Acquired haemophilia A in southern Switzerland from 2013 to 2019: a case series
DOI: https://doi.org/https://doi.org/10.57187/smw.2023.40048
Andrea
Rubertiab, Johanna A
Kremer Hovingac, Federico
Nappia, Aurora
Vettesed, Elena
Bianchie, Eliana
Fernandese, Elena
Galfettie, Rita
Monottia, Pamella
Paule, Stefano
Regazzonif, Daniela
Valenteg, Davide
Rossiehi, Georg
Stussiei, Bernhard
Gerberdej
aDepartment
of Internal Medicine, Ospedale La Carità, Locarno, Switzerland
bDepartment
of Cardiology, Inselspital, Bern University Hospital, Bern, Switzerland
cDepartment of Haematology and
Central Haematology Laboratory, Inselspital, Bern University Hospital,
University of Bern, Switzerland
dDepartment
of Laboratory Medicine EOLAB, Bellinzona, Switzerland
eClinic
of Haematology, Oncology Institute of Southern Switzerland, Bellinzona,
Switzerland
fDepartment of Internal Medicine, Ospedale Civico,
Lugano, Switzerland
gPrivate practice, Chiasso, Switzerland
hLaboratory of Experimental Haematology, Institute of Oncology Research,
Bellinzona, Switzerland
iFaculty of Biomedical Sciences, Università della Svizzera Italiana, Lugano,
Switzerland
jUniversity of Zurich, Switzerland
Summary
AIMS OF THE
STUDY: Acquired haemophilia A is a rare disease with an annual incidence of
1.48 per million. Based on clinical observations, we suspect a higher incidence
in southern Switzerland, and aimed at providing local epidemiological data, and
clinical information regarding diagnosis, treatment and outcome in our region.
METHODS: All
adult patients with acquired haemophilia A treated between 2013 and 2019 in our
facility were included in the present retrospective analysis.
RESULTS: We
treated 11 patients with acquired haemophilia A between 2013 and 2019,
resulting in an annual incidence of 4.5 per million (95% confidence interval
[CI] 0–9.0). Median delay from first symptoms to diagnosis was 4.5 days, and
the median age at diagnosis was 79 years (range 23–87). Possible causative
conditions were: pregnancy (n = 1), polyarteritis nodosa (n = 1),
myelodysplastic syndrome (n = 1), chronic human immunodeficiency virus (HIV) (n
= 1), and HIV postexposure prophylaxis (n = 1). In five patients no underlying
or associated condition was identified. Median activated partial thromboplastin
time (aPTT)) at baseline was 79 seconds (65–117; ref. value <38 sec), and
FVIII:C 2.15% (<1–3.75%). A FVIII:C <1% was present in 4/10 patients.
Median FVIII-inhibitor titre was 10.3 BU/ml (2.4–75.0 BU/ml). All patients had
bleeding symptoms, 5/10 patients had major bleedings, and 7/10 patients were
treated with bypassing agents. All patients received corticosteroids; 7/10
patients received immunosuppressive combination therapy. FVIII levels of ≥50%
were achieved after a median of 40 days (8–62). One patient had a severe immunosuppressive
therapy-related
infection. An 87-years-old woman died for reasons not related to acquired haemophilia
A or immunosuppressive therapy.
CONCLUSIONS:
Acquired haemophilia A is a rare disease, but manageable despite the advanced
patient age and comorbidities. Its incidence in Southern Switzerland is higher
than previously suspected.
Introduction
Acquired haemophilia
A is a rare autoimmune disorder with a reported annual incidence of 1.48
patients per 1 million. Mortality of acquired haemophilia
A can be as high as
33%, with deaths directly related to bleeds ranging from 3%–9% [1–4]. Infections related to immunosuppressive
therapy (IST) and underlying conditions make up for the rest of the mortality [1, 2, 4–6]. There are two peaks in onset of acquired haemophilia A, one in younger women aged 20–40 years, mostly related to pregnancy, and a
second associated with age (>65 years) [7–9].
The reasons for antibody development against coagulation factor VIII (FVIII)
are still largely unknown [10, 11]. There
is an association with certain HLA class II alleles, with single nucleotide polymorphisms of the
CTLA-4 gene, as well as with mutations in the FVIII gene [12, 13].Neutralising anti-factor VIII (anti-FVIII) antibodies in
patients with acquired haemophilia A are
primarily directed to the A2, C1 and C2 domains
[14].
Anti-factor VIII antibodies of the IgA subtype are associated with a higher rate
of recurrence [15]. About half of the
patients have no identifiable trigger (“idiopathic acquired
haemophilia A”), whereas
the other half of the patients have underlying or associated neoplasia, autoimmune
diseases or a history of exposure to certain drugs at the time of haemophilia diagnosis [1, 2, 4, 16–20]. The bleeding pattern in
patients with acquired haemophilia A is different from that in patients
with severe congenital haemophilia A. Joint bleedings are rare, but large skin
haematomas, gastrointestinal and urogenital as well as muscle and
retroperitoneal bleedings are common (figure 1)
[4, 21]. Overall, the bleeding phenotype seems to be more severe in acquired than
in congenital haemophilia A, and is possibly related to age, concomitant
medication and the specificity of the anti-FVIII antibody [22].
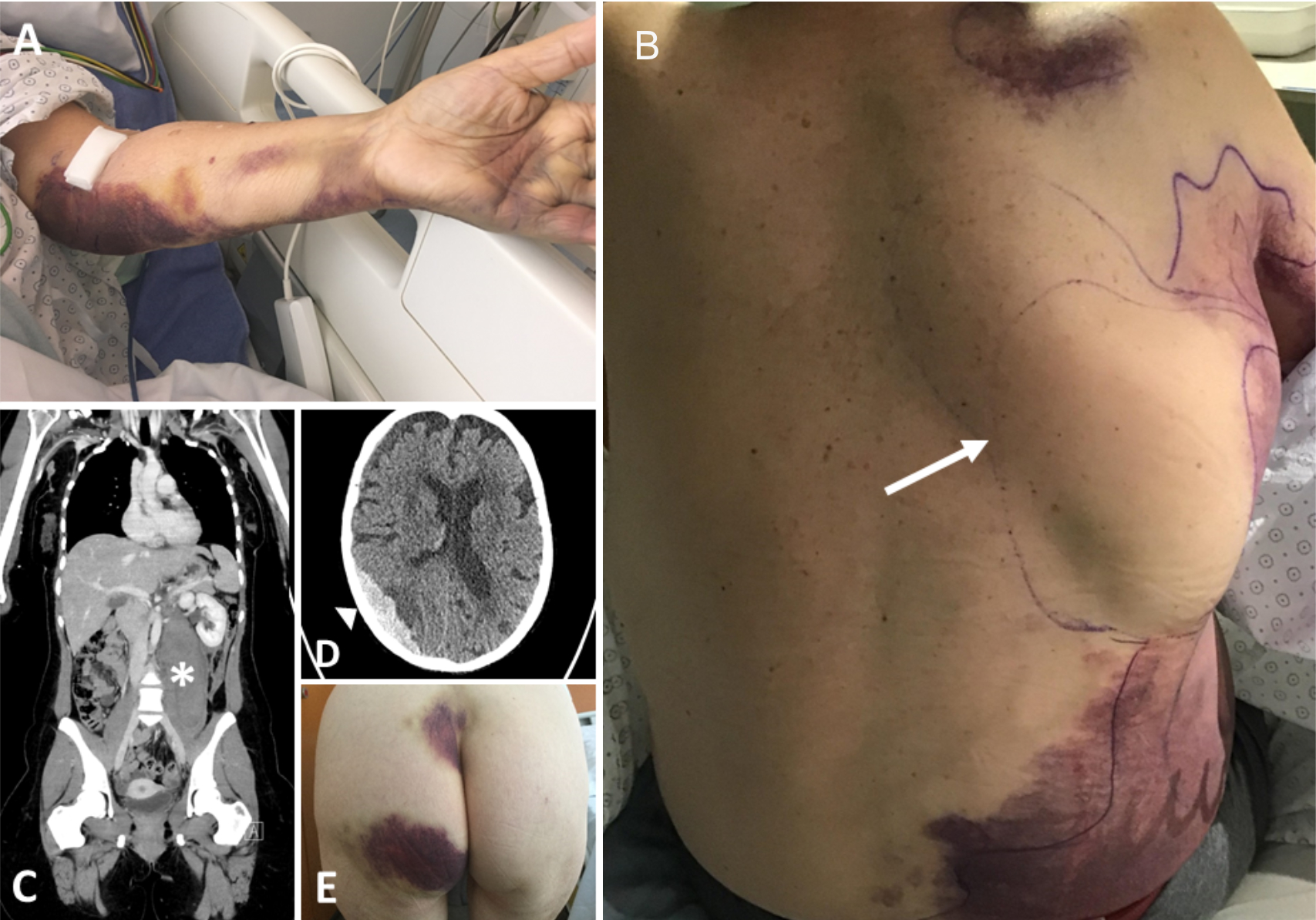
Figure 1 Bleeding pattern in patients with acquired haemophilia
A; (A) subcutaneous bleeding after venipuncture (patient 9). (B) large
spontaneous bruises and bleeding into the latissimus dorsi muscle (arrow;
patient 8). (C) spontaneous retroperitoneal bleeding in the psoas muscle
(asterisk; patient 3); (D) subdural haematoma following frequent falls while on
direct oral anticoagulant therapy (arrowhead; patient 4); (E) large skin haematomas
on the backside under low-dose heparin therapy (patient 5).
Once acquired haemophilia A is suspected, the diagnostic process is straightforward [4]. The prothrombin time (Quick/INR) is normal
and patients have a markedly prolonged activated partial thromboplastin time
(aPTT). In this situation aPTT mixing studies, which can be done in
non-specialised routine laboratories, are an important first diagnostic step (fig.
2) [23–26]. Further analyses include
measurement of FVIII coagulation activity (FVIII:C) and confirmation of the
FVIII inhibitor in specialised laboratories [8].
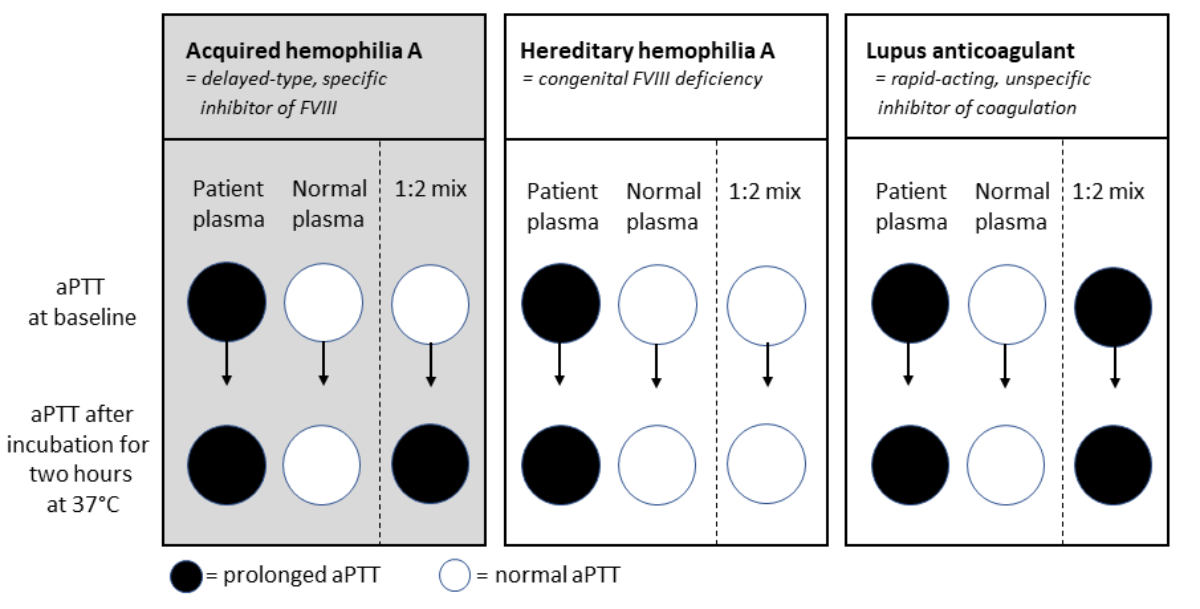
Figure 2 Typical
examples of activated
partial thromboplastin time (aPTT)
mixing studies for patients with normal prothrombin time (Quick/INR) and
prolonged aPTT at baseline. Patients with acquired haemophilia A have an
increased bleeding risk, and a prolonged aPTT at baseline, which normalizes
when adding normal plasma (1:2 mix), but increases after 2 hour incubation at
37 °C due to a delayed-type inhibitor; Patients with hereditary haemophilia A
have an increased bleeding risk, and a prolonged aPTT at baseline, which
normalizes when adding normal plasma (1:2 mix), and remains normal after 2 hour
incubation at 37 °C; Patients with positive lupus anticoagulant can have an
increased risk for thrombosis, and have a prolonged aPTT, which does not
normalize when adding normal plasma (1:2 mix), and stays prolonged after 2 hour
incubation at 37 °C due to a rapid-acting inhibitor.
Up to one
third of the patients will have spontaneous remission of acquired haemophilia A, but given the devastating consequences of severe haemorrhage, most experts
agree that immediate treatment is necessary for all patients [8, 27]. There are three major pillars of acquired haemophilia A treatment. First, unnecessary interventions must be avoided, including
venous punctures, frequent blood pressure measurements, endoscopy or surgery.
Second, haemorrhage should be controlled using one of the two currently
available FVIII bypassing agents, recombinant activated factor VII (rFVIIa;
NovoSeven®) and activated prothrombin complex concentrate (aPCC; FEIBA®), or recombinant porcine FVIII susoctocog alfa
(rpFVIII; Obizur®) [8].Due to
inactivation by circulating anti-FVIII antibodies, conventional FVIII
concentrates are generally not effective and should be avoided. Third,
immunosuppressive treatment should be started immediately to halt autoantibody
production. The backbone of immunosuppressive treatment are corticosteroids,
although timely addition of the anti-CD20 antibody rituximab is now
recommended, especially in the case of low FVIII activity (<1%), high
inhibitor titres (>20 Bethesda units (BU)/ml)
or corticosteroid intolerance [8].
Cyclophosphamide can be added as a corticosteroid-sparing agent, or in case of
treatment failure [5].
We report incidence, clinical
presentation and outcome of acquired haemophilia A diagnosed in Southern Switzerland between 2013 and
2019.
Patients and methods
Adult
patients diagnosed with acquired haemophilia A at the Ente Ospedaliero Cantonale
(EOC) between 2013 and 2019 were included. The end of follow-up was 31 March
2020. Clinical data were extracted retrospectively from the electronic patient
chart. Bleeding was graded according to the current International Society on Thrombosis
and Hemostasis (ISTH) criteria [28].
Hospital and
laboratory setting
Ente
Ospedaliero Cantonale is the only public healthcare provider in the Italian
speaking part of Southern Switzerland (Ticino), a region with 353,500
inhabitants. It is a tertiary care referral centre with 903 beds and 370,000
outpatient visits per year and consists of five small- to medium-sized
acute-care hospitals, two chronic-care facilities and one rehabilitation centre.
The five acute care facilities are geographically separated, and each has its
own laboratory facility, offering basic coagulation assays including
prothrombin time, activated partial thromboplastin time, fibrinogen and d-dimer
measurement. Specialised coagulation testing is centralised in two main
laboratory facilities.
Coagulation
assays
The coagulation tests are performed using
citrated plasma (0.109 M/3.2% Vacutainer tubes from Becton Dickinson). Prothrombin time (ref. value Quick 70–130%,
ReadiPlasTin®, HemosIL), aPTT (ref. value 25–37 seconds; SynthASil®, HemosIL),
coagulation factor VIII (ref. value 50–150%, SynthASil®, HemosIL), and anti‐Xa
activity (Liquid Anti‐Xa®, HemosIL) are determined on the ACL TOP 350 and ACL
TOP 550 coagulometers (Axonlab). Normal
pooled plasma is used for aPTT mixing studies. The mixing studies are performed by adding to the
patient’s plasma an equal volume of NPP (1:2 mix). aPTT in patient plasma, in normal pooled plasma and in the 1:2
mix is measured immediately and after a two-hour incubation at 37 °C in a water
bath (fig. 2). The Rosner index at 2 hours (RI = [aPTT mix – aPTT NPP) / aPTT
patient × 100]) is
calculated automatically. For patients with an RI ≥11% (inhibitor possible) a
senior haematologist is informed by the laboratory technician, whereas in
patients with an RI <11% (inhibitor unlikely) only a consultation with a haematologist
is recommended. The presence of a FVIII inhibitor is assessed in a modified
Nijmegen assay on an Atellica COAG 360 (Siemens, Germany).
All
patients signed a written informed consent for publication, and the competent
local ethical committee waived the need for formal review board approval of the
study (Req-2019-00726).
Statistical
analysis
Statistical
analyses were performed with Microsoft Excel Software or R statistics (version
4.1.3). Baseline characteristics are displayed as median with range and
interquartile range (IQR) or absolute numbers with percentages, as appropriate.
For the incidence calculations, the cases of acquired haemophilia
A were used as
numerators and the total population of the Ticino as denominator. The
population data was by the Swiss Federal Statistical Office. The mean annual
incidence rate and the 95% confidence interval [CI]was calculated over the
whole 7-year observation period.
Results
Incidence and underlying diseases
Eleven cases of acquired haemophilia A were
diagnosed between 2013 and 2019, resulting in an annual incidence of 4.5 cases
per 1 million inhabitants (95% CI 0–9.0). One patient did not give consent to participate,
and 10 patients were included in the final analysis.
Median age at diagnosis was 79 years (range
23–87), 7/10 patients were female (table 1). One patient had acquired haemophilia
A related to pregnancy; a 23-year-old female healthcare worker developed acquired
haemophilia A after HIV postexposure prophylaxis, one patient had polyarteritis
nodosa, one patient hat myelodysplastic syndrome, and one patient chronic HIV
infection with antiretroviral treatment. No associated or underlying condition
was identified in the other five patients.
Table 1Patient
baseline characteristics.
|
Patient-ID
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
Median (IQR)
|
| Gender |
f |
m |
f |
f |
f |
f |
f |
m |
f |
m |
|
| Age (years) |
41 |
68 |
23 |
87 |
77 |
83 |
82 |
71 |
84 |
87 |
79 (69–84) |
| Year of diagnosis |
2013 |
2016 |
2017 |
2017 |
2017 |
2017 |
2018 |
2019 |
2019 |
2019 |
|
| Comorbidities |
– |
Ischaemic cardiopathy, DM, aHT |
HIV-PEP |
MDS |
Ischaemic cardiopathy |
aHT |
Ischaemic cardiopathy |
Polyarteritis nodosa |
DM, aHT |
Ischaemic cardiopathy, aortic valve stenosis |
|
| Anti-aggregation / anticoagulation |
– |
ASS |
– |
DOAC |
Heparin |
– |
ASS |
VKA |
– |
ASS |
|
| AHA trigger |
Pregnancy |
HIV (on ART) |
Idiopathic (PEP) |
MDS |
Idiopathic |
Idiopathic |
Idiopathic |
PAN |
Idiopathic |
Idiopathic |
|
| Diagnostic delay (days) |
5 |
22 |
0 |
4 |
1 |
89 |
9 |
4 |
0 |
14 |
4.5 (2–13) |
| Type of bleeding |
Obstetrical |
Cutaneous |
Cutaneous Muscular |
CutaneousCNS |
Cutaneous |
Muco-cutaneous |
Cutaneous |
Cutaneous Muscular |
Cutaneous |
Cutaneous |
|
| Cause of bleeding |
Iatrogenic |
Minor Trauma |
Major Trauma |
Minor Trauma |
Spontaneous |
Spontaneous |
Spontaneous |
Spontaneous |
Minor Trauma |
Spontaneous |
|
| ISTH bleeding grade |
Major |
Minor |
Major |
Major |
Minor |
Minor |
Minor |
Major |
Minor |
Major |
|
Coagulation tests
Median aPTT at baseline was 79 seconds (IQR
65–117); Quick/INR was abnormal in one patient receiving vitamin K antagonists
(table 2). Median Rosner index at 2 hours was 30 (IQR 26.6–41.0), median
FVIII:C 2.15% (IQR <1–3.75%). A FVIII:C <1% was present in 4/10 patients.
Median FVIII inhibitor was 10.3 BU/ml (IQR 2.4–75.0 BU/ml), and 4/10 patients
had a titre >20 BU/ml.
Table 2Coagulation tests at diagnosis and during follow-up, coagulation factor
consumption and transfusion of blood products.
|
Patient-ID
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
Median (IQR)
|
|
Baseline
|
| aPTT (sec) |
64 |
118 |
49 |
112 |
44 |
76 |
67 |
200 |
200 |
82 |
79 (65–117)
|
| aPTT mix 1:2 (sec) |
34 |
45 |
31 |
51 |
37 |
37 |
37 |
121 |
47 |
41 |
39 (37–47)
|
|
After 2 h at 37 °C
|
| aPTT (sec) |
74 |
108 |
68 |
118 |
68 |
82 |
84 |
172 |
200 |
89 |
87 (76–116)
|
| aPTT mix 1:2 (sec) |
55 |
75 |
47 |
95 |
45 |
51 |
54 |
201 |
85 |
60 |
58 (52–83)
|
| Rosner index (2 h) |
41.9 |
38.9 |
23.5 |
46.6 |
16.2 |
26.8 |
28.6 |
98.3 |
26.5 |
31.5 |
30 (26.6–41)
|
| FVIII:C (%) |
2.3 |
<1 |
10 |
<1 |
11 |
4 |
3 |
<1 |
<1 |
2 |
2.15(<1–3.75)
|
| FVIII-Inhibitor (BU) |
1.8 |
103 |
0.8 |
53 |
1.6 |
8.4 |
2.4 |
330 |
75 |
10.3 |
10.3 (2.4–75)
|
| rFVIIa (mg/kg) |
3.9 |
0.3 |
0.1 |
0 |
0 |
0 |
0 |
2.5 |
0.1 |
0.2 |
0.1 (0–0.3)
|
| aPCC (IU/kg) |
0 |
1611 |
744 |
1750 |
0 |
0 |
0 |
1703 |
665 |
53 |
359 (0–1395)
|
| FFP (U) |
6 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0
|
| RBC (U) |
9 |
2 |
4 |
20 |
2 |
0 |
0 |
9 |
1 |
2 |
2 (1–8)
|
| Platelet transfusion (U) |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
0
|
| Immunosuppression |
P / C |
P / C |
P / R / C |
P / R |
P |
P |
P |
P / R / C / IA |
P / R |
P / R |
|
| Prednisone (days) |
151 |
157 |
907 |
64 |
74 |
132 |
203 |
ongoing |
108 |
ongoing |
|
|
Time to FVIII:C
|
| ≥5% (days) |
1 |
31 |
NA
|
28 |
NA
|
5 |
2 |
11 |
7 |
2 |
6 (2–15)
|
| ≥50% (days) |
39 |
52 |
51 |
46 |
8 |
41 |
14 |
62 |
24 |
18 |
40 (20–50)
|
Bleeding manifestations and treatment of bleeds
In half of the patients, the first haemorrhagic
manifestation was a major bleeding (gynaecological, muscular, intracranial,
cutaneous); two out of these five events were unprovoked (table 1). The
remaining five patients had minor, mostly unprovoked mucocutaneous bleedings.
FVIII bypassing agents were given in 7/10 patients (table 2), and most patients
received first rFVIIa, and switched to prothrombin
complex concentrate (aPCC) during the course of the
disease. No patient was treated with recombinant porcine FVIII (rpFVIII). Red
blood cell transfusions were necessary in 8/10 patients.
Immunosuppressive therapy
All patients were treated with
corticosteroids, seven of them received in addition rituximab and/or
cyclophosphamide (table 2). One patient was treated with immunoadsorption of
the FVIII inhibitor.
Disease course and side effects
FVIII:C levels ≥50% were achieved after a
median of 40 days (range 8–62 days) (table 2). One patient had asymptomatic
disease recurrence one month after corticosteroid suspension. Four patients had
side effects of corticosteroid treatment (arterial hypertension, hyperglycaemia,
insomnia, agitation, and myopathy) and one patient had erysipelas during
rituximab-induced neutropenia. One 87-year-old female patient (tables 1 and 2,
patient 4) died 17 months after the diagnosis of acquired haemophilia A of
heart failure, cause of death was unrelated to the haemophilia or immunosuppressive
therapy.
Discussion
The annual incidence of acquired haemophilia
A in Ticino between 2013 and 2019 was 4.5 cases per million inhabitants. Although
the low absolute number of in our study leads to uncertainty regarding the
incidence estimates, acquired haemophilia A seems to be more frequent in
Southern Switzerland than in the UK (1.48 cases per million per year), and more
in line with recent data reported by German colleagues (5–6 cases per million
per year) [1, 29]. Our data are likely
incomplete, thus underestimating the true incidence, as only patients with acquired
haemophilia A referred to our public hospital could be included. In 2018, we
implemented an acquired haemophilia A screening algorithm for all patients with
a prolonged aPTT, and a normal prothrombin time on a first occasion.
With a median age of 79 years in Southern
Switzerland, and 74–78 years in large registries, acquired haemophilia A is
clearly a disease of the elderly [1, 4].
The late onset of disease is challenging, as many patients suffer from
comorbidities at the time of haemophilia diagnosis [30, 31]. Anti-platelet therapy or anticoagulation are prevalent
in this age group and expose the patients to an additional bleeding risk at the
onset of acquired haemophilia A. Conversely, the discontinuation of
anti-platelet therapy or anticoagulation together with the use of haemostatic
bypassing agents exposes the patients to an elevated thrombotic risk. Diabetes
mellitus is a concern, as corticosteroids will lead to difficult-to-control
blood glucose levels. In our cohort of ten patients, four had coronary heart
disease and two diabetes mellitus.
We identified possible triggers for FVIII inhibitor development in 5/10 patients. However, with the exception of the one
patient with acquired haemophilia A in the peripartum period, the other four
conditions could be causative as well as coincidental.
The median diagnostic delay from first
symptoms prior to admission to diagnosis was 4.5 days, which is similar to the
three days reported in larger studies [4].
Most common presentations at diagnosis were large skin haematomas with or
without trauma, and a markedly prolonged activated partial thromboplastin time
of more than twice the upper limit of normal, with a typical delayed-type
inhibitor in the aPTT mixing studies. All of our patients had bleedings, but no
patient died due to haemorrhagic complications, which is in line with the
bleeding mortality of 3% observed in the EACH registry [4]. The majority of our patients needed treatment with FVIII
bypassing agents. The treatments of choice in our institution are rFVIIa and
aPCC (prothrombin complex concentrate) [1, 32, 33].
We did not use the recombinant
porcine FVIII (rpFVIII), susoctocog alfa or the
bispecific antibody emicizumab in our patients, as both drugs were not yet
reimbursed for this indication in Switzerland. Susoctocog alfa is a therapeutic
alternative, but cross-reacting antibodies may reduce FVIII recovery, and antibody
detection and frequent FVIII measurements with a suitable laboratory test are
mandatory [8, 34, 35]. Emicizumab, a
recombinant, humanised bispecific antibody with FVIII-like activity has been
used in a few cases of acquired haemophilia A so far, but no high-quality data
in acquired haemophilia A is available [36, 37].
All of our patients received immunosuppressive
treatment. Reasons for initiating immunosuppressive
combination therapy were low FVIII:C, high inhibitor
titre, major bleeding, and corticosteroid-related side effects. Initially, and
in accordance with the GTH-AH 2010/01 study protocol cyclophosphamide was our
preferred second-line immunosuppressive therapy [38]. In 2019, we
changed our policy by adding rituximab early in the treatment process, if a
poor response to corticosteroids was predicted (FVIII:C <1%, and/or
inhibitor titre >20 BU/ml), or if tolerability of corticosteroid therapy was
a concern [8]. Only one of our patients
experienced a severe infectious complication, and no patient died because of immunosuppressive
therapy. FVIII:C restored to values >50% within a
median of 40 days, which is in line with previous reports using similar immunosuppressive
therapy approaches, but shorter than reduced-intensity
schemes [38, 39]. Our relatively small
patient cohort does not allow for statistical subgroup analysis, but
individuals with low initial FVIII activity (<1%), and a high FVIII
inhibitor titre, had delayed FVIII normalisation and higher consumption of bypassing
agent, than patients with an initial FVIII:C ≥1%. All patients
achieved complete remission (normal FVIII activity, clearance of the FVIII
inhibitor), and only one patient experienced an asymptomatic relapse.
Our present report has limitations, mainly
the relatively short observation period of seven years, which might be subject
to a sampling bias, the low patient number, and the retrospective data
collection, that does not allow for more detailed statistical analysis.
Nevertheless, our data are representative for the disease and in line with data
obtained from large registries (table S1) [1, 4,
5, 38, 40]. Also, the true incidence of acquired haemophilia A in Ticino
is very likely underestimated, as our analysis includes only the patients
treated in the public hospitals.
In conclusion, we report an annual
incidence of acquired haemophilia A of 4.5 cases per million inhabitants in our
region. The outcome of acquired haemophilia A was favourable despite the
advanced age of the patients.
Parts
of this work have been presented at the Annual Meeting of the Society of
Thrombosis and Hemostasis Research (GTH) 2020, Bremen, Germany.
Acknowledgments
We would like to thank Maurizio
Petrilli, Tiziana Conte, and Damiana Carluccio for their technical assistance,
as well as Dr. Franco Keller and all technicians of the Department of
Laboratory medicine (EOLAB) for their ongoing commitment for early diagnosis of
acquired haemophilia A.
Authorship
contributions: AR and BG designed the study; BG, AV,
AR and FN collected and analysed data, BG and AR wrote the first draft of the
manuscript; AR and BG performed statistical analyses; all authors critically
read, discussed and corrected the manuscript.
Bernhard Gerber, MD
Clinic of Haematology
Oncology Institute of Southern Switzerland
CH-6500 Bellinzona
bernhard.gerber[at]eoc.ch
References
1.
Collins PW
,
Hirsch S
,
Baglin TP
,
Dolan G
,
Hanley J
,
Makris M
, et al.; UK Haemophilia Centre Doctors’ Organisation
. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007 Mar;109(5):1870–7. https://doi.org/10.1182/blood-2006-06-029850
2.
Borg JY
,
Guillet B
,
Le Cam-Duchez V
,
Goudemand J
,
Lévesque H
,
Group SS
; SACHA Study Group
. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia. 2013 Jul;19(4):564–70. https://doi.org/10.1111/hae.12138
3.
Green D
,
Lechner K
. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb Haemost. 1981 Jun;45(3):200–3. https://doi.org/10.1055/s-0038-1650169
4.
Knoebl P
,
Marco P
,
Baudo F
,
Collins P
,
Huth-Kühne A
,
Nemes L
, et al.; EACH2 Registry Contributors
. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012 Apr;10(4):622–31. https://doi.org/10.1111/j.1538-7836.2012.04654.x
5.
Tiede A
,
Klamroth R
,
Scharf RE
,
Trappe RU
,
Holstein K
,
Huth-Kühne A
, et al.
Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH-AH 01/2010 study. Blood. 2015 Feb;125(7):1091–7. https://doi.org/10.1182/blood-2014-07-587089
6.
Delgado J
,
Jimenez-Yuste V
,
Hernandez-Navarro F
,
Villar A
. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003 Apr;121(1):21–35. https://doi.org/10.1046/j.1365-2141.2003.04162.x
7.
Tengborn L
,
Baudo F
,
Huth-Kühne A
,
Knoebl P
,
Lévesque H
,
Marco P
, et al.; EACH2 registry contributors
. Pregnancy-associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG. 2012 Nov;119(12):1529–37. https://doi.org/10.1111/j.1471-0528.2012.03469.x
8.
Tiede A
,
Collins P
,
Knoebl P
,
Teitel J
,
Kessler C
,
Shima M
, et al.
International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020 Jul;105(7):1791–801. https://doi.org/10.3324/haematol.2019.230771
9.
Lapalud P
,
Ali T
,
Cayzac C
,
Mathieu-Dupas E
,
Levesque H
,
Pfeiffer C
, et al.
The IgG autoimmune response in postpartum acquired hemophilia A targets mainly the A1a1 domain of FVIII. J Thromb Haemost. 2012 Sep;10(9):1814–22. https://doi.org/10.1111/j.1538-7836.2012.04850.x
10.
Reding MT
. Immunological aspects of inhibitor development. Haemophilia. 2006 Dec;12(s6 Suppl 6):30–5. https://doi.org/10.1111/j.1365-2516.2006.01363.x
11.
Tiede A
,
Zieger B
,
Lisman T
. Acquired bleeding disorders. Haemophilia. 2021 Feb;27(S3 Suppl 3):5–13. https://doi.org/10.1111/hae.14033
12.
Oldenburg J
,
Zeitler H
,
Pavlova A
. Genetic markers in acquired haemophilia. Haemophilia. 2010 May;16 Suppl 3:41–5. https://doi.org/10.1111/j.1365-2516.2010.02259.x
13.
Tiede A
,
Eisert R
,
Czwalinna A
,
Miesbach W
,
Scharrer I
,
Ganser A
. Acquired haemophilia caused by non-haemophilic factor VIII gene variants. Ann Hematol. 2010 Jun;89(6):607–12. https://doi.org/10.1007/s00277-009-0887-3
14.
Kahle J
,
Orlowski A
,
Stichel D
,
Healey JF
,
Parker ET
,
Jacquemin M
, et al.
Frequency and epitope specificity of anti-factor VIII C1 domain antibodies in acquired and congenital hemophilia A. Blood. 2017 Aug;130(6):808–16. https://doi.org/10.1182/blood-2016-11-751347
15.
Tiede A
,
Hofbauer CJ
,
Werwitzke S
,
Knöbl P
,
Gottstein S
,
Scharf RE
, et al.
Anti-factor VIII IgA as a potential marker of poor prognosis in acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood. 2016 May;127(19):2289–97. https://doi.org/10.1182/blood-2015-09-672774
16.
Kessler CM
,
Ma AD
,
Al-Mondhiry HA
,
Gut RZ
,
Cooper DL
. Assessment of acquired hemophilia patient demographics in the United States: the Hemostasis and Thrombosis Research Society Registry. Blood Coagul Fibrinolysis. 2016 Oct;27(7):761–9. https://doi.org/10.1097/MBC.0000000000000582
17.
Hirsiger JR
,
Martinez M
,
Tsakiris DA
,
Cittone MG
,
Graf L
,
Oldenburg J
, et al.
Investigating potential mechanisms underlying FVIII inhibition in acquired hemophilia A associated with mRNA COVID-19 vaccines. J Thromb Haemost. 2022 Apr;20(4):1015–8. https://doi.org/10.1111/jth.15665
18.
Cittone MG
,
Battegay R
,
Condoluci A
,
Terzi di Bergamo L
,
Fernandes E
,
Galfetti E
, et al.
The statistical risk of diagnosing coincidental acquired hemophilia A following anti-SARS-CoV-2 vaccination. J Thromb Haemost. 2021 Sep;19(9):2360–2. https://doi.org/10.1111/jth.15421
19.
Sanges S
,
Jeanpierre E
,
Lopez B
,
Russick J
,
Delignat S
,
Carpentier B
, et al.
Acquired Hemophilia A in IgG4-Related Disease: Case Report, Immunopathogenic Study, and Review of the Literature. Front Immunol. 2020 Dec;11:558811. https://doi.org/10.3389/fimmu.2020.558811
20.
Mizrahi T
,
Doyon K
,
Dubé E
,
Bonnefoy A
,
Warner M
,
Cloutier S
, et al.
Relapse pattern and long-term outcomes in subjects with acquired haemophilia A. Haemophilia. 2019 Mar;25(2):252–7. https://doi.org/10.1111/hae.13685
21.
Tiede A
,
Giangrande P
,
Teitel J
,
Amano K
,
Benson G
,
Nemes L
, et al.
Clinical evaluation of bleeds and response to haemostatic treatment in patients with acquired haemophilia: A global expert consensus statement. Haemophilia. 2019 Nov;25(6):969–78. https://doi.org/10.1111/hae.13844
22.
Matsumoto T
,
Nogami K
,
Ogiwara K
,
Shima M
. A putative inhibitory mechanism in the tenase complex responsible for loss of coagulation function in acquired haemophilia A patients with anti-C2 autoantibodies. Thromb Haemost. 2012 Feb;107(2):288–301. https://doi.org/10.1160/TH11-05-0331
23.
Lossing TS
,
Kasper CK
,
Feinstein DI
. Detection of factor VIII inhibitors with the partial thromboplastin time. Blood. 1977 May;49(5):793–7. https://doi.org/10.1182/blood.V49.5.793.793
24.
Rosner E
,
Pauzner R
,
Lusky A
,
Modan M
,
Many A
. Detection and quantitative evaluation of lupus circulating anticoagulant activity. Thromb Haemost. 1987 Apr;57(2):144–7. https://doi.org/10.1055/s-0038-1651083
25.
Kumano O
,
Ieko M
,
Naito S
,
Yoshida M
,
Takahashi N
,
Suzuki T
, et al.
New formulas for mixing test to discriminate between lupus anticoagulant and acquired hemophilia A. Thromb Res. 2016 Jul;143:53–7. https://doi.org/10.1016/j.thromres.2016.05.004
26.
Rasmussen KL
,
Philips M
,
Tripodi A
,
Goetze JP
. Unexpected, isolated activated partial thromboplastin time prolongation: A practical mini-review. Eur J Haematol. 2020 Jun;104(6):519–25. https://doi.org/10.1111/ejh.13394
27.
Lottenberg R
,
Kentro TB
,
Kitchens CS
. Acquired hemophilia. A natural history study of 16 patients with factor VIII inhibitors receiving little or no therapy. Arch Intern Med. 1987 Jun;147(6):1077–81. https://doi.org/10.1001/archinte.1987.00370060073014
28.
Schulman S
,
Kearon C
; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis
. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005 Apr;3(4):692–4. https://doi.org/10.1111/j.1538-7836.2005.01204.x
29.
Tiede A
,
Wahler S
. The rising incidence of acquired haemophilia A in Germany. Haemophilia. 2020.
30.
Christensen LD
,
Reilev M
,
Juul-Larsen HG
,
Jørgensen LM
,
Kaae S
,
Andersen O
, et al.
Use of prescription drugs in the older adult population-a nationwide pharmacoepidemiological study. Eur J Clin Pharmacol. 2019 Aug;75(8):1125–33. https://doi.org/10.1007/s00228-019-02669-2
31.
Saeedi P
,
Petersohn I
,
Salpea P
,
Malanda B
,
Karuranga S
,
Unwin N
, et al.; IDF Diabetes Atlas Committee
. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019 Nov;157:107843. https://doi.org/10.1016/j.diabres.2019.107843
32.
Zanon E
,
Pasca S
,
Siragusa S
,
Napolitano M
,
Santoro C
,
Mameli L
, et al.; FAIR Study Group
. Low dose of aPCC after the initial treatment in acquired haemophilia A is useful to reduce bleeding relapses: data from the FAIR registry. Thromb Res. 2019 Feb;174:24–6. https://doi.org/10.1016/j.thromres.2018.12.006
33.
Baudo F
,
Collins P
,
Huth-Kühne A
,
Lévesque H
,
Marco P
,
Nemes L
, et al.; EACH2 registry contributors
. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012 Jul;120(1):39–46. https://doi.org/10.1182/blood-2012-02-408930
34.
Kruse-Jarres R
,
St-Louis J
,
Greist A
,
Shapiro A
,
Smith H
,
Chowdary P
, et al.
Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015 Mar;21(2):162–70. https://doi.org/10.1111/hae.12627
35.
Türkantoz H
,
Königs C
,
Knöbl P
,
Klamroth R
,
Holstein K
,
Huth-Kühne A
, et al.
Cross-reacting inhibitors against recombinant porcine factor VIII in acquired hemophilia A: data from the GTH-AH 01/2010 Study. J Thromb Haemost. 2020 Jan;18(1):36–43. https://doi.org/10.1111/jth.14618
36.
Tiede A
,
Kemkes-Matthes B
,
Knobl P
. Should Emicizumab Be Used in Patients with Acquired Haemophilia A? J Thromb Haemost. 2020.
37.
Fontana P
,
Alberio L
,
Albisetti M
,
Angelillo-Scherrer A
,
Asmis LM
,
Casini A
, et al.
Management of bleeding events and invasive procedures in patients with haemophilia A without inhibitors treated with emicizumab. Swiss Med Wkly. 2020 Dec;150(5153):w20422. https://doi.org/10.4414/smw.2020.20422
38.
Collins P
,
Baudo F
,
Knoebl P
,
Lévesque H
,
Nemes L
,
Pellegrini F
, et al.; EACH2 registry collaborators
. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012 Jul;120(1):47–55. https://doi.org/10.1182/blood-2012-02-409185
39.
Dobbelstein C
,
Moschovakis GL
,
Tiede A
. Reduced-intensity, risk factor-stratified immunosuppression for acquired hemophilia A: single-center observational study. Ann Hematol. 2020 Sep;99(9):2105–12. https://doi.org/10.1007/s00277-020-04150-y
40.
Holstein K
,
Liu X
,
Smith A
,
Knöbl P
,
Klamroth R
,
Geisen U
, et al.
Bleeding and response to hemostatic therapy in acquired hemophilia A: results from the GTH-AH 01/2010 study. Blood. 2020 Jul;136(3):279–87. https://doi.org/10.1182/blood.2019003639
41.
Collins PW
,
Hirsch S
,
Baglin TP
,
Dolan G
,
Hanley J
,
Makris M
, et al.; UK Haemophilia Centre Doctors’ Organisation
. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007 Mar;109(5):1870–7. https://doi.org/10.1182/blood-2006-06-029850
42.
Knoebl P
,
Marco P
,
Baudo F
,
Collins P
,
Huth-Kühne A
,
Nemes L
, et al.; EACH2 Registry Contributors
. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012 Apr;10(4):622–31. https://doi.org/10.1111/j.1538-7836.2012.04654.x
43.
Borg JY
,
Guillet B
,
Le Cam-Duchez V
,
Goudemand J
,
Lévesque H
,
Group SS
; SACHA Study Group
. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hémophilie Acquise) registry. Haemophilia. 2013 Jul;19(4):564–70. https://doi.org/10.1111/hae.12138
44.
Jayakar JP
,
O’Neill N
,
Yan M
,
Nisenbaum R
,
Garvey MB
,
Teitel J
, et al.
Retrospective review of Acquired Haemophilia A from the largest Canadian Haemophilia treatment centre. Haemophilia. 2018 Sep;24(5):e383–7. https://doi.org/10.1111/hae.13598
45.
Huang SY
,
Tsay W
,
Lin SY
,
Hsu SC
,
Hung MH
,
Shen MC
. A study of 65 patients with acquired hemophilia A in Taiwan. J Formos Med Assoc. 2015 Apr;114(4):321–7. https://doi.org/10.1016/j.jfma.2013.01.006
Appendix
Table S1Comparison of large collections of patients
with acquired haemophilia A.
|
Reference
|
Inclusion
period/region
|
Number of patients / study design
|
Incidence per year
|
Female sex
|
Median age, years
(range)
|
Days to diagnosis,
(range)
|
idiopathic
|
Median FVIII:C (%)
(IQR)
|
Median FVIII
-inhibitor, (BU/ml) (IQR)
|
Major bleeding
|
Hemostatic agents
needed
|
IST
|
CR / time to remission
(days)
|
Mortality
|
Relapse
|
| Collins et al. 2007 [41] |
2001–2003 UK |
172 / retrospective multicentre questionnaire |
1.48/mio |
57% |
78 (2–98) |
ND |
63% |
3 (1.7–7) |
13 (4–28) |
66% |
67% |
95% |
71%/57a
|
9.1% due to bleeding |
20% |
| Knoebl et al. (EACH2) [42] |
2003–2008 Europe |
501 / prospective multicentre registry |
ND |
47% |
74 (61–80) |
3 (0–12) |
52% |
2 (1–5) |
12.8 (4.3–42.4) |
70.3% |
70% |
95% |
72.6%/NDb
|
20% (in total), 3% due to bleeding |
ND |
| Borg et al. 2013 (SACHA) [43] |
2001–2005 France |
82 / prospective multicentre registry |
ND |
39% |
77 (25–103) |
ND |
55% |
2 (<1–30) |
16 (1–2800) |
35% |
46% |
88% |
61% /ND |
33% (in total), 3.5% due to bleeding, 12% treatment
related |
0% at 1 year |
| Jayakar et al. 2018 [44] |
1990–2016 Canada |
40 / retrospective single centre |
ND |
32% |
68 (ND) |
ND |
63% |
ND |
ND |
40% |
74% |
100% |
73%/ND |
4.5% due to bleeding |
ND |
| Huang et al. 2015 [45] |
1987–2010 Taiwan |
65 / retrospective two centres |
ND |
35% |
64 (18-94) |
ND |
52% |
65% (<1%
FVIII:C), 35% mean 3.8 (1.1–17)% |
19.4 (0.7–2414) |
14% |
ND |
86% |
60%/16 weeks |
29% due to bleeding, 20% treatment related |
12% |
| Ruberti et al. 2023 (this article) |
2013–2019 Switzerland |
10 / retrospective single centre |
4.5/mio |
70% |
79 (23–87) |
4.5 (2–13) |
50% |
2.15 (<1–3.75) |
10.3 (2.4–75) |
50% |
70% |
100% |
100%/40c
|
10% (in total), 0% due to bleeding |
10% |