
Figure 1 Network topology for different soft-thresholding powers. (A) Numbers in the plots indicate the corresponding soft-thresholding powers. (B) The approximate scale-free topology can be attained at a soft-thresholding power of 16.
DOI: https://doi.org/10.57187/smw.2022.40033
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the latest member of the Coronaviridae family, which caused a pandemic of coronavirus disease (COVID-19), endangering global health [1–5]. The genome of SARS-CoV-2 is most closely related to that of SARS-CoV, which emerged in 2003 [3, 6, 7]. Although numerous coronaviruses cause pneumonia, SARS-CoV-2 causes the most devastating form of pneumonia that has been noted globally to date. This virus mainly infects lower respiratory tract cells, leading to the development of SARS that could progress to pneumonia and death in severe cases [8]. SARS-CoV-2 consists of 20 different proteins containing four main structural proteins (S: spike; E: envelope; M: membrane; N: nucleocapsid), and several non-structural proteins such as RNA-dependent RNA polymerase, coronavirus main protease, and papain-like protease (PLpro) may serve as viable antiviral drug targets [9–11]. Although lopinavir and ritonavir have been used as treatment in preliminary clinical studies, there is still no consensus on the effectiveness of these drugs in treating patients with COVID-19 [12]. Therefore, gaining a better understanding of the molecular mechanism by which SARS-CoV-2 interacts with human proteins will play a critical role in identifying appropriate antiviral drug targets [13]. Towards this goal, we adopted a bioinformatics approach using publicly available clinical and genetic data. Weighted gene co-expression network analysis (WGCNA) is a common method for investigating gene–phenotype relationships, including in the context of diseases [14, 15]. The main advantage of WGCNA is that gene expression data are converted to a co-expression module, which is conducive for providing insights into phenotypic traits of interest [16, 17]. A comprehensive R package collection has been developed to interpret various aspects of WGCNA [18–20].
SARS-CoV-2 consists of 20 different proteins containing four main structural proteins (S: spike; E: envelope; M: membrane; N: nucleocapsid), and several non-structural proteins such as RNA-dependent RNA polymerase, coronavirus main protease, and papain-like protease (PLpro) may serve as viable antiviral drug targets [9–11]. Although lopinavir and ritonavir have been used as treatment in preliminary clinical studies, there is still no consensus on the effectiveness of these drugs in treating patients with COVID-19 [12]. Therefore, gaining a better understanding of the molecular mechanism by which SARS-CoV-2 interacts with human proteins will play a critical role in identifying appropriate antiviral drug targets [13]. Towards this goal, we adopted a bioinformatics approach using publicly available clinical and genetic data.
Weighted gene co-expression network analysis (WGCNA) is a common method for investigating gene–phenotype relationships, including in the context of diseases [14, 15]. The main advantage of WGCNA is that gene expression data are converted to a co-expression module, which is conducive for providing insights into phenotypic traits of interest [16, 17]. A comprehensive R package collection has been developed to interpret various aspects of WGCNA [18–20].
The increasing morbidity and mortality of COVID-19 cause adverse consequences. However, the performance of vaccine or drug treatment in combating infection with the causative SARS-CoV-2 is not satisfactory. To further understand SARS-CoV-2 infection-induced alterations of host gene expressions and pathways, we aimed to construct co-expression modules from publicly available data of patients with COVID-19. In particular, we screened the modules of co-expressed genes among differentially expressed genes (DEGs) between SARS-CoV-2-infected and non-infected (normal) samples. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were further performed to identify the principal functions of core genes in each module related to SARS-CoV-2 infection. The identified core genes and signalling pathways associated with SARS-CoV-2 infection are expected to significantly supplement the current understanding of COVID-19. The findings are also anticipated to serve as a basis for determining a new approach and creating a hypothetical model for future experimental studies regarding SARS-CoV-2 pathogenesis.
The expression profiles from the GSE152075 dataset, including the mRNA levels from nasopharyngeal swabs and clinical data from 430 and 54 patients positive and negative for SARS-CoV-2 infection, respectively, were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo) [21]. The baseline information of patients is presented in table 1. We excluded data from patients without available information on age or sex. Finally, data from 377 SARS-CoV-2-infected samples and 54 normal samples with available associated clinical information and mRNA level were used for further analysis. Normalisation, batch effect correction, and differential expression were performed using the R package "DEseq2".
Table 1The baseline characteristics data.
| Variables | SARS-CoV-2 positive | SARS-CoV-2 negative | |
| Sex | Male | 176 | 24 |
| Female | 201 | 30 | |
| Age (years) | ≤60 | 223 | 39 |
| >60 | 155 | 14 | |
The raw microarray data files between the SARS-CoV-2-infected and normal samples were subsequently analysed using the R and Bioconductor package "edgeR". We used a classical t test to identify the DEGs, with P values of <0.05 and fold change values of ≥2 being statistically significant. Aberrantly expressed genes between the SARS-CoV-2-infected and normal samples were selected for subsequent analyses using the WGCNA algorithm to calculate the gene expression levels [22].
The WGCNA algorithm was utilised to discern the power value in the construction of modules related to SARS-CoV-2 infection [22]. The independence and average connectivity degree of the power value were assessed using the gradient method. The applicable power value was determined as a degree of independence above 0.8. The WGCNA algorithm was then utilised to construct the modules after the power value was determined, and gene information was extracted from each module. The genes that were not differently expressed following the WGCNA were not used for further analysis because they revealed no or very low discrepancy in expression between the researched samples.
The WGCNA algorithm was applied to screen out the co-expression modules using the R software. The module-trait interactions were assessed on the basis of the relationship between the module eigengenes and phenotypes (clinical traits) to screen out gene expression sets strongly associated with the phenotype. Gene significance was estimated as the outright value of the relationship between the expression profile and each clinical trait; the module membership was identified on the basis of the relationship between the expression profile and each module eigengene [23, 24].
GO term enrichment and KEGG pathway analyses were implemented to explore a full-scale set of functionally annotated core genes. Thereby, GO functional and KEGG pathway enrichment analyses were utilised to assess the modules significantly associated with SARS-CoV-2 infection using the R packages ‘cluster Profiler’ and ‘ggplot2’ [25, 26]. GO annotation outcomes can generally be sorted into three main categories: molecular function, biological process, and cellular component [27, 28]. Following multiple test calibration, a false discovery rate of ≤0.05 was set as the threshold for significance [28]. The core genes in the modules were then screened out on the basis of the protein–protein interaction (PPI) network constructed using the Cytoscape software.
The GEO database is freely available to all investigators worldwide. Thus, supplementary approval by an ethics committee was not necessary for this study.
A total of 1444 DEGs were identified between the 430 SARS-CoV-2-infected samples and 54 normal samples, which were utilised to build the co-expression modules using the WGCNA package tools. One of the most vital factors in the WGCNA is the power value, which has a substantial impact on the independence and average connectivity degree of co-expression modules. Therefore, the power value was identified using the gradient method herein (figure 1).
Figure 1 Network topology for different soft-thresholding powers. (A) Numbers in the plots indicate the corresponding soft-thresholding powers. (B) The approximate scale-free topology can be attained at a soft-thresholding power of 16.
The power value of our module was calculated to be 16; the independence degree was >0.8; and the average connectivity degree was higher. Accordingly, the power value was used to construct the co-expression modules. Eleven co-expression modules related to SARS-CoV-2 positivity were screened out in the WGCNA; these modules are visualised in distinctive colours in figure 2.
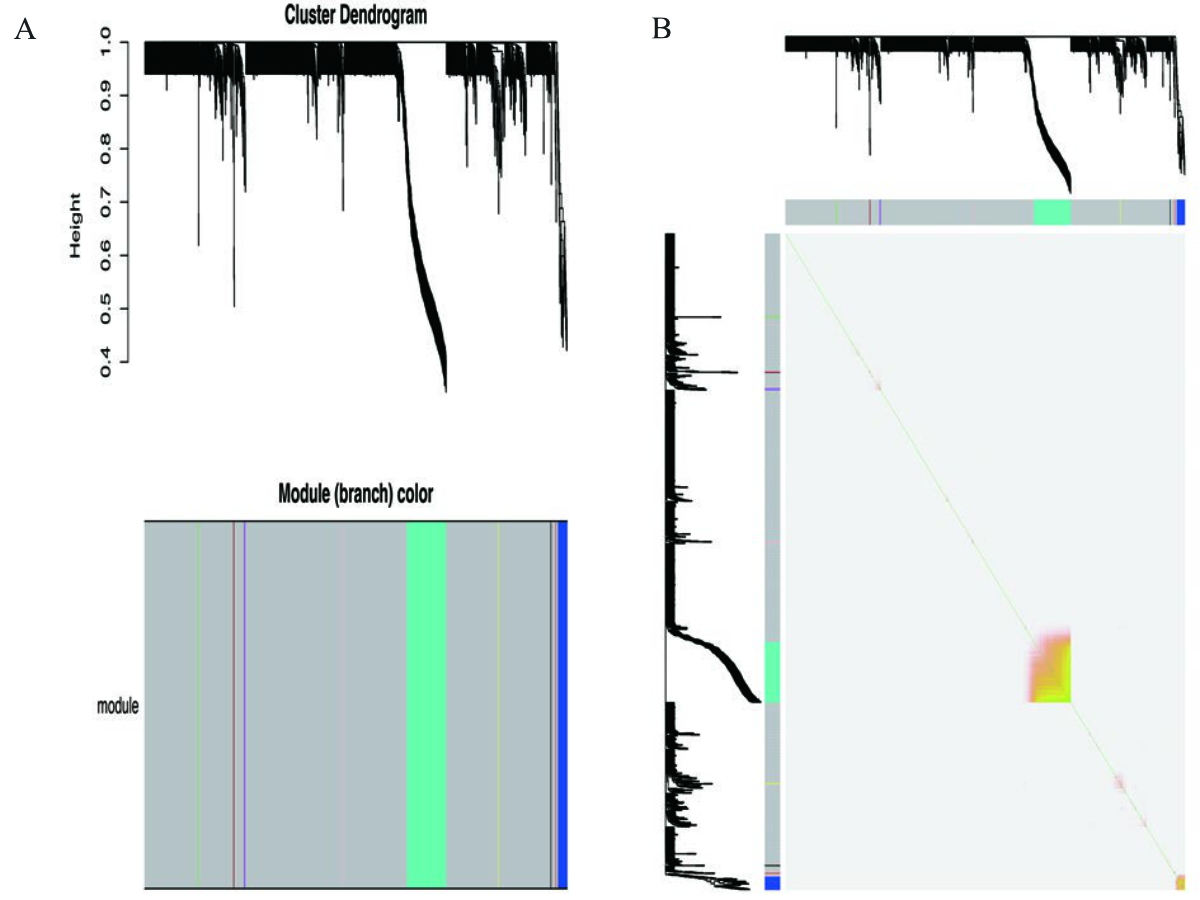
Figure 2 Gene modules identified in the WGCNA. (A) Gene dendrogram obtained by clustering the dissimilarity based on consensus Topological Overlap. The colour rows indicate the corresponding modules. (B) Visualisation of the gene network using a heatmap plot. The heatmap depicts the TOM DEGs in the yellow and grey modules. Abbreviations: WGCNA, weighted gene co-expression network analysis; DEG, differentially expressed gene; TOM, Topological Overlap Matrix.
The gene co-expression modules correlated with SARS-CoV-2 positivity, age, and sex were screened out to evaluate the module-trait associations (figure 3). The yellow and grey modules had the greatest correlation with SARS-CoV-2 positivity. Similarly, these two modules were greatly correlated with age. Therefore, the yellow and grey modules were selected for further analysis.

Figure 3 Relationships between gene co-expression modules and different clinical data.
The DEGs in the yellow and grey modules associated with SARS-COV-2 positivity were subjected to GO enrichment analysis. The top three enrichment results of the GO biological process terms were viral gene expression, viral transcription, and protein targeting the membrane. The top three enrichment results of the GO cellular component terms were mitochondrial inner membrane, ribosome, and cytosol, while those of the GO molecular function terms were structural constituent of the ribosome, electron transfer activity, and oxidoreductase activity, acting on NAD(P)H. The overall top five enrichment results of the GO analysis for the DEGs in the yellow and grey modules are shown in figure 4.

Figure 4 Top five enrichment results of the GO analysis of the DEGs in the yellow and grey modules. Abbreviations: GO, gene ontology; DEG, differentially expressed gene.
The most significantly enriched pathways of the DEGs in the yellow and grey modules are shown in figure 5. The KEGG analysis revealed that the upregulated DEGs in the two modules were commonly enriched in oxidative phosphorylation, ribosome, and thermogenesis pathways.
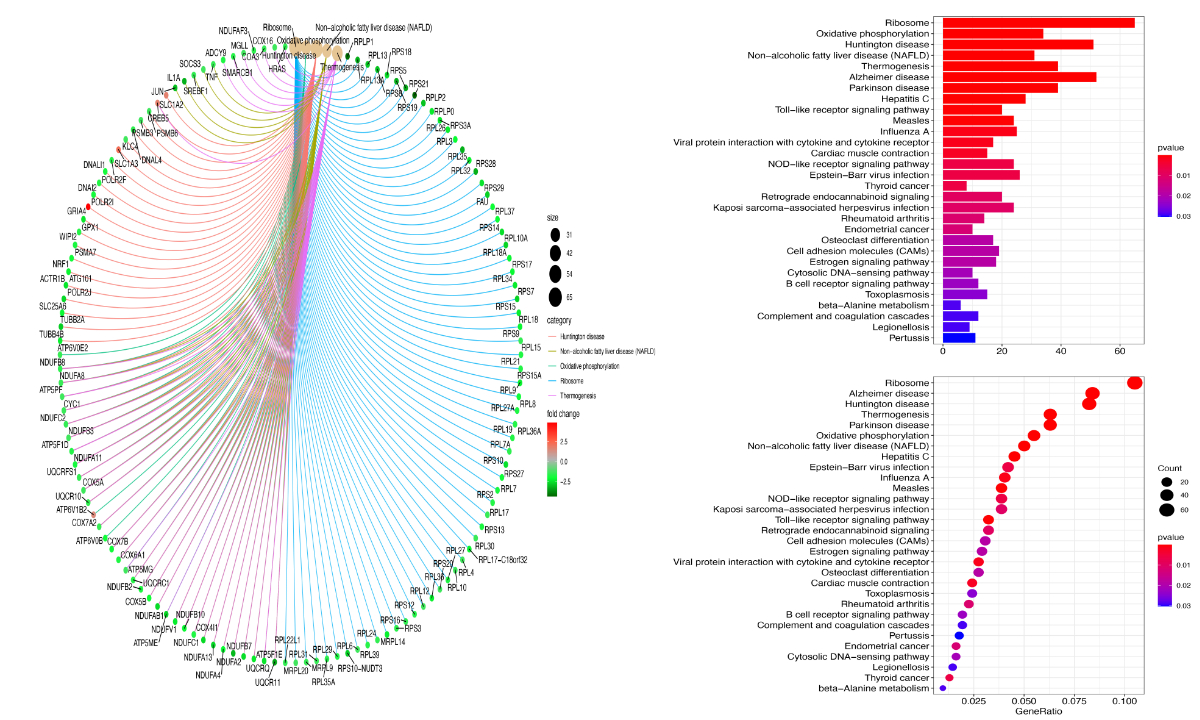
Figure 5 Top five enrichment results of the canonical pathway analysis of the DEGs in the yellow and grey modules. Abbreviations: DEG, differentially expressed gene.
The PPI network is shown in figure 6. The intramodular connectivity was analysed for all DEGs in the yellow and grey SARS-CoV-2 infection modules. Among the DEGs, the core genes in these two modules showed a high intramodular connectivity. The five core host response genes were RPL35A, RPL7A, RPS15, RPS20, and RPL17, which all encode ribosomal proteins.

Figure 6 Protein–protein interaction network of the DEGs in the yellow and grey modules. Abbreviations: DEG, differentially expressed gene.
Given the rapid spread of SARS-CoV-2 infection, jeopardising global health with no established effective treatment, it is indispensable to identify the molecular mechanisms associated with this infection and determine appropriate treatment and prevention methods. To this end, we employed WGCNA and other systems biology methods to explore the expression profiles from the GSE152075 dataset, including SARS-CoV-2-infected and normal samples. The WGCNA identified several modules, two of which (yellow and grey modules) were significantly correlated with SARS-CoV-2 positivity. Since SARS-CoV-2 consists of PLpro, RNA polymerase, 3CL protease, helicase protein, membrane protein, spike protein, envelope protein, and nucleocapsid protein, we identified RPL35A, RPL7A, RPS15, RPS20, and RPL17 as the five core genes with top connectivity with other genes in these modules. This finding indicates that the transcription of these genes encoding ribosomal proteins may be indispensable in viral protein biosynthesis after SARS-CoV-2 infection.
Ribosomal proteins and viral mRNA are well known to be involved in viral protein biosynthesis [29, 30]. Reduction in the transcription of ribosomal proteins has been associated with a robust antiviral response to SARS-CoV-2 infection [31]. Ribosomal proteins associate with rRNA to regulate the cellular translation cycle and have been shown to be involved in two key antiviral mechanisms. First, these proteins can serve as immune elements to trigger signalling pathways for antiviral defence. For instance, RPS20 has been shown to inhibit viral reproduction in cells by regulating Toll-like receptor 3 (TLR3), which can trigger the immune response [32, 33]. Totura and colleagues further disclosed that TLR3 signalling via TIR domain-containing adapter-inducing interferon-beta serves as a defensive innate immune response to SARS-CoV infection [33]. In the present study, we found that RPS20 expression was significantly downregulated in the patients positive for SARS-CoV-2 infection. This indicates that a reduction in RPS20 levels may regulate TLR3 expression to suppress the immune response after SARS-CoV-2 infection. Second, ribosomal proteins directly interact with viral proteins to inhibit the transcription or translation of viral genes. For example, RPL9 binds to phosphoprotein P to inhibit the transcription of viral genes [29, 34, 35]. Similarly, RPS10 has been shown to integrate HIV-1 to Nef protein, thereby weakening the production of viral proteins [36]. Accordingly, these proteins can be utilised to inhibit viral copying by integrating definite phosphoproteins or can serve as activators for host immune elements. Although RPL9 and RPS10 were not identified as core genes in the SARS-CoV-2 infection modules in the present study, these genes showed relatively low expression levels in the SARS-CoV-2-infected samples compared with those in the normal samples (figure 6). Robledo and colleagues disclosed that RPS15 is indispensable to the late maturation step of the 40S subunit, the large subunit correlated with rRNA processing and ribosome production [37]. Human RPL7a contains eight exons and seven introns spread over 3179 bp [38]. Fumagilli and colleagues revealed that knock off RPL7a leads to a G1 block and a decreased percentage of cells in the S phase [39].
Downregulating RPL17 modifies the diversity of mature ribosomes by upregulating 5.8 S rRNA in mice [40]. RPL17 is a tunnel wall in eukaryotes and archaebacteria and regulates the outcome of a nascent polypeptide [41]. Consequently, these genes can be potential targets for developing vaccines or drugs targeting SARS-CoV-2 in further studies.
The GO analysis demonstrated viral infection strategies (viral translation) as activated biological processes in the yellow and grey SARS-CoV-2 infection modules. Li et al. [42] and Kuba et al. [43] respectively reported that angiotensin-converting enzyme 2 (ACE2) is an efficient SARS-CoV receptor in vitro and in vivo. In addition, SARS-CoV-2 and SARS-CoV have nearly identical three-dimensional structures of spike proteins [44, 45]. This suggests that binding to ACE2 is a key step for entry of the virus to human cells, leading to SARS-CoV-2 infection. Indeed, some studies have shown that upregulated ACE2 expression was associated with augmented disease severity and was a critical factor for viral entry to cells in establishing SARS-CoV-2 infection [46, 47]. These studies provide important insights into the critical step of SARS-CoV-2 infection, suggesting blocking ACE2 receptors as a potential therapeutic approach.
Some limitations of our study are as follows: First, while we performed a general analysis using the GSE152075 dataset containing clinical information and mRNA expression levels, we could not obtain complete clinical data (e.g. illness severity and ethnicity). This study only discusses the difference in the abnormal gene expression between the SARS-CoV-2-infected and normal samples. The correlation between other modules and the remaining clinical traits was not evaluated. Second, although we utilised WGCNA to identify the difference in the abnormal gene expression between the SARS-CoV-2-infected and normal samples, the role of the identified five-core gene set in SARS-CoV-2 infection needs further validation through an external set or a series of experiments.
In conclusion, the core genes and signalling pathways identified in the SARS-CoV-2 infection modules can significantly supplement the current understanding of COVID-19. In particular, the study findings can serve as a basis for creating a hypothetical model for future experimental work with respect to the association of SARS-CoV-2 infection with ribosomal protein function. Moreover, the core genes and signalling pathways may be used as therapeutic targets for COVID-19 treatment.
The study received no financial support.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest. No potential conflict of interest was disclosed.
1. Li X, Zai J, Wang X, Li Y. Potential of large "first generation" human-to-human transmission of 2019-nCoV. J Med Virol. 2020 Apr;92(4):448-454. eng. Epub 2020/01/31. doi:https://doi.org/10.1002/jmv.25693. Cited in: Pubmed; PMID 31997390.
2. Hemmat N, Derakhshani A, Bannazadeh Baghi H, Silvestris N, Baradaran B, De Summa S. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front Genet. 2020 Jun;11:641. https://doi.org/10.3389/fgene.2020.00641
3. van Doremalen N, Bushmaker T, Morris DH, Holbrook MG, Gamble A, Williamson BN, et al. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl J Med. 2020 Apr;382(16):1564–7. https://doi.org/10.1056/NEJMc2004973
4. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020 Mar;382(12):1177–9. https://doi.org/10.1056/NEJMc2001737
5. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents. 2020 Mar;55(3):105924. https://doi.org/10.1016/j.ijantimicag.2020.105924
6. Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, Zhang Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020 Jun;92(6):667-674. eng. Epub 2020/03/14. doi:https://doi.org/10.1002/jmv.25762. Cited in: Pubmed; PMID 32167180.
7. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020 Apr 16;19:100682. eng. Epub 2020/04/18. doi:https://doi.org/10.1016/j.genrep.2020.100682. Cited in: Pubmed; PMID 32300673.
8. Hui DS, E IA, Madani TA, Ntoumi F, Kock R, Dar O, et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health - The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020 Feb;91:264-266. eng. Epub 2020/01/19. doi:https://doi.org/10.1016/j.ijid.2020.01.009. Cited in: Pubmed; PMID 31953166.
9. Calligari P, Bobone S, Ricci G, Bocedi A. Molecular Investigation of SARS-CoV-2 Proteins and Their Interactions with Antiviral Drugs. Viruses. 2020 Apr 14;12(4). eng. Epub 2020/04/17. doi:https://doi.org/10.3390/v12040445. Cited in: Pubmed; PMID 32295237.
10. Prajapat M, Sarma P, Shekhar N, Avti P, Sinha S, Kaur H, Kumar S, Bhattacharyya A, Kumar H, Bansal S, Medhi B. Drug targets for corona virus: A systematic review. Indian J Pharmacol. 2020 Jan-Feb;52(1):56-65. eng. Epub 2020/03/24. doi:https://doi.org/10.4103/ijp.IJP_115_20. Cited in: Pubmed; PMID 32201449.
11. Chen Y, Guo Y, Pan Y, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020 Feb;525(1):S0006-291X(20)30339-9. https://doi.org/10.1016/j.bbrc.2020.02.071
12. Zhao S, Lin Q, Ran J, Musa SS, Yang G, Wang W, Lou Y, Gao D, Yang L, He D, Wang MH. Preliminary estimation of the basic reproduction number of novel coronavirus (2019-nCoV) in China, from 2019 to 2020: A data-driven analysis in the early phase of the outbreak. Int J Infect Dis. 2020 Mar;92:214-217. eng. Epub 2020/02/03. doi:https://doi.org/10.1016/j.ijid.2020.01.050. Cited in: Pubmed; PMID 32007643.
13. Guzzi PH, Mercatelli D, Ceraolo C, Giorgi FM. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. J Clin Med. 2020 Apr 1;9(4). eng. Epub 2020/04/05. doi:https://doi.org/10.3390/jcm9040982. Cited in: Pubmed; PMID 32244779.
14. Pei G, Chen L, Zhang W. WGCNA Application to Proteomic and Metabolomic Data Analysis. Methods Enzymol. 2017;585:135-158. eng. Epub 2017/01/23. doi:https://doi.org/10.1016/bs.mie.2016.09.016. Cited in: Pubmed; PMID 28109426.
15. Yin L, Cai Z, Zhu B, Xu C. Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes (Basel). 2018 Feb 14;9(2). eng. Epub 2018/02/15. doi:https://doi.org/10.3390/genes9020092. Cited in: Pubmed; PMID 29443924.
16. Udyavar AR, Hoeksema MD, Clark JE, Zou Y, Tang Z, Li Z, Li M, Chen H, Statnikov A, Shyr Y, Liebler DC, Field J, Eisenberg R, Estrada L, Massion PP, Quaranta V. Co-expression network analysis identifies Spleen Tyrosine Kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst Biol. 2013;7 Suppl 5(Suppl 5):S1. eng. Epub 2014/02/26. doi:https://doi.org/10.1186/1752-0509-7-s5-s1. Cited in: Pubmed; PMID 24564859. https://doi.org/10.1186/1752-0509-7-S5-S1
17. Shi Z, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol. 2010 May 27;4:74. eng. Epub 2010/05/29. doi:https://doi.org/10.1186/1752-0509-4-74. Cited in: Pubmed; PMID 20507583.
18. Liu X, Hu AX, Zhao JL, Chen FL. Identification of Key Gene Modules in Human Osteosarcoma by Co-Expression Analysis Weighted Gene Co-Expression Network Analysis (WGCNA). J Cell Biochem. 2017 Nov;118(11):3953-3959. eng. Epub 2017/04/12. doi:https://doi.org/10.1002/jcb.26050. Cited in: Pubmed; PMID 28398605.
19. Chen PY, Cripps AW, West NP, Cox AJ, Zhang P. A correlation-based network for biomarker discovery in obesity with metabolic syndrome. BMC Bioinformatics. 2019 Dec 10;20(Suppl 6):477. eng. Epub 2019/12/12. doi:https://doi.org/10.1186/s12859-019-3064-2. Cited in: Pubmed; PMID 31823713.
20. Xia WX, Yu Q, Li GH, Liu YW, Xiao FH, Yang LQ, Rahman ZU, Wang HT, Kong QP. Identification of four hub genes associated with adrenocortical carcinoma progression by WGCNA. PeerJ. 2019;7:e6555. eng. Epub 2019/03/20. doi:https://doi.org/10.7717/peerj.6555. Cited in: Pubmed; PMID 30886771.
21. Lieberman NA, Peddu V, Xie H, Shrestha L, Huang ML, Mears MC, et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020 Sep;18(9):e3000849. https://doi.org/10.1371/journal.pbio.3000849
22. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008 Dec 29;9:559. eng. Epub 2008/12/31. doi:https://doi.org/10.1186/1471-2105-9-559. Cited in: Pubmed; PMID 19114008.
23. Shi K, Bing ZT, Cao GQ, Guo L, Cao YN, Jiang HO, Zhang MX. Identify the signature genes for diagnose of uveal melanoma by weight gene co-expression network analysis. Int J Ophthalmol. 2015;8(2):269-74. eng. Epub 2015/05/06. doi:https://doi.org/10.3980/j.issn.2222-3959.2015.02.10. Cited in: Pubmed; PMID 25938039.
24. Li X, He Y, Hao C, Li X, Li X. Weighted gene correlation network analysis reveals novel regulatory modules associated with recurrent early pregnancy loss. Biosci Rep. 2020 Jun 26;40(6). eng. Epub 2020/05/14. doi:https://doi.org/10.1042/bsr20193938. Cited in: Pubmed; PMID 32401299. https://doi.org/10.1042/BSR20193938
25. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012 May;16(5):284-7. eng. Epub 2012/03/30. doi:https://doi.org/10.1089/omi.2011.0118. Cited in: Pubmed; PMID 22455463.
26. Villanueva RA, Chen ZJ. ggplot2: Elegant graphics for data analysis. Taylor & Francis; 2019.
27. Zhang W, Xu J, Li Y, Zou X. Integrating network topology, gene expression data and GO annotation information for protein complex prediction. J Bioinform Comput Biol. 2019 Feb;17(1):1950001. eng. Epub 2019/02/26. doi:https://doi.org/10.1142/s021972001950001x. Cited in: Pubmed; PMID 30803297. https://doi.org/10.1142/S021972001950001X
28. Thomas PD, Hill DP, Mi H, Osumi-Sutherland D, Van Auken K, Carbon S, Balhoff JP, Albou LP, Good B, Gaudet P, Lewis SE, Mungall CJ. Gene Ontology Causal Activity Modeling (GO-CAM) moves beyond GO annotations to structured descriptions of biological functions and systems. Nat Genet. 2019 Oct;51(10):1429-1433. eng. Epub 2019/09/25. doi:https://doi.org/10.1038/s41588-019-0500-1. Cited in: Pubmed; PMID 31548717.
29. Li Y, Dong W, Shi Y, Deng F, Chen X, Wan C, Zhou M, Zhao L, Fu ZF, Peng G. Rabies virus phosphoprotein interacts with ribosomal protein L9 and affects rabies virus replication. Virology. 2016 Jan 15;488:216-24. eng. Epub 2015/12/15. doi:https://doi.org/10.1016/j.virol.2015.11.018. Cited in: Pubmed; PMID 26655239.
30. Abbas W, Dichamp I, Herbein G. The HIV-1 Nef protein interacts with two components of the 40S small ribosomal subunit, the RPS10 protein and the 18S rRNA. Virol J. 2012 Jul 10;9:103. eng. Epub 2012/06/08. doi:https://doi.org/10.1186/1743-422x-9-103. Cited in: Pubmed; PMID 22672539. https://doi.org/10.1186/1743-422X-9-103
31. Rofeal M, Abd El-Malek F. Ribosomal proteins as a possible tool for blocking SARS-COV 2 virus replication for a potential prospective treatment. Elsevier; 2020. https://doi.org/10.1016/j.mehy.2020.109904
32. Lv H, Dong W, Qian G, Wang J, Li X, Cao Z, Lv Q, Wang C, Guo K, Zhang Y. uS10, a novel Npro-interacting protein, inhibits classical swine fever virus replication. J Gen Virol. 2017 Jul;98(7):1679-1692. eng. Epub 2017/07/20. doi:https://doi.org/10.1099/jgv.0.000867. Cited in: Pubmed; PMID 28721853.
33. Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, Baric RS. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio. 2015 May 26;6(3):e00638-15. eng. Epub 2015/05/28. doi:https://doi.org/10.1128/mBio.00638-15. Cited in: Pubmed; PMID 26015500.
34. Horng JC, Moroz V, Rigotti DJ, Fairman R, Raleigh DP. Characterization of large peptide fragments derived from the N-terminal domain of the ribosomal protein L9: definition of the minimum folding motif and characterization of local electrostatic interactions. Biochemistry. 2002 Nov 12;41(45):13360-9. eng. Epub 2002/11/06. doi:https://doi.org/10.1021/bi026410c. Cited in: Pubmed; PMID 12416980.
35. Beyer AR, Bann DV, Rice B, Pultz IS, Kane M, Goff SP, Golovkina TV, Parent LJ. Nucleolar trafficking of the mouse mammary tumor virus gag protein induced by interaction with ribosomal protein L9. J Virol. 2013 Jan;87(2):1069-82. eng. Epub 2012/11/09. doi:https://doi.org/10.1128/jvi.02463-12. Cited in: Pubmed; PMID 23135726. https://doi.org/10.1128/JVI.02463-12
36. Abbas W, Dichamp I, Herbein G. The HIV-1 Nef Protein Interacts with two components of the 40S small ribosomal subunit, the RPS10 protein and the 18S rRNA. Virology Journal. 2012 2012/07/10;9(1):103. doi:https://doi.org/10.1186/1743-422X-9-103.
37. Robledo S, Idol RA, Crimmins DL, Ladenson JH, Mason PJ, Bessler M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA. 2008 Sep;14(9):1918–29. https://doi.org/10.1261/rna.1132008
38. Colombo P, Yon J, Fried M. The organization and expression of the human L7a ribosomal protein gene. Biochim Biophys Acta. 1991 Dec;1129(1):93–5. https://doi.org/10.1016/0167-4781(91)90218-B
39. Fumagalli S, Ivanenkov VV, Teng T, Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012 May;26(10):1028–40. https://doi.org/10.1101/gad.189951.112
40. Wang M, Parshin AV, Shcherbik N, Pestov DG. Reduced expression of the mouse ribosomal protein Rpl17 alters the diversity of mature ribosomes by enhancing production of shortened 5.8S rRNA. RNA. 2015 Jul;21(7):1240–8. https://doi.org/10.1261/rna.051169.115
41. Rospert S. Ribosome function: governing the fate of a nascent polypeptide. Curr Biol. 2004 May;14(10):R386–8. https://doi.org/10.1016/j.cub.2004.05.013
42. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003 Nov 27;426(6965):450-4. eng. Epub 2003/12/04. doi:https://doi.org/10.1038/nature02145. Cited in: Pubmed; PMID 14647384.
43. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005 Aug;11(8):875-9. eng. Epub 2005/07/12. doi:https://doi.org/10.1038/nm1267. Cited in: Pubmed; PMID 16007097.
44. Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Science China Life Sciences. 2020 2020/03/01;63(3):457-460. doi:https://doi.org/10.1007/s11427-020-1637-5.
45. Rabaan AA, Al-Ahmed SH, Haque S, Sah R, Tiwari R, Malik YS, Dhama K, Yatoo MI, Bonilla-Aldana DK, Rodriguez-Morales AJ. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez Med. 2020 Ahead Of Print Jun 1;28(2):174-184. eng. Epub 2020/04/11. Cited in: Pubmed; PMID 32275259.
46. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 Mar;579(7798):270-273. eng. Epub 2020/02/06. doi:https://doi.org/10.1038/s41586-020-2012-7. Cited in: Pubmed; PMID 32015507.
47. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. bioRxiv. 2020:2020.01.26.919985. doi:https://doi.org/10.1101/2020.01.26.919985