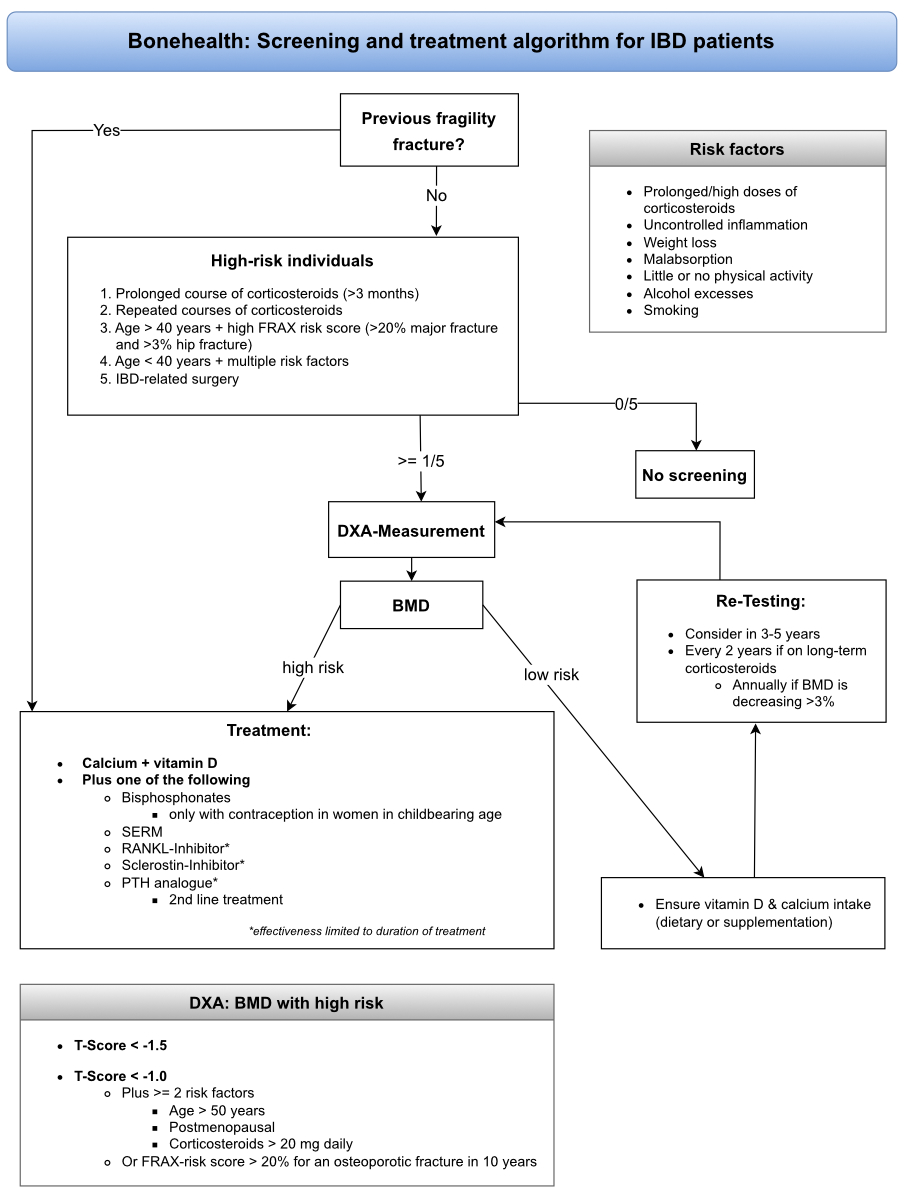
Figure 1 The screening and treatment algorithm for patients with IBD regarding bone health. This algorithm summarises and simplifies the above screening recommendations and is intended as a practical aid.
DOI: https://doi.org/https://doi.org/10.57187/s.3407
Patients with inflammatory bowel disease (IBD) are prone to reduced bone mineral density and have a >38% higher overall risk of spine fractures than the general population [1, 2]. Besides disease activity, IBD specialists must consider possible side effects of medication and the presence of associated diseases and extraintestinal manifestations.
Osteopenia and osteoporosis are complications of IBD that may not receive sufficient attention from the treating physicians. Malnutrition, chronic intestinal inflammation and corticosteroid intake are the major pathophysiological factors contributing to osteoporosis. The primary mechanism involved is an imbalance of the receptor activator of nuclear factor kappa-B ligand (RANKL)/osteoprotegerin (OPG) ratio at the molecular level or bone anabolic and catabolic activity at the cellular level.
The established standard diagnostic procedure to detect IBD-associated bone deterioration is dual-energy X-ray absorptiometry (DXA). From a therapeutic perspective, besides good disease control, vitamin D supplementation and glucocorticoid sparing, several specific osteological options are available: bisphosphonates, RANKL inhibitors (denosumab), parathyroid hormone (PTH) analogues and selective estrogen receptor modulators (SERMs).
The pathophysiology of low bone mineral density in patients with IBD is multifactorial. It involves malabsorption, corticosteroid treatment and the systemic inflammatory process itself, all of which may adversely affect the adequate formation of peak bone mass (up to the age of about 30 years), age-related loss of bone mass or both. Malnutrition might occur in patients with IBD from reduced dietary intake, malabsorption due to intestinal inflammation, short bowel syndrome after intestinal resection or increased energy requirements due to hypercatabolism [3]. In particular, calcium and vitamin D deficiencies are detrimental to bone health and mineralisation, especially in combination with physical inactivity and decreased exposure to sunshine in patients with active disease. In patients with IBD, bone mineral density correlates more with disease severity than disease location and extent [4]. Chronic gut inflammation is an independent risk factor promoting loss of bone mineral density [5].
From an osteoimmunological perspective, the high prevalence of reduced bone mineral density and osteoporosis in patients with IBD can be explained by pro-inflammatory and pro-apoptotic mechanisms. Much of the evidence regarding the effects of pro-inflammatory mechanisms on bone loss originates from studies on rheumatoid arthritis [6–8]. Because rheumatoid arthritis causes bone loss not only at sites prone to inflammation, such as the joints of the hands and fingers, but also systemically throughout the skeleton, a comparison of rheumatoid arthritis and IBD is helpful and valid.
At the cellular level, the key effector cells at the interface between inflammation and bone are the osteoclasts, bone-forming osteoblasts, regulatory osteocytes, and T cells. At the cytokine level, secreted cytokine levels are elevated systemically in both rheumatoid arthritis and IBD: TNF-α, IL-1β, IL-6, IL-8, IL-12/IL-23, IL-17, IL-18, IL-32, and interferon-gamma (IFN-γ) [9].
The release of these inflammatory cytokines, particularly TNF-α, IL-1, IL-6 and IFN-y, modulates the receptor activator of nuclear factor kappa-B (RANK)/RANKL/OPG pathway, inducing bone loss [9]. The RANK and its ligand (RANKL) and decoy receptor OPG regulate bone-resorbing cells (osteoclasts) and lead to bone resorption [10]. RANK is a transmembrane protein belonging to the TNF receptor family and is the primary activator of osteoclastogenesis [11]. Osteoclast maturation is necessary to initiate the bone resorption process. RANKL secreted by osteoblasts binds to RANK on the surface of pre-osteoclasts and mature osteoclasts, inducing their proliferation, activity, and survival. Osteoprotegerin is produced by osteoblasts and competes with RANKL to bind to the RANK receptor, thus blocking osteoclastogenesis.
In addition, increased levels of IL-2, IL-17, IFN-y and TNF-α have been documented in Crohn’s disease. In contrast, ulcerative colitis is associated with cytokines of the Th2 profile, such as IL-4, IL-5, and IL-13 [12]. IL-12, a T-cell mediator, is also an important inflammatory cytokine found in IBD and interacts with TNF-α to inhibit osteoclastogenesis [13–15].
Moreover, there is sufficient evidence that TNF-α plays an important role in bone loss in Crohn’s disease. TNF-α blockade improves bone formation and reduces bone resorption by suppressing T cells [16, 17]. Infliximab maintenance therapy also leads to about a 14% increase in lumbar bone density [18, 19]. The Swiss IBD cohort study (SIBDCS), launched in 2006 with the aim of enrolling and following patients with IBD throughout Switzerland [20], demonstrated that early administration of anti-TNF-α was associated with a lower osteoporosis risk.
TNF-α inhibition appears to have a beneficial effect on bone turnover, especially when administered in combination with bisphosphonates [21]. The fact that this was observed in infliximab responders and non-responders and during the maintenance phase in patients with or without corticosteroid co-administration suggests an independent mode of action [22, 23]. Of course, this effect might also be secondary to reducing active inflammation and decreased glucocorticoid exposure.
While some cytokines directly act through their receptors (IL-1, IL-6, IL-17 and TNF-α), others act indirectly through the OPG/RANKL/RANK pathway via T cells or fibroblasts [24]. Importantly, the upregulated cytokines are not only bone destructive (e.g. IL-6, TNF-α and IL-1β) but also bone protective and anti-osteoclastogenic (e.g. IL-12/IL-23, IL-17, IL-18, IL-4, IL-10 and IFN-y) [25]. This observation is of interest since the therapeutic effects of anti-IL12/23 inhibitors were recently found to lower both the need for glucocorticoids and direct bone-specific harm [26]. However, reliable data for vedolizumab or ustekinumab are currently lacking.
Regarding the apoptotic mechanisms leading to bone loss, the mode of action of systemic glucocorticoids (GCs) must be elucidated. Glucocorticoids decrease the ability of osteoblasts to rebuild eroded bone and compensate for bone loss by dose-dependently activating caspases and the Wingless-related integration site (WNT) pathway in osteoblasts [27]. In addition, dying osteoblasts and systemic glucocorticoids activate bone resorption via the OPG/RANKL/RANK pathway, resulting in a double negative bone balance [28]. In addition, apoptosis of osteocytes also leads to a reduced load-bearing capacity of the bone, since on one hand, microfractures are no longer recognized and repaired, and on the other hand, adaptations no longer occur.
Corticosteroids also increase the expression of RANKL and decrease OPG receptors in stem cells and osteoblastic cells, thus inhibiting stem cell differentiation into osteoblasts, inducing osteoblast apoptosis, and favouring the differentiation of precursors into osteoclasts [29]. This mechanism will decrease the secretion of the osteoid matrix and enhances bone reabsorption. However, the increase in RANKL is only transient, and the failure of bone formation seems to be more important than bone resorption [30].
In summary, an imbalance of the RANKL/OPG ratio plays an essential role in developing low bone mineral density in patients with IBD.
Endocrine manifestations are frequent in IBD including metabolic bone disease with bone loss, growth failure and hypogonadism with pubertal delay in patients with childhood onset of disease. The pathogenesis of growth failure is multifactorial and include disease-related inflammation, malnutrition, glucocorticoid use, and hypogonadism [31, 32] and is related to an impairment of the GH-IGF1 axis with relative growth hormone resistance [33]. In parallel to growth failure, the presence of delayed puberty, weight loss and glucocorticoid treatment impair an increase of bone mass in adolescence. Therefore, patients with childhood onset of IBD may present with low peak bone mass in early adulthood, an important predisposing factor for low bone mass and increased fracture risk later in life.
One of the key hormones involved in bone metabolism is vitamin D. Vitamin D levels are lower in adult and paediatric patients with IBD as compared to healthy controls [34], which is due to impaired intestinal absorption (specifically in patients with small-bowel resection), reduced dietary or supplemental intake, low sun exposure and vitamin D loss in the presence of protein-losing enteropathies. As malabsorptive conditions severely impair vitamin D status with hypocalcaemia, impaired bone mineralisation and increased risk of fractures, biochemical assessment and supplementation should be routinely considered in all patients with IBD. Furthermore, recent data suggest that poor vitamin D status may negatively impact the clinical course of IBD due to its immune-modulating effect [35].
Adipokines are cytokines secreted by adipose tissue and have been implicated in the pathogenesis of IBD. Recent research has suggested that adipokines (such as leptin, adiponectin, apelin, and chemerin) may be involved in the development of IBD-induced bone loss. Specifically visceral adipose tissue exhibits pro-inflammatory, immunoregulatory, and endocrine activity [36, 37]. While the exact mechanisms regarding the crosstalk between fat and bone tissues are still unclear, dysregulation of adipokine levels has been shown to play an important role in the regulation of osteoblastic and osteoclastic cell functions.
In summary, the chronic inflammation and malnutrition associated with IBD can disrupt the normal function of several hormones involved in bone metabolism resulting in decreased bone density and increased risk of fractures. In clinical practice, preventive measures to ascertain bone accrual in adolescence and prevention of accelerated bone loss in adulthood include assessment of calcium/vitamin D status as well as exclusion of sex hormone deficiencies, and consider replacement therapies if needed.
Another, maybe underestimated mode of action associated with bone deterioration in IBD patients is the iron-induced hypophosphataemia, an emerging complication first been reported in 1982 in a patient receiving repeated intravenous therapy with saccharated iron oxide [38], iron polymaltose [39] or ferric carboxymaltose [40]. The application of iron isomaltose is not associated with hypophosphataemia [41] and therefore an option in patients with low bone density or osteoporosis.
A recent systematic review including 3661 IBD patients and 12,789 healthy controls identified a prevalence of osteoporosis of 2–16% in the former population (7–15% for Crohn’s disease and 2–9% for ulcerative colitis) and of 3–10% in the later. These results are consistent with observations made in a 10 years follow-up population-based inception Danish cohort, where the prevalence of osteoporosis in 513 IBD patients was 14%, which was also significantly increased compared to controls [42, 43].
Because of widely differing screening and treatment practices and a lack of awareness, osteopenia and osteoporosis are often underestimated, particularly in patients with IBD. In the Swiss IBD Cohort Study (SIBDCS), 19% of patients had low bone mineral density [44]. Other studies estimate that osteopenia and osteoporosis affect 35–40% and up to 15% of patients with IBD, respectively [45–47], with a high regional variability.
The lack of awareness in patients with IBD might be explained by young age and the lack of a direct burden from musculoskeletal pain.
Bone mineral density is a surrogate for bone health and a predictor of fracture risk. It is measured by dual-energy X-ray absorptiometry (DXA) [48] in the femur (femoral neck or total hip) and/or lumbar spine [49]. In postmenopausal women and men aged ≥50 years, osteoporosis is defined as a bone mineral density value at least 2.5 standard deviations (SDs) below the peak bone mass of healthy adults (T-score ≤−2.5), and osteopenia, a precursor of osteoporosis, is defined as a bone mineral density value between 1.0 and 2.5 SDs below normal [2] (table 1). In individuals aged <50 years, low bone mineral density (osteoporosis) is defined as a bone mineral density value “below the expected range for age” (Z-score <–2 SDs) [50].
Table 1 The World Health Organization’s definition of osteoporosis based on DXA measurement [51].
| Interpretation | T-score |
| Normal | –1.0 and higher |
| Osteopenia | –1.0 to –2.5 |
| Osteoporosis | –2.5 and lower |
| Severe osteoporosis | –2.5 and lower with one or more fragility fractures |
Patients aged >40 years without a previous history of prolonged corticosteroid therapy should be assessed using the fracture risk assessment tool (FRAX; https://frax.shef.ac.uk/FRAX/tool.aspx?country=15), which assesses the 10-year probability of a major osteoporotic fracture (MOF). FRAX is a proven tool but does not discriminate past versus current corticosteroid use or stratify risk according to corticosteroid doses >7.5 mg [52].
According to current guidelines (British Society of Gastroenterology, European Crohn’s and Colitis Organisation and American Gastroenterological Association), we suggest screening for osteopenia or osteoporosis using DXA in high-risk individuals and after IBD-related surgery [26, 50, 53]. At-risk individuals are patients with IBD with a prolonged course (>3 months) or repeated corticosteroid courses, those aged >40 years with a high FRAX risk score (>20% major fracture and >3% hip fracture), and those <40 years with multiple risk factors (prolonged/high doses corticosteroids, uncontrolled inflammation, weight loss, malabsorption, little or no physical activity, alcohol excesses and smoking; figure 1 and table 2).
Figure 1 The screening and treatment algorithm for patients with IBD regarding bone health. This algorithm summarises and simplifies the above screening recommendations and is intended as a practical aid.
Table 2 Etiologic factors associated with altered bone health in inflammatory bowel disease [48].
| General factors | Decreased vitamin D concentration |
| Low calcium intake | |
| Low magnesium and potassium concentrations | |
| Low body mass index | |
| Decreased physical activity | |
| Smoking | |
| Excessive alcohol consumption | |
| IBD-associated factors | IBD therapy: corticosteroids (damaging) and anti-TNF-α therapy (beneficial) |
| IBD-specific factors: sex hormone deficiency | |
| IBD-associated inflammatory processes: disbalance of cytokines and other immunological networks | |
| Gastrointestinal damage: intestinal insufficiency (short bowel syndrome) and intestinal leakage |
An antiresorptive treatment is recommended for patients with a previous fragility fracture, and DXA measurements may be used for treatment monitoring.
There is currently no evidence that budesonide causes reduced bone mineral density in patients with IBD. Studies on microscopic colitis – where budesonide is the state-of-the-art treatment – did not show significant differences in the incidence of osteopenia/osteoporosis [54]. Nonetheless, these two clinical conditions differ in particular regarding the severity of secondary malabsorption and consequential deficiencies. Therefore, it cannot be excluded that budesonide may have more adverse effects in patients with IBD.
Smoking is a major risk factor for patients with Crohn’s disease. It is associated with relapses, more frequent postoperative complications due to impaired microcirculation and reduced bone mineral density with an increased fracture risk. Therefore, patients should make every effort to stop smoking immediately [26, 55, 56].
DXA measurement is still the standard diagnostic modality for assessing bone mineral density/bone health. However, since the two-dimensional nature of DXA imposes limitations, other imaging modalities (currently mainly used in research settings) should be mentioned to complete the diagnostic spectrum [57]:
Only a few studies have examined adults with IBD using HR-pQCT, demonstrating decreased trabecular and volumetric bone mineral density and possibly a decreased cortical bone area [58]. In 2017, Pepe et al. evaluated the SWISS IBD Cohort using HR-pQCT (Xtreme I; Scanco, Switzerland). They demonstrated a predominantly altered trabecular bone microstructure compared to healthy controls, which is associated with fractures during childhood/adolescence and may predispose to fragility fractures throughout life [43]. Further prospective studies with HR-pQCT are needed to elucidate its clinical impact and possible advantages compared to DXA.
Patients on long-term corticosteroids should undergo bone densitometry every second year. However, yearly DXA measurements are recommended for those showing a >3% decrease in bone mineral density annually. However, in Switzerland, health insurance currently does not cover repeated DXA measurements after one year, and corresponding cost reimbursement requests must be submitted.
A patient is considered high-risk, and treatment to prevent osteoporosis should be initiated, if their bone mineral densitometry indicates a T-score <−1.5, a T-Score <−1.0 and they have at least two risk factors (age >50 years, postmenopausal or corticosteroids >20 mg daily) or a FRAX score with a >20% risk for an osteoporotic fracture within 10 years [59].
Additionally, patients with past fragility fractures, aged >69 years, taking corticosteroids (>7.5 mg/day) or with a high-risk FRAX score should be evaluated for bone protective therapy before the start of corticosteroid treatment [60]. Newer guidelines from the American College of Rheumatology strongly recommend an initial clinical fracture risk assessment for all adults initiating or continuing corticosteroid therapy ≥2.5 mg/day for >3 months, those who have never had fracture risk assessment, and those previously treated with osteoporotic therapy. For those taking glucocorticoids at >7.5 mg/day, the FRAX score should be multiplied by 1.15 to determine the 10-year risk of major osteoporotic fracture and by 1.2 to determine the hip fracture risk [61].
Adequate medical treatment can partially reverse bone deterioration and prevent osteoporotic fractures even in high-risk individuals such as postmenopausal women, older men with osteoporosis and patients taking glucocorticoids [49,62–64]. Risk factors for increased bone loss should be limited, and adequate dietary intake or supplementation of calcium or vitamin D should be maintained.
More than 50% of patients with ulcerative colitis and Crohn’s disease in Northern Europe and the United Kingdom suffer from vitamin D deficiency (25-hydroxyvitamin D [25OHD] <50 nmol/l) [65, 66]. Therefore, we recommend quantifying/measuring vitamin D levels (25OHD) in patients with IBD at least once per year. However, patients should be informed that the health insurance company only reimburses vitamin D supplementation for a severe deficiency (25OHD <25 nmol/l).
High-risk patients with IBD and osteopenia (not meeting the criteria for pharmacological treatment) and all patients starting corticosteroid therapy should be adequately supplemented with vitamin D (800–1000 IU/day) and calcium (500–1000 mg/day, dietary or supplement). We suggest determining the patient’s daily calcium intake to assess their supplementation needs (https://www.rheumaliga.ch/calciumrechner). Patients should be prevented from ingesting >1400 mg of calcium daily since this may increase their all-cause mortality [67]. British guidelines recommend that those with vitamin D deficiency receive an initial treatment of 50,000 IU weekly for six weeks [26].
In addition to vitamin D and/or calcium supplementation, individuals with osteoporosis or increased fracture risk should receive a specific osteological treatment with antiresorptives (bisphosphonates, denosumab or selective estrogen receptor modulators [SERMs]), an osteoanabolic agent (teriparatide) or a dual effect agent (romosozumab; table 3).
Table 3 An overview of osteoprotective medication.
| Substance class | Substance | Dosage | Contraindications |
| Bisphosphonates | Alendronate (Fosamax®) | 70 mg p.o. weekly | GFR <30 ml/min, hypocalcaemia, gastrointestinal ulceration |
| Risedronate (Actonel®) | 30–35 mg p.o. weekly | GFR <30 ml/min, hypocalcaemia, gastrointestinal ulceration | |
| Zoledronate (Aclasta®) | 5 mg i.v. 1×/year | GFR <35 ml/min, hypocalcaemia | |
| PTH analogue | Teriparatide (Forsteo®) | 20 µg s.c. daily | Hypercalcaemia |
| SERM | Raloxifene (Evista®) | 60 mg p.o. daily | Venous thromboembolism, restricted liver function, cholestasis, uterine bleeding |
| RANKL inhibitor | Denosumab (Prolia®) | 60 mg s.c./6 months | Hypocalcaemia |
| Sclerostin inhibitor | Romosozumab (Evenity®) | 210 mg s.c./month | Hypocalcaemia, history of myocardial or cerebrovascular incidence |
| PTH = parathyroid hormone, SERM = selective estrogen receptor modulator | |||
The treatment duration of antiresorptive drugs depends on their mode of action and long-term efficacy and safety. Data on reducing fracture risk in patients with IBD are primarily available for bisphosphonates (alendronate, risedronate and zoledronate). Intravenous zoledronate may be preferred due to the potential gastrointestinal side effects of oral bisphosphonates. For women of childbearing age, bisphosphonates should only be prescribed if they are taking effective contraception. Because of an elevated risk, osteonecrosis of the jaw or atypical femoral fractures should be ruled out before starting bisphosphonate therapy.
Unlike bisphosphonates, which are deposited on the mineralised bone surface and taken up by the mature osteoclasts, denosumab, a RANKL inhibitor, inhibits the proliferation and differentiation of osteoclast precursor cells and impairs the action of mature osteoclasts. Notably, the effectiveness of denosumab is limited to the duration of treatment, unlike bisphosphonates, which are known for their residual anti-resorptive efficacy after treatment is discontinued. Sequential bisphosphonate treatment is mandatory after discontinuing denosumab to prevent accelerated bone loss and vertebral fractures [68, 69]. Romoszumab can increase bone density via another mode of action. By inhibiting sclerostin, romosozumab strongly stimulates bone formation and, to a lesser extent, inhibits bone resorption. Like with denosumab, sequential therapy with an antiresorptive agent is mandatory after discontinuing romosozumab.
Antiresorptive, second-line therapy with teriparatide is suggested in cases of severe osteoporosis (bone mineral density at the spine <–3.5 SDs) or treatment failure (incident vertebral fractures during bisphosphonate treatment). Teriparatide (a PTH analogue) stimulates osteoblastic bone formation, resulting in bone accrual, specifically in the trabecular bone. After treatment with teriparatide (usually for 24 months), sequential antiresorptive therapy is necessary to support the secondary mineralisation of the newly formed bone and maintain the bone mineral content [70,71].
Since most studies on treating and preventing osteoporosis have been conducted in postmenopausal women, evidence for treating young patients with IBD is limited. In these patients, the primary focus should be on controlling the risk factors.
Of note are new guidelines by the American College of Rheumatology on preventing and treating glucocorticoid-induced osteoporosis. They suggest treating adults aged ≥40 years receiving high-dose glucocorticoids (initial dose ≥30 mg/day for 30 days or cumulative dose ≥5 gm in one year) with oral/intravenous bisphosphonate, PTH analogue or RANKL inhibitor [61].
Sequential treatments are recommended when initial osteoporotic therapy and glucocorticoids are discontinued and at low (10-year risk of MOF <10%, hip ≤1%) or moderate (10-year risk of MOF 10–20%, hip 1–3%) FRAX risk.
No subsequent osteoprotective therapy is needed if the initial osteoporotic therapy consists of oral/intravenous bisphosphonate or SERM. In cases initially treated with PTH or romosozumab, subsequent treatment with oral/intravenous bisphosphonate or denosumab followed by bisphosphonate is mandatory. Additionally, in cases given denosumab therapy, an oral/intravenous bisphosphonate treatment is indicated for 1–2 years until the rebound after denosumab is finished (table 4) [61].
Table 4 The sequential treatments recommended when initial osteoporotic and glucocorticoid therapies are discontinued and patients are at low or moderate risk.
| Initial osteoporotic therapy | Subsequent osteoporotic therapy options |
| Oral/intravenous bisphosphonate | No subsequent osteoporotic therapy is needed |
| SERM | No subsequent osteoporotic therapy is needed |
| PTH analogue | Oral or intravenous bisphosphonate or denosumab followed by bisphosphonate |
| Denosumab | Oral or intravenous bisphosphonate |
| Romosozumab | Oral or intravenous bisphosphonate or denosumab followed by bisphosphonate |
| PTH = parathyroid hormone, SERM = selective estrogen receptor modulator | |
When initial osteoporotic therapy and glucocorticoids are discontinued, and the patient remains at high risk, the continuation of current therapy or a switch to intravenous bisphosphonate, denosumab, PTH, SERM or romosozumab is indicated [61].
We thank Prof. Dr. med. Hans Jörg Häuselmann for his support.
We also thank Sandoz Pharmaceuticals AG for providing an initial platform for the scientific exchange.
The authors received no financial support for writing this review.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. The following relationships or activities outside of the submitted manuscript have been reported: L. Biedermann: consulting fees, honoraria or travel support from Abbvie, Amgen, Bristol Myers Squibb, Esocap, Falk, Janssen, Lilly, Pfizer, Sanofi and Takeda. P. Hruz: participation on a data safety board or advisory board for Abbvie, Bristol Myers Squibb, Falk, iQone, Lilly, MSD, Sandoz and Takeda. P. Juillerat: consulting fees or honoraria from AbbVie, Arena, Amgen, Bristol Myers Squibb, Ferring, Gilead, Janssen, Lilly, MSD, Pfizer, Pierre Fabre, Roche, Sandoz, Takeda, Tillots and UCB Pharma. A. Kreienbühl: honoraria or travel support from Amgen, Janssen-Cilag and Takeda and participation on a data safety board or advisory board for Takeda. S. Restellini: consulting fees, honoraria or travel support from AbbVie, Bristol Myers Squibb, Falk, iQone, Janssen, Sandoz, Takeda, UCB and Vifor. G. Rogler: grants, consulting fees or honoraria from Abbvie, Ardeypharm, Arena, Augurix, Bristol Myers Squibb, Boehringer, Calypso, Celgene, Falk, Ferring, Fisher, Flamentera, Genentech, Gilead, Janssen, Lilly, MSD, Novartis, Pfizer, Phadia, Roche, UCB, Takeda, Tillots, Vifor, Vital Solutions, USB and Zeller. F. W. Seibold: consulting fees from Abbvie, Amgen, Bristol Myers Squibb, Falk, Lilly, Takeda S. Vavricka: Participation on a Data Safety Monitoring Board or Advisory Board for Abbvie, Bristol Myers Squibb, Falk, iQone, Lilly, MSD, Sandoz and Takeda.
1. Szafors P, Che H, Barnetche T, Morel J, Gaujoux-Viala C, Combe B, et al. Risk of fracture and low bone mineral density in adults with inflammatory bowel diseases. A systematic literature review with meta-analysis. Osteoporos Int. 2018 Nov;29(11):2389–97. 10.1007/s00198-018-4586-6
2. Chedid VG, Kane SV. Bone Health in Patients With Inflammatory Bowel Diseases. J Clin Densitom. 2020;23(2):182–9. 10.1016/j.jocd.2019.07.009
3. Prieto JM, Andrade AR, Magro DO, Imbrizi M, Nishitokukado I, Ortiz-Agostinho CL, et al. Nutritional Global Status and Its Impact in Crohn’s Disease. J Can Assoc Gastroenterol. 2021 Mar;4(6):290–5. 10.1093/jcag/gwab006
4. Cravo M, Guerreiro CS, dos Santos PM, Brito M, Ferreira P, Fidalgo C, et al. Risk factors for metabolic bone disease in Crohn’s disease patients. Inflamm Bowel Dis. 2010 Dec;16(12):2117–24. 10.1002/ibd.21297
5. Reinshagen M. Osteoporosis in inflammatory bowel disease. J Crohns Colitis. 2008 Sep;2(3):202–7. 10.1016/j.crohns.2008.01.005
6. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007 Apr;7(4):292–304. 10.1038/nri2062
7. Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019 Jun;570(7760):246–51. 10.1038/s41586-019-1263-7
8. Shim JH, Stavre Z, Gravallese EM. Bone Loss in Rheumatoid Arthritis: Basic Mechanisms and Clinical Implications. Calcif Tissue Int. 2018 May;102(5):533–46. 10.1007/s00223-017-0373-1
9. Moschen AR, Kaser A, Enrich B, Ludwiczek O, Gabriel M, Obrist P, et al. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut. 2005 Apr;54(4):479–87. 10.1136/gut.2004.044370
10. Ke K, Arra M, Abu-Amer Y. Mechanisms Underlying Bone Loss Associated with Gut Inflammation. Int J Mol Sci. 2019 Dec;20(24):6323. 10.3390/ijms20246323
11. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008 May;473(2):139–46. 10.1016/j.abb.2008.03.018
12. Nemeth ZH, Bogdanovski DA, Barratt-Stopper P, Paglinco SR, Antonioli L, Rolandelli RH. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus. 2017 Apr;9(4):e1177. 10.7759/cureus.1177
13. Kitaura H, Nagata N, Fujimura Y, Hotokezaka H, Yoshida N, Nakayama K. Effect of IL-12 on TNF-alpha-mediated osteoclast formation in bone marrow cells: apoptosis mediated by Fas/Fas ligand interaction. J Immunol. 2002 Nov;169(9):4732–8. 10.4049/jimmunol.169.9.4732
14. Yoshimatsu M, Kitaura H, Fujimura Y, Eguchi T, Kohara H, Morita Y, et al. IL-12 inhibits TNF-alpha induced osteoclastogenesis via a T cell-independent mechanism in vivo. Bone. 2009 Nov;45(5):1010–6. 10.1016/j.bone.2009.07.079
15. Bravenboer N, Oostlander AE, van Bodegraven AA. Bone loss in patients with inflammatory bowel disease: cause, detection and treatment. Curr Opin Gastroenterol. 2021 Mar;37(2):128–34. 10.1097/MOG.0000000000000710
16. Abreu MT, Geller JL, Vasiliauskas EA, Kam LY, Vora P, Martyak LA, et al. Treatment with infliximab is associated with increased markers of bone formation in patients with Crohn’s disease. J Clin Gastroenterol. 2006 Jan;40(1):55–63. 10.1097/01.mcg.0000190762.80615.d4
17. Mauro M, Radovic V, Armstrong D. Improvement of lumbar bone mass after infliximab therapy in Crohn’s disease patients. Can J Gastroenterol. 2007 Oct;21(10):637–42. 10.1155/2007/216162
18. Baban YN, Edicheria CM, Joseph J, Kaur P, Mostafa JA. Osteoporosis Complications in Crohn’s Disease Patients: Factors, Pathogenesis, and Treatment Outlines. Cureus. 2021 Dec;13(12):e20564. 10.7759/cureus.20564
19. Frei R, Fournier N, Zeitz J, Scharl M, Morell B, Greuter T, et al. Early Initiation of Anti-TNF is Associated with Favourable Long-term Outcome in Crohn’s Disease: 10-Year-Follow-up Data from the Swiss IBD Cohort Study. J Crohns Colitis. 2019 Sep;13(10):1292–301. 10.1093/ecco-jcc/jjz057
20. Pittet V, Michetti P, Mueller C, Braegger CP, von Känel R, Schoepfer A, et al.; Swiss IBD Cohort Study Group. Cohort Profile Update: The Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS). Int J Epidemiol. 2019 Apr;48(2):385–386f. 10.1093/ije/dyy298
21. Pazianas M, Rhim AD, Weinberg AM, Su C, Lichtenstein GR. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn’s disease. Ann N Y Acad Sci. 2006 Apr;1068(1):543–56. 10.1196/annals.1346.055
22. Bernstein M, Irwin S, Greenberg GR. Maintenance infliximab treatment is associated with improved bone mineral density in Crohn’s disease. Am J Gastroenterol. 2005 Sep;100(9):2031–5. 10.1111/j.1572-0241.2005.50219.x
23. Veerappan SG, O’Morain CA, Daly JS, Ryan BM. Review article: the effects of antitumour necrosis factor-α on bone metabolism in inflammatory bowel disease. Aliment Pharmacol Ther. 2011 Jun;33(12):1261–72. 10.1111/j.1365-2036.2011.04667.x
24. Gravallese EM, Goldring SR, Schett G. The Role of the Immune System in the Local and Systemic Bone Loss of Inflammatory Arthritis. Osteoimmunology: Interactions of the Immune and Skeletal Systems: Second Edition. 2016 Jan 1;241–56. 10.1016/B978-0-12-800571-2.00013-X
25. Aeberli D. Skeleton, Inflammatory Diseases of. Reference Module in Biomedical Sciences. 2014 Jan 1;
26. Lamb CA, Kennedy NA, Raine T, Hendy PA, Smith PJ, Limdi JK, et al.; IBD guidelines eDelphi consensus group. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019 Dec;68 Suppl 3:s1–106. 10.1136/gutjnl-2019-318484
27. Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol. 2010 Feb;6(2):82–8. 10.1038/nrrheum.2009.259
28. Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008 Jun;23(6):915–27. 10.1359/jbmr.080207
29. Compston J. Glucocorticoid-induced osteoporosis: an update. Endocrine. 2018 Jul;61(1):7–16. 10.1007/s12020-018-1588-2
30. Gado M, Baschant U, Hofbauer LC, Henneicke H. Bad to the Bone: The Effects of Therapeutic Glucocorticoids on Osteoblasts and Osteocytes. Front Endocrinol (Lausanne). 2022 Mar;13:835720. 10.3389/fendo.2022.835720
31. Shamir R, Phillip M, Levine A. Growth retardation in pediatric Crohn’s disease: pathogenesis and interventions. Inflamm Bowel Dis. 2007 May;13(5):620–8. 10.1002/ibd.20115
32. Tigas S, Tsatsoulis A. Endocrine and metabolic manifestations in inflammatory bowel disease. Ann Gastroenterol. 2012;25(1):37–44.
33. Katsanos KH, Tsatsoulis A, Christodoulou D, Challa A, Katsaraki A, Tsianos EV. Reduced serum insulin-like growth factor-1 (IGF-1) and IGF-binding protein-3 levels in adults with inflammatory bowel disease. Growth Horm IGF Res. 2001 Dec;11(6):364–7. 10.1054/ghir.2001.0248
34. Pappa HM, Grand RJ, Gordon CM. Report on the vitamin D status of adult and pediatric patients with inflammatory bowel disease and its significance for bone health and disease. Inflamm Bowel Dis. 2006 Dec;12(12):1162–74. 10.1097/01.mib.0000236929.74040.b0
35. Giustina A, di Filippo L, Allora A, Bikle DD, Cavestro GM, Feldman D, et al. Vitamin D and malabsorptive gastrointestinal conditions: A bidirectional relationship? Rev Endocr Metab Disord. 2023 Apr;24(2):121–38. 10.1007/s11154-023-09792-7
36. Weidinger C, Ziegler JF, Letizia M, Schmidt F, Siegmund B. Adipokines and Their Role in Intestinal Inflammation. Front Immunol. 2018 Oct;9(OCT):1974. 10.3389/fimmu.2018.01974
37. Karaskova E, Velganova-Veghova M, Geryk M, Foltenova H, Kucerova V, Karasek D. Role of Adipose Tissue in Inflammatory Bowel Disease. Int J Mol Sci. 2021 Apr;22(8):4226. 10.3390/ijms22084226
38. Okada M, Imamura K, Fuchigami T, Omae T, Iida M, Nanishi F, et al. [2 cases of nonspecific multiple ulcers of the small intestine associated with osteomalacia caused by long-term intravenous administration of saccharated ferric oxide]. Nihon Naika Gakkai Zasshi. 1982 Nov;71(11):1566–72. 10.2169/naika.71.1566
39. Schouten BJ, Doogue MP, Soule SG, Hunt PJ. Iron polymaltose-induced FGF23 elevation complicated by hypophosphataemic osteomalacia. Ann Clin Biochem. 2009 Mar;46(Pt 2):167–9. 10.1258/acb.2008.008151
40. Mani LY, Nseir G, Venetz JP, Pascual M. Severe hypophosphatemia after intravenous administration of iron carboxymaltose in a stable renal transplant recipient. Transplantation. 2010 Oct;90(7):804–5. 10.1097/TP.0b013e3181f00a18
41. Kalra PA, Bhandari S. Efficacy and safety of iron isomaltoside (Monofer®) in the management of patients with iron deficiency anemia. Int J Nephrol Renovasc Dis. 2016 Mar;9:53–64. 10.2147/IJNRD.S89704
42. Kärnsund S, Lo B, Bendtsen F, Holm J, Burisch J. Systematic review of the prevalence and development of osteoporosis or low bone mineral density and its risk factors in patients with inflammatory bowel disease. World J Gastroenterol. 2020 Sep;26(35):5362–74. 10.3748/wjg.v26.i35.5362
43. Pepe J, Zawadynski S, Herrmann FR, Juillerat P, Michetti P, Ferrari-Lacraz S, et al. Structural Basis of Bone Fragility in Young Subjects with Inflammatory Bowel Disease: A High-resolution pQCT Study of the SWISS IBD Cohort (SIBDC). Inflamm Bowel Dis. 2017 Aug;23(8):1410–7. 10.1097/MIB.0000000000001139
44. Schüle S, Rossel JB, Frey D, Biedermann L, Scharl M, Zeitz J, et al.; Swiss IBD cohort study. Prediction of low bone mineral density in patients with inflammatory bowel diseases. United European Gastroenterol J. 2016 Oct;4(5):669–76. 10.1177/2050640616658224
45. Piodi LP, Poloni A, Ulivieri FM. Managing osteoporosis in ulcerative colitis: something new? World J Gastroenterol. 2014 Oct;20(39):14087–98. 10.3748/wjg.v20.i39.14087
46. Agrawal M, Arora S, Li J, Rahmani R, Sun L, Steinlauf AF, et al. Bone, inflammation, and inflammatory bowel disease. Curr Osteoporos Rep. 2011 Dec;9(4):251–7. 10.1007/s11914-011-0077-9
47. Lee N, Radford-Smith G, Taaffe DR. Bone loss in Crohn’s disease: exercise as a potential countermeasure. Inflamm Bowel Dis. 2005 Dec;11(12):1108–18. 10.1097/01.MIB.0000192325.28168.08
48. van Bodegraven AA, Bravenboer N. Perspective on skeletal health in inflammatory bowel disease. Osteoporos Int. 2020 Apr;31(4):637–46. 10.1007/s00198-019-05234-w
49. Cosman F, de Beur SJ, LeBoff MS, Lewiecki EM, Tanner B, Randall S, et al.; National Osteoporosis Foundation. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014 Oct;25(10):2359–81. 10.1007/s00198-014-2794-2
50. Harbord M, Annese V, Vavricka SR, Allez M, Barreiro-de Acosta M, Boberg KM, et al.; European Crohn’s and Colitis Organisation. The First European Evidence-based Consensus on Extra-intestinal Manifestations in Inflammatory Bowel Disease. J Crohns Colitis. 2016 Mar;10(3):239–54. 10.1093/ecco-jcc/jjv213
51. World Health Organization. WHO SCIENTIFIC GROUP ON THE ASSESSMENT OF OSTEOPOROSIS AT PRIMARY HEALTH CARE LEVEL Summary Meeting Report. 2004;
52. Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int. 2011 Mar;22(3):809–16. 10.1007/s00198-010-1524-7
53. Farraye FA, Melmed GY, Lichtenstein GR, Kane SV. ACG Clinical Guideline: Preventive Care in Inflammatory Bowel Disease. Am J Gastroenterol. 2017 Feb;112(2):241–58. 10.1038/ajg.2016.537
54. Tome J, Sehgal K, Kamboj AK, Comstock B, Harmsen WS, Khanna S, et al. Budesonide Maintenance in Microscopic Colitis: Clinical Outcomes and Safety Profile From a Population-Based Study. Am J Gastroenterol. 2022 Aug;117(8):1311–5. 10.14309/ajg.0000000000001774
55. Yoon V, Maalouf NM, Sakhaee K. The effects of smoking on bone metabolism. Osteoporos Int. 2012 Aug;23(8):2081–92. 10.1007/s00198-012-1940-y
56. Karban A, Eliakim R. Effect of smoking on inflammatory bowel disease: Is it disease or organ specific? World Journal of Gastroenterology: WJG. 2007 Apr 4.
57. Williams KM, Darukhanavala A, Hicks R, Kelly A. An update on methods for assessing bone quality and health in Cystic fibrosis. J Clin Transl Endocrinol. 2021 Dec;27:100281. 10.1016/j.jcte.2021.100281
58. Dubner SE, Shults J, Baldassano RN, Zemel BS, Thayu M, Burnham JM, et al. Longitudinal assessment of bone density and structure in an incident cohort of children with Crohn’s disease. Gastroenterology. 2009 Jan;136(1):123–30. 10.1053/j.gastro.2008.09.072
59. Schweizerische Gesellschaft für Rheumatologie. corticosteroid-OSTEOPOROSE: Vorbeugung und Behandlung. 2023.
60. Maricic M, Deal C, Dore R, Laster A. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis: Comment on the Article by Buckley et al. Arthritis Care Res (Hoboken). 2018 Jun;70(6):949–50. 10.1002/acr.23424
61. Glucocorticoid-Induced Osteoporosis Clinical Practice Guidelines.
62. Cranney A, Wells G, Willan A, Griffith L, Zytaruk N, Robinson V, et al.; Osteoporosis Methodology Group and The Osteoporosis Research Advisory Group. Meta-analyses of therapies for postmenopausal osteoporosis. II. Meta-analysis of alendronate for the treatment of postmenopausal women. Endocr Rev. 2002 Aug;23(4):508–16. 10.1210/er.2001-2002
63. Melek J, Sakuraba A. Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clin Gastroenterol Hepatol. 2014 Jan;12(1):32–44.e5. 10.1016/j.cgh.2013.08.024
64. Bakker SF, Dik VK, Witte BI, Lips P, Roos JC, Van Bodegraven AA. Increase in bone mineral density in strictly treated Crohn’s disease patients with concomitant calcium and vitamin D supplementation. J Crohns Colitis. 2013 Jun;7(5):377–84. 10.1016/j.crohns.2012.06.003
65. Frigstad SO, Høivik M, Jahnsen J, Dahl SR, Cvancarova M, Grimstad T, et al. Vitamin D deficiency in inflammatory bowel disease: prevalence and predictors in a Norwegian outpatient population. Scand J Gastroenterol. 2017 Jan;52(1):100–6. 10.1080/00365521.2016.1233577
66. Chatu S, Chhaya V, Holmes R, Neild P, Kang JY, Pollok RC, et al. Factors associated with vitamin D deficiency in a multicultural inflammatory bowel disease cohort. Frontline Gastroenterol. 2013 Jan;4(1):51–6. 10.1136/flgastro-2012-100231
67. Michaëlsson K, Melhus H, Warensjö Lemming E, Wolk A, Byberg L. Long term calcium intake and rates of all cause and cardiovascular mortality: community based prospective longitudinal cohort study. BMJ. 2013 Feb;346(7895):f228. 10.1136/bmj.f228
68. Lamy O, Stoll D, Aubry-Rozier B, Gonzalez Rodriguez E. Correction to: stopping Denosumab. Curr Osteoporos Rep. 2022 Oct;20(5):363. 10.1007/s11914-020-00565-8
69. Fernández Fernández E, Benavent Núñez D, Bonilla Hernán G, Monjo Henry I, García Carazo S, Bernad Pineda M, et al. Multiple vertebral fractures following discontinuation of denosumab treatment: ten clinical cases report. Reumatol Clin (Engl Ed). 2020;16(6):480–4. 10.1016/j.reumae.2018.11.021
70. Kurland ES, Heller SL, Diamond B, McMahon DJ, Cosman F, Bilezikian JP. The importance of bisphosphonate therapy in maintaining bone mass in men after therapy with teriparatide [human parathyroid hormone(1–34)]. Osteoporosis International 2004 15:12. 2004 Jun 3;15(12):992–7.
71. Kaufman JM, Orwoll E, Goemaere S, San Martin J, Hossain A, Dalsky GP, et al. Teriparatide effects on vertebral fractures and bone mineral density in men with osteoporosis: treatment and discontinuation of therapy. Osteoporos Int. 2005 May;16(5):510–6. 10.1007/s00198-004-1713-3