Epidemic and Non-epidemic
Vibrio cholerae Strains Isolated in Russia and Certain Countries of Commonwealth of Independent States
(CIS). Epidemiological and Molecular Aspects on Cholera. New York (NY): Springer New
York; 2011. pp. 51–78. 10.1007/978-1-60327-265-0_4
20.Das B, Verma J, Kumar P, Ghosh A, Ramamurthy T. Antibiotic resistance in Vibrio cholerae: understanding the ecology of resistance genes and mechanisms. Vaccine. 2020 Feb;38 Suppl
1:A83–92. 10.1016/j.vaccine.2019.06.031
21.Gutierrez-Rodarte M, Kolappan S, Burrell BA, Craig L. The Vibrio cholerae minor pilin TcpB mediates uptake of the cholera toxin phage CTXφ. J Biol Chem. 2019 Oct;294(43):15698–710.
10.1074/jbc.RA119.009980
22.Udden SM, Zahid MS, Biswas K, Ahmad QS, Cravioto A, Nair GB, et al. Acquisition of
classical CTX prophage from Vibrio cholerae O141 by El Tor strains aided by lytic phages and chitin-induced competence. Proc
Natl Acad Sci USA. 2008 Aug;105(33):11951–6. 10.1073/pnas.0805560105
23.BAG. Zahlen zu Infektionskrankheiten - Cholera; Bundesamt für Gesundheit BAG [Internet].
2023 [cited 2023 Jun 6]. Available from: https://www.bag.admin.ch/bag/de/home/zahlen-und-statistiken/zahlen-zu-infektionskrankheiten.exturl.html/aHR0cHM6Ly9tZWxkZXN5c3RlbWUuYmFnYXBwcy5jaC9pbmZyZX/BvcnRpbmcvZGF0ZW5kZXRhaWxzL2QvY2hvbGVyYS5odG1sP3dl/YmdyYWI9aWdub3Jl.html
24.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor.
Bioinformatics. 2018 Sep;34(17):i884–90. 10.1093/bioinformatics/bty560
25.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes:
a new genome assembly algorithm and its applications to single-cell sequencing. J
Comput Biol. 2012 May;19(5):455–77. 10.1089/cmb.2012.0021
26.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the
quality of microbial genomes recovered from isolates, single cells, and metagenomes.
Genome Res. 2015 Jul;25(7):1043–55. 10.1101/gr.186072.114
27.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome
alignment and visualization of thousands of intraspecific microbial genomes. Genome
Biol. 2014;15(11):524. 10.1186/s13059-014-0524-x
28.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput.
Nucleic Acids Res. 2004 Mar;32(5):1792–7. 10.1093/nar/gkh340
29.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic
algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015 Jan;32(1):268–74.
10.1093/molbev/msu300
30.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic
tree display and annotation. Nucleic Acids Res. 2021 Jul;49 W1:W293–6. 10.1093/nar/gkab301
31.Inkscape Project. Inkscape 1.2.2 [Internet]. 2022 [cited 2023 Jul 6]. Available from:
https://inkscape.org
32.Jolley KA, Bray JE, Maiden MC. Open-access bacterial population genomics: BIGSdb software,
the PubMLST.org website and their applications. Wellcome Open Res. 2018 Sep;3:124.
10.12688/wellcomeopenres.14826.1
33.Lee I, Ha SM, Baek MG, Kim DW, Yi H, Chun J. VicPred: A Vibrio cholerae Genotype Prediction Tool. Front Microbiol. 2021 Sep;12:691895. 10.3389/fmicb.2021.691895
34.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014 Jul;30(14):2068–9.
10.1093/bioinformatics/btu153
35.Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset
for big data analysis—10 years on. Nucleic Acids Res. 2016 Jan;44 D1:D694–7. 10.1093/nar/gkv1239
36.Feldgarden M, Brover V, Gonzalez-Escalona N, Frye JG, Haendiges J, Haft DH, et al. AMRFinderPlus
and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial
resistance, stress response, and virulence. Sci Rep. 2021 Jun;11(1):12728. 10.1038/s41598-021-91456-0
37.Bortolaia V, Kaas RS, Ruppe E, Roberts MC, Schwarz S, Cattoir V, et al. ResFinder
4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. 2020 Dec;75(12):3491–500.
10.1093/jac/dkaa345
38.Vinothkumar K, Kumar GN, Bhardwaj AK. Characterization of Vibrio fluvialis qnrVC5 Gene in Native and Heterologous Hosts: Synergy of qnrVC5 with other Determinants in Conferring Quinolone Resistance. Front Microbiol. 2016 Feb;7:146.
10.3389/fmicb.2016.00146
39.Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, et al. Vibrio cholerae
produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl
Acad Sci USA. 1991 Jun;88(12):5242–6. 10.1073/pnas.88.12.5242
40.Trucksis M, Galen JE, Michalski J, Fasano A, Kaper JB. Accessory cholera enterotoxin
(Ace), the third toxin of a Vibrio cholerae virulence cassette. Proc Natl Acad Sci USA. 1993 Jun;90(11):5267–71. 10.1073/pnas.90.11.5267
41.Purdy A, Rohwer F, Edwards R, Azam F, Bartlett DH. A glimpse into the expanded genome
content of Vibrio cholerae through identification of genes present in environmental strains. J Bacteriol. 2005 May;187(9):2992–3001.
10.1128/JB.187.9.2992-3001.2005
42.Fleischmann S, Herrig I, Wesp J, Stiedl J, Reifferscheid G, Strauch E, et al. Prevalence
and Distribution of Potentially Human Pathogenic Vibrio spp. on German North and Baltic Sea Coasts. Front Cell Infect Microbiol. 2022 Jul;12:846819.
10.3389/fcimb.2022.846819
43.Greig DR, Schaefer U, Octavia S, Hunter E, Chattaway MA, Dallman TJ, et al. Evaluation
of Whole-Genome Sequencing for Identification and Typing of Vibrio cholerae. J Clin
Microbiol. 2018 Oct;56(11):e00831-18. 10.1128/JCM.00831-18
44.Ottaviani D, Leoni F, Rocchegiani E, Santarelli S, Masini L, Di Trani V, et al. Prevalence
and virulence properties of non-O1 non-O139 Vibrio cholerae strains from seafood and
clinical samples collected in Italy. Int J Food Microbiol. 2009 Jun;132(1):47–53.
10.1016/j.ijfoodmicro.2009.03.014
45.Robert-Pillot A, Copin S, Himber C, Gay M, Quilici ML. Occurrence of the three major
Vibrio species pathogenic for human in seafood products consumed in France using real-time
PCR. Int J Food Microbiol. 2014 Oct;189:75–81. 10.1016/j.ijfoodmicro.2014.07.014
46.Vu TT, Alter T, Huehn S. Prevalence of Vibrio spp. in Retail Seafood in Berlin, Germany.
J Food Prot. 2018 Apr;81(4):593–7. 10.4315/0362-028X.JFP-17-366
47.Schärer K, Savioz S, Cernela N, Saegesser G, Stephan R. Occurrence of Vibrio spp.
in fish and shellfish collected from the Swiss market. J Food Prot. 2011 Aug;74(8):1345–7.
10.4315/0362-028X.JFP-11-001
48.Schwartz K, Hammerl JA, Göllner C, Strauch E. Environmental and Clinical Strains of
Vibrio cholerae Non-O1, Non-O139 From Germany Possess Similar Virulence Gene Profiles. Front Microbiol.
2019 Apr;10:733. 10.3389/fmicb.2019.00733
49.Greig DR, Schaefer U, Octavia S, Hunter E, Chattaway MA, Dallman TJ, et al. Evaluation
of Whole-Genome Sequencing for Identification and Typing of Vibrio cholerae. J Clin
Microbiol. 2018 Oct;56(11):e00831-18. 10.1128/JCM.00831-18
50.Rijal N, Acharya J, Adhikari S, Upadhaya BP, Shakya G, Kansakar P, et al. Changing
epidemiology and antimicrobial resistance in Vibrio cholerae: AMR surveillance findings (2006-2016) from Nepal. BMC Infect Dis. 2019 Sep;19(1):801.
10.1186/s12879-019-4432-2
51.Sack RB, Rahman M, Yunus M, Khan EH. Antimicrobial resistance in organisms causing
diarrheal disease. Clin Infect Dis. 1997 Jan;24 Suppl 1:S102–5. 10.1093/clinids/24.Supplement_1.S102
52.Sack DA, Lyke C, Mclaughlin C, Suwanvanichkij V. Antimicrobial resistance in shigellosis,
cholera and campylobacteriosis - World Health Organization [Internet]. 2001 [cited
2023 Oct 31]. Available from: https://iris.who.int/handle/10665/66875
53.Garg P, Sinha S, Chakraborty R, Bhattacharya SK, Nair GB, Ramamurthy T, et al. Emergence
of fluoroquinolone-resistant strains of Vibrio cholerae O1 biotype El Tor among hospitalized patients with cholera in Calcutta, India. Antimicrob
Agents Chemother. 2001 May;45(5):1605–6. 10.1128/AAC.45.5.1605-1606.2001
54.Verma J, Bag S, Saha B, Kumar P, Ghosh TS, Dayal M, et al. Genomic plasticity associated
with antimicrobial resistance in Vibrio cholerae. Proc Natl Acad Sci USA. 2019 Mar;116(13):6226–31. 10.1073/pnas.1900141116
55.Morita D, Takahashi E, Morita M, Ohnishi M, Mizuno T, Miyoshi SI, et al. Genomic characterization
of antibiotic resistance-encoding genes in clinical isolates of Vibrio cholerae non-O1/non-O139 strains from Kolkata, India: generation of novel types of genomic
islands containing plural antibiotic resistance genes. Microbiol Immunol. 2020 Jun;64(6):435–44.
10.1111/1348-0421.12790
56.Bhandari M, Rathnayake IU, Huygens F, Jennison AV. Clinical and Environmental Vibrio
cholerae Non-O1, Non-O139 Strains from Australia Have Similar Virulence and Antimicrobial
Resistance Gene Profiles. Microbiol Spectr. 2023 Feb;11(1):e0263122. 10.1128/spectrum.02631-22
57.Lepuschitz S, Baron S, Larvor E, Granier SA, Pretzer C, Mach RL, et al. Phenotypic
and Genotypic Antimicrobial Resistance Traits of Vibrio cholerae Non-O1/Non-O139 Isolated From a Large Austrian Lake Frequently Associated With Cases
of Human Infection. Front Microbiol. 2019 Nov;10:2600. 10.3389/fmicb.2019.02600
58.Marshall BM, Levy SB. Food animals and antimicrobials: impacts on human health. Clin
Microbiol Rev. 2011 Oct;24(4):718–33. 10.1128/CMR.00002-11
59.Schar D, Klein EY, Laxminarayan R, Gilbert M, Van Boeckel TP. Global trends in antimicrobial
use in aquaculture. Scientific Reports 2020 10:1 [Internet]. 2020 Dec 14 [cited 2023
Aug 24];10(1):1–9. Available from: https://www.nature.com/articles/s41598-020-78849-3 10.1038/s41598-020-78849-3
60.Maggiore A, Afonso A, Barrucci F, De Sanctis G. Climate change as a driver of emerging
risks for food and feed safety, plant, animal health and nutritional quality. EFSA
Support Publ. 2020 Jun;17(6).
61.Vezzulli L, Baker-Austin C, Kirschner A, Pruzzo C, Martinez-Urtaza J. Global emergence
of environmental non-O1/O139 Vibrio cholerae infections linked with climate change: a neglected research field? Environ Microbiol.
2020 Oct;22(10):4342–55. 10.1111/1462-2920.15040
62.Constantin de Magny G, Colwell RR. Cholera and climate: a demonstrated relationship.
Trans Am Clin Climatol Assoc. 2009;120:119–28.
Appendix

Figure S1Maximum-likelihood phylogenetic tree of 122
L3b isolates. The country of isolation is indicated (outer ring). The tree is
based on 39,702 polymorphic sites identified in a multi-alignment-derived 3.1
Mbp core genome and was visualised using iTOL. The scale bar indicates the
number of substitutions per site in the core genome alignment.