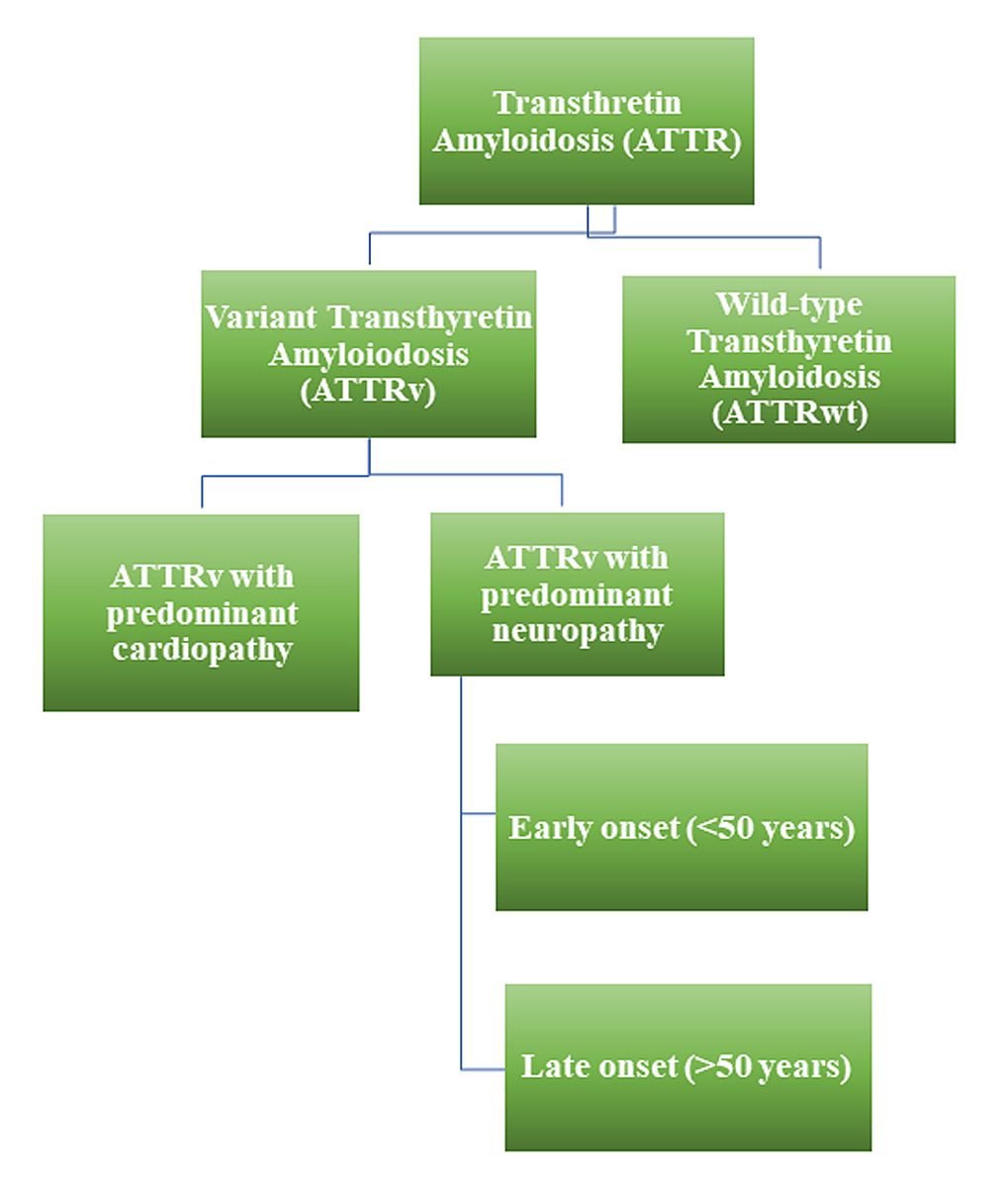
Figure 1 Schematic representation of different types of transthyretin amyloidosis. ATTR: transthyretin amyloidosis; ATTRv: variant transthyretin amyloidosis; ATTRwt: wild-type transthyretin amyloidosis.
DOI: https://doi.org/10.4414/SMW.2021.w30053
These guidelines were written after an extensive literature search using MedLine publications from January 1980 to April 2021, under the terms “ATTR” AND “transthyretin” AND “amyloidosis”. Only full articles in English were considered. Following the literature search and review, the steering committee developed a summary of relevant statements that were evaluated by all the participants. Consensus was established using online, single point-per-point voting, through a nominal group technique. The level of evidence and the strength of recommendation for specific management options were weighed and graded according to predefined scales, as outlined in tables 1 and 2.
Table 1Classes of recommendations.
| Class | Definition | Wording |
| I | Evidence and/or general agreement that a given treatment or procedure is beneficial, useful, effective. | Is recommended |
| II | Conflicting evidence and/or a divergence of opinion about usefulness/efficacy of the given treatment or procedure. | |
| IIa | Weight of evidence/opinion is in favour of usefulness/efficacy. | Should be considered |
| IIb | Usefulness/efficacy is less well established by evidence/opinion. | May be considered |
| III | Evidence or general agreement that the given treatment or procedure is not useful/effective and in some cases may be harmful | Is not recommended |
Table 2Levels of evidence.
| Level of evidence | Definition |
| A | Data derived from multiple randomised clinical trials or meta-analyses. |
| B | Data derived from a single randomised clinical trial or large nonrandomised studies. |
| C | Consensus of opinion of the experts and/or small studies, retrospective studies, registries. |
The final guidelines were reviewed according to the Appraisal of Guidelines for Research and Evaluation (AGREE) II tool, to guarantee methodological rigour and transparency. A summary of the resulting recommendations is presented in table 3.
Table 3Summary of Swiss Amyloidosis Network (SAN) recommendations.
| Recommendation | Level |
| Hereditary transthyretin amyloidosis (ATTRv) | |
| A comprehensive initial work-up in a centre experienced in diagnosis and treatment of patients with ATTRv amyloidosis is recommended. | I, C |
| A low threshold for genetic testing in patients with suspected ATTRv amyloidosis is recommended. | I, C |
| A biopsy of the organ mainly involved, the gastrointestinal tract, the accessory salivary glands or an abdominal fat aspirate for detection of amyloid should be considered in suspected systemic ATTR amyloidosis. | IIa, C |
| For patients with systemic ATTR amyloidosis, TTR amyloid detection and characterisation in one anatomical site is sufficient to make the diagnosis. | I, C |
| A positive bone scintigraphy (Perugini score grade 2 or higher) can substitute tissue biopsy for diagnosing ATTRv amyloidosis with cardiac involvement in the absence of concomitant monoclonal gammopathy, but false negative results can occur. | I, B |
| Genetic counselling for all patients with hereditary amyloidosis is recommended | I, C |
| Genetic counselling should be coordinated by amyloidosis specialists together with a certified genetic counselling centre. | I, C |
| Pre-symptomatic testing should be considered. | IIa, C |
| Cardiac and neurological staging of all patients with ATTRv amyloidosis is recommended. | I, B |
| Disease-modifying treatment is recommended according to current drug approval status in Switzerland. | I, B |
| Liver transplantation should be considered as second-line treatment option for patients with early-onset ATTRv amyloidosis with a primarily neurological phenotype and RNAi; patisiran, Onpattro®) treatment failure or intolerance. | IIa, C |
| Heart transplantation should be considered as an option for younger patients with predominant and advanced cardiac involvement not responding to disease-modifying drugs, or for whom disease-modifying drugs are not available or unlikely to be effective | IIa, C |
| Clinical and biological follow-up in specialised centres every 3–6 months is recommended, including a comprehensive follow-up with disability and QOL questionnaires, and a standardised cardiological and neurological work-up | I, C |
| A standardised ophthalmological assessment is recommended every 1–2 years. | I, C |
| SAN recommends that the lead for patient follow-up should be cardiological for patients with predominantly cardiac manifestations, and neurological for patients with predominantly neurological manifestations. | I, C |
| Wild-type transthyretin amyloidosis (ATTRwt) | |
| Interdisciplinary discussion of the results obtained during the screening for monoclonal gammopathy with specialists in haemato-oncology should be considered. | IIa, C |
| The following screening panel for monoclonal gammopathy should be considered: serum protein electrophoresis, serum immunofixation, serum free light chain measurement and urine immunofixation. | IIa, B |
| TTR genetic testing should be considered in all patients with cardiac ATTR amyloidosis regardless of patient age. | IIa, C |
| A visit to a neurologist may be considered at baseline for patients with ATTRwt amyloidosis and neuropathy. | IIb, C |
| A cardiological work-up is recommended at baseline, including ECG, 24-hour ECG, blood pressure and TTE. The need for bone scintigraphy, cardiac MRI and myocardial biopsy should be discussed by an interdisciplinary team on a case-by-case basis. | I, B |
| Initial staging of all patients with ATTRwt amyloidosis should be considered. | IIa, B |
| SAN recommends treatment guided by experienced centres, using a multidisciplinary approach (cardiology, haemato-oncology, neurology, nephrology, gastroenterology). | I, C |
| Treatment with tafamidis 61 mg should be considered for patients with cardiac ATTRwt amyloidosis and dyspnoea NYHA class I–III | IIa, B |
| Case-by-case discussions should be considered prior to tafamidis initiation for patients with significant comorbidities that interfere with intermediate-term survival. | IIa, C |
| Case-by-case discussions should be considered for patients with complex cardiac situations (e.g., concomitant aortic stenosis, concomitant severe coronary artery disease), patients with a formal indication for implantation of an internal cardioverter defibrillator, and patients with typical angina due to microvascular disease | IIa, C |
| Cardiological follow-up should be considered every 3–6-months, including clinical and laboratory evaluation (NT-proBNP, troponin T, creatinine, proteinuria, albuminuria), and a comprehensive evaluation including ECG, 24-hour ECG, TTE every 612 months, ergometry, depending on disease severity and treatment. | IIa, C |
| Regular blood pressure and body weight home monitoring may be considered in order to adapt diuretic dose targeting euvolaemia | IIb, C |
| Patients at risk for developing systemic ATTR amyloidosis | |
| Asymptomatic carriers: based on the specific TTR mutation and the onset age in other affected family members, PADO can be estimated. Systemic monitoring of asymptomatic carriers should be considered at least 10 years prior to PADO | IIa, C |
| Domino Liver TPL: 6-monthly or yearly follow-up for signs of neuropathy (including BMI, autonomic dysfunction) should be considered. | IIa, B |
| ATTR in tissue biopsies (lumbar spine, CTS, etc.):a cardiac work-up including ECG, TTE (including speckle-tracking echocardiography based left ventricular strain analysis) and genetic testing for ATTRv amyloidosis should be considered | IIa, C |
ATTRv amyloidosis: variant transthyretin amyloidosis; ATTRwt amyloidosis: wild-type transthyretin amyloidosis; ATTRv: variant transthyretin amyloidosis; BMI: body mass index; ECG: electrocardiography; CTS: carpal tunnel syndrome; NYHA: New York Heart Association; NT-proBNP: N-terminal pro-B-type natriuretic peptid; MRI: magnetic resonance imaging; PADO: predicted age at disease onset; QOL: quality of life; RNAi: RNA interference; TPL: transplantation; TTE: transthoracic echocardiography; TTR: transthyretin.
Systemic amyloidosis is a term for a rare and underdiagnosed group of diseases, characterised by progressive tissue infiltration of insoluble fibrillary proteins, derived from a misfolded precursor protein, which normally circulates in blood as a constituent of plasma [1]. The precursor protein defines the amyloid subtype and the associated clinical manifestations. Free immunoglobulin light chains and variant or wild-type transthyretin (TTR) are the main amyloidogenic precursor proteins, causing light-chain (AL) amyloidosis, and transthyretin (ATTR) amyloidosis, respectively [1–3].
ATTR amyloidosis can be subdivided based on the transthyretin protein involved, wild-type or variant (fig. 1) [1–3]. Even though ATTR amyloidosis is a rare disease, its prevalence is higher than current data suggest, in particular, cases with predominantly cardiac involvement, due to wild-type ATTR deposition [4–7]. According to an analysis of Centers for Medicare and Medicaid Services data, the prevalence of cardiac ATTR amyloidosis increased from 18 to 55 per 100,000 between 2000 and 2012, likely due to growing awareness and improved diagnostics, rather than due to an actual increase in prevalence [8]. It can be expected that the number of patients being diagnosed with cardiac ATTR amyloidosis will continue to rise in the future.
Figure 1 Schematic representation of different types of transthyretin amyloidosis. ATTR: transthyretin amyloidosis; ATTRv: variant transthyretin amyloidosis; ATTRwt: wild-type transthyretin amyloidosis.
Delays in the diagnosis of ATTR amyloidosis remain common, likely because of: the perceived rarity of the disease, clinical heterogeneity, scarcity of centres and specialists dedicated to disease management and, ultimately, equivocal histological diagnosis.
Transthyretin (TTR), also known as prealbumin, is a 127 amino acid soluble and typically non-glycosylated protein, mainly synthesised and secreted by hepatocytes in the liver, the choroid plexus ependymal cells (in the brain) and retinal epithelium (in the eye) [9]. TTR binds to retinol-binding protein, which is involved in vitamin A transport. TTR is also responsible for the transport of thyroxine (T4) in plasma and cerebrospinal fluid (CSF), and circulates as a tetramer [10].
In both wild-type ATTR amyloidosis (ATTRwt), formerly known as "senile" systemic amyloidosis, and in the hereditary form, known as variant ATTR amyloidosis (ATTRv), the disease is a consequence of transthyretin tetramer dissociation into folded monomers, which then undergo conformational changes, leading to the formation of non-native oligomers and amyloid fibrils [11]. Truncated C-terminal fragments can initiate aggregation and fibrillogenesis from either unstable variants or wild-type TTR [12]. This mechanism is promoted by mechanical shear stress, in combination with hydrophobic forces generated by extracellular matrix components (such as serum amyloid P and glycosaminoglycans) [12], and also involves proteostasis processes, which misfold and aggregate proteins [13, 14]. Amyloid-related tissue changes can be displayed as mechanical disarrangement, disruption of cell membrane integrity, inflammatory reactions, hypoxic cell damage and/or alteration of the structure of the extracellular matrix [14, 15].
Depending on the absence or presence of a pathogenic transthyretin mutation, ATTR amyloidosis can be further subdivided into ATTR amyloidosis caused by deposits of the wild-type TTR protein (ATTRwt), and ATTR amyloidosis caused by a genetically determined variant TTR protein (ATTRv).
Overall, ATTRv amyloidosis is a rare disease, with a global prevalence of 1:1,000,000 people [16, 17], though it is endemic in certain areas of Portugal, Japan, Sweden and Brazil, where the prevalence may be as high as 1:1000 [16, 17].
ATTRwt amyloidosis is more common than ATTRv amyloidosis, although the true prevalence is unknown. Amyloid is formed by wild-type TTR deposits in the heart and ligaments during aging, particularly in male subjects [18, 19]. The reason(s) behind the observed tissue tropism and gender difference are not yet clear. In autopsy studies, ≈25% of people at least 80 years old had wild-type TTR fibrils in their hearts, regardless of the presence of symptoms [19, 20]. Studies using non-biopsy diagnostic approaches demonstrated TTR prevalences of 10–13% among patients undergoing percutaneous aortic valve replacement for severe aortic stenosis, 13% among patients with heart failure with preserved ejection fraction (HFpEF) and 5% among patients with presumed hypertrophic cardiomyopathy [5, 7, 21–23]. About 60% of patients with ATTR amyloidosis have a personal history of carpal tunnel syndrome, and 5–14% have a personal history of lumbar spinal stenosis [24]. The prevalence of ATTR in tissue biopsies from unselected patients with lumbar spinal stenosis is as high as 38.5%, and is 10.2% in patients with bilateral carpal tunnel syndrome [25, 26]. Most of these patients do not have systemic ATTR amyloidosis with cardiac and/or neurological involvement at the time of surgery, and the percentage of patients who will eventually progress and develop systemic ATTR amyloidosis is not yet known.
The main clinical features of ATTR amyloidosis are cardiomyopathy and neuropathy.
ATTRwt amyloidosis usually presents as heart failure (HFpEF) in elderly male patients, but bilateral carpal tunnel syndrome, lumbar spinal stenosis and biceps tendon rupture can precede the onset of cardiac symptoms by a decade or more [24]. In ATTRwt amyloidosis, peripheral and autonomic neuropathy manifestations are less severe than in ATTRv amyloidosis (table 4).
Table 4Main clinical characteristics of ATTRv and ATTRwt amyloidosis.
| ATTRv amyloidosis | ATTRwt amyloidosis | ||
| Prevalence | <1:100000 | Unknown | |
| Age at presentation | Variable | >60 years | |
| Gender prevalence | Male | Male | |
| Common disease manifestation | Polyneuropathy | Heart | |
| Carpal tunnel syndrome | Extracardiac involvement: | Carpal tunnel syndrome | |
| Dysautonomia | Lumbar spinal stenosis | ||
| Eye involvement | Biceps tendon rupture | ||
ATTRv amyloidosis: variant transthyretin amyloidosis; ATTRwt amyloidosis: wild-type transthyretin amyloidosis.
ATTRv amyloidosis usually presents as peripheral small-fibre polyneuropathy with dysautonomia, but can also manifest as a primary cardiomyopathy, depending on the genotype (see section 2 and fig. 2) [3]. The natural history of ATTRv amyloidosis, including age of onset, phenotype and clinical course varies with: mutation, fibril type (full length vs fragments) and within families and/or regions.
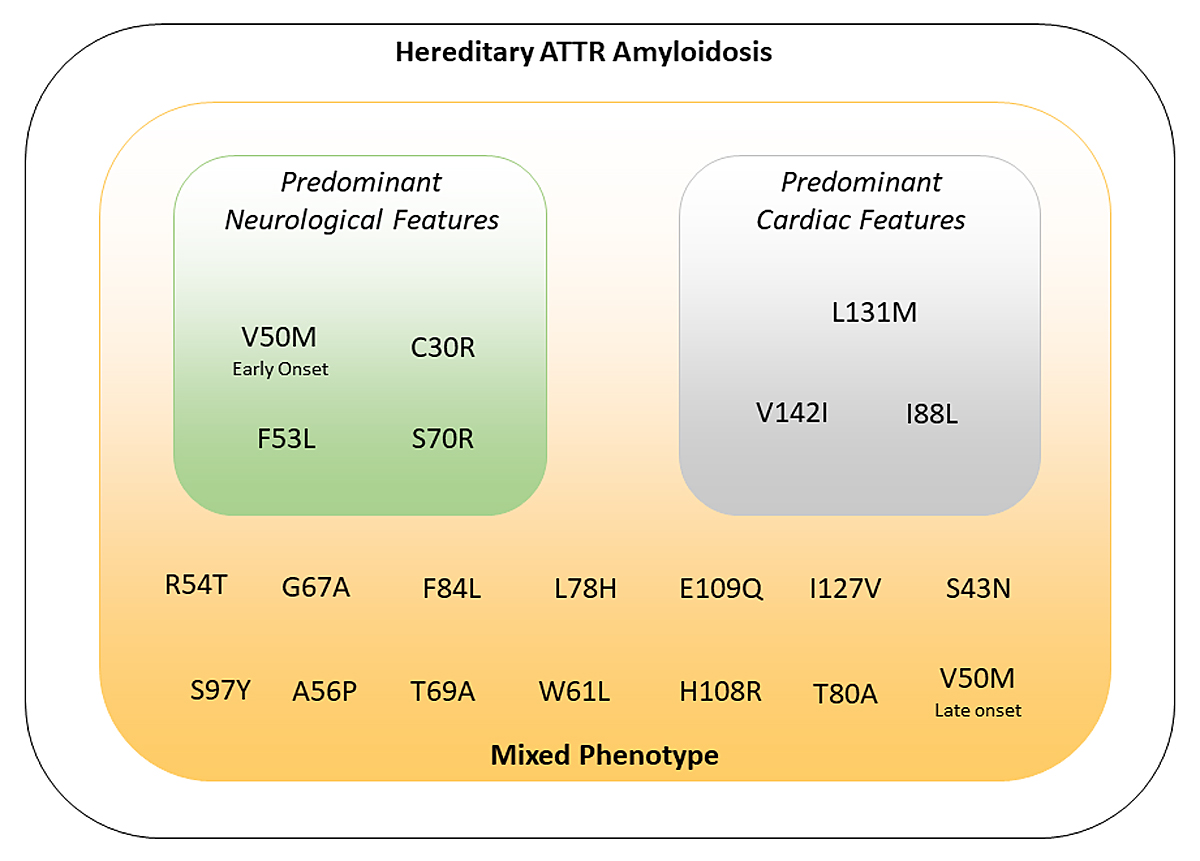
Figure 2 Correlations between genotype and phenotype in variant transthyretin amyloidosis. ATTR: transthyretin amyloidosis.
Diagnosis of ATTRv amyloidosis neuropathy is usually straight-forward where the disease is endemic (Portugal, Japan, Sweden, Brazil), thanks to higher awareness, typical phenotype and known family history. In non-endemic areas, where ATTRv amyloidosis is rare and mostly presents as a late-onset disease, the diagnosis can be challenging. Unexplained sensorimotor neuropathy or autonomic symptoms (orthostatic dysfunction, erectile dysfunction, sweating abnormalities, unexplained weight loss and diarrhoea), are typical but unspecific symptoms of ATTRv amyloidosis, and may delay diagnosis [27].
The main objective of the initial assessment is to confirm the diagnosis and rule out other amyloidosis subtypes, mainly light-chain amyloidosis. Once a diagnosis of ATTR amyloidosis is made, some of the main tasks of a comprehensive amyloidosis management are: determination of the extent of systemic involvement (staging); treatment indication and strategy; definition of follow-up intervals and tests; education of patients and their families (including genetic counselling for ATTRv amyloidosis).
At diagnosis, each patient should undergo a complete evaluation of organ involvement. The cardiac evaluation includes: functional class, fluid balance, orthostatic function and heart rhythm. Assessment of the nervous system includes: functional capacity, quantification of nerve damage, assessment of autonomic dysfunction (including gastrointestinal and urogenital systems) and weight changes. Renal function should be assessed (estimated glomerular filtration rate [eGFR], albuminuria, proteinuria), as should the eyes (keratoconjunctivitis sicca, vitreal deposits, secondary glaucoma or retinal angiopathy) [6].
ATTRv amyloidosis is an autosomal-dominant disease with variable penetrance. Based on the underlying mutation in the TTR gene, ATTRv amyloidosis can present as predominantly neurological or predominantly cardiological disease, although cardiomyopathy and polyneuropathy manifestations may coexist (fig. 2).
The most common TTR mutation, p.V142I (also known as “V122I”), occurs mainly as cardiomyopathy in 3–10% of black Americans older than 60 years, with undefined phenotypic penetrance [28].
The p.V50M (also known as “V30M”) mutation is the most common and prototypical form of amyloid polyneuropathy. Two different clinical forms called "early-onset", and "late-onset" ATTRv related to p.V50M can be distinguished.
Early-onset p.V50M: Early-onset ATTRv amyloidosis usually presents in Portuguese and Japanese patients under 50 years of age, with a peak between the ages of 25 and 35 years. Initial symptoms include paraesthesia of the lower limbs (40%), neuropathic pain (5%), autonomic dysfunction with diarrhoea and/or constipation (40%), erectile dysfunction (9%), hypotension and unexplained weight loss (16% at presentation) [29–32]. With advancing disease, the involvement of large-diameter nerve fibres eventually manifests with impairment of all sensory modalities and motor dysfunction, with progressive walking imbalance until a wheelchair in needed [29–32]. Without disease-modifying treatment, the mean survival time of early-onset ATTRv amyloidosis ranges from 10 to 15 years from diagnosis [33].
Late-onset p.V50M: Late-onset ATTRv amyloidosis is more common in central Europe and Sweden and usually affects individuals (mostly men) older than 50 years of age, with a positive family history in 50–60% of cases [32, 34]. The median delay to diagnosis is 3 years, likely because of the clinical phenotype variability. Sensorimotor symptoms can begin distally, in the lower extremities, with disturbance of both superficial and deep sensation [29, 32]. However, a great proportion of patients present with symptoms mimicking other forms of peripheral neuropathy (upper limb onset neuropathy, ataxic neuropathy, multiplex neuritis and motor neuropathy) [35–37]. Autonomic dysfunction is usually milder than in early-onset patients, whereas neuropathic pain is present in almost 50% of the patients. The rate of neuropathy progression is higher than in early-onset cases [38, 39]. The most common misdiagnoses are chronic inflammatory demyelinating polyneuropathy followed by idiopathic axonal polyneuropathy and lumbar canal stenosis [40, 41]. Cardiomyopathy may also occur.
Additional differences between early-onset and late-onset p.V50M include a higher rate of renal involvement, with proteinuria and progressive renal failure in the former and a significant association with bilateral carpal tunnel syndrome in the latter. Atrioventricular conduction blocks requiring pace-maker implantation are very common in early-onset p.V50M. Biochemically, amyloid fibrils consist of only full-length TTR in early-onset patients’ tissues, and of both C-terminal fragmented and full-length TTR in late-onset cases [42].
Outside endemic areas, disease clusters have been reported in many other countries, mainly related to migratory movements [34].
A comprehensive initial work-up in a centre experienced in diagnosis and treatment of patients with ATTRv amyloidosis is recommended (table 5) [I, C]. The initial work-up aims at securing the diagnosis and provides the basis for treatment indication, and for treatment response assessments.
A low threshold for genetic testing in patients with suspected ATTRv amyloidosis is recommended [I, C]. Cost coverage for genetic testing by the healthcare insurance is granted either under the position for amyloidotic polyneuropathy or under the "orphan disease" provisions in the case of non-neurological manifestations. For the latter, a request for coverage has to be obtained from the health insurance beforehand, which we also recommend for the amyloidotic polyneuropathy provisions, although it is not mandatory there.
Suspicion of ATTRv amyloidosis should be high for patients with progressive idiopathic, axonal polyneuropathy (i.e., affecting temperature and pain sensation), or atypical chronic inflammatory demyelinating polyneuropathy, especially if refractory to treatment, particularly in elderly patients who also present with symptoms of systemic involvement, even in the absence of a suggestive family history [27]. For patients with a known family history of ATTRv amyloidosis, any onset of any type of axonal polyneuropathy, autonomic dysfunction or cardiac arrhythmia warrants an assessment for organ involvement. Of note, individuals with a known family history may be offered presymptomatic testing, and should be regularly followed to detect amyloidosis onset (see sections 2.2.3 and 4.1).
Initial work-up in ATTRv amyloidosis aims at confirming amyloid deposits with a tissue biopsy (gastrointestinal, salivary gland, skin, peripheral nerve, heart), including typing of the amyloid by immunohistochemistry or spectrometry, and screening for TTR mutations by TTR gene sequencing. In the absence of monoclonal gammopathy, scintigraphy using bone-avid radiotracers can diagnose ATTR amyloidosis with cardiac involvement without the need for biopsies, but false negative scintigraphy results can occur, especially in patients with cardiac ATTRv amyloidosis (see section 3.1.1). In a second step, the extent of organ involvement has to be determined.
During the initial assessment, autonomic dysfunction, gait disorders, gastrointestinal disorders, weight loss, carpal tunnel syndrome or previous surgery for bilateral carpal tunnel syndrome, and concurrent cardiac abnormalities should be checked. A detailed family history for ATTRv must be obtained, and any severe neuropathy of a family member, especially when leading to death within a few years of symptom onset, should raise suspicion for ATTRv [43, 44].
A biopsy of the main involved organ, the gastrointestinal tract, the accessory salivary glands or an abdominal fat aspirate for amyloid detection should be considered in suspected systemic ATTR amyloidosis [ IIa, C]. Skin biopsy can be useful in centres experienced in amyloid detection in ATTRv amyloidosis with mainly neurological manifestations. For patients with systemic ATTR amyloidosis, TTR amyloid detection and characterisation in one anatomical site is sufficient to make the diagnosis [ I, C]. A positive bone scintigraphy (Perugini score grade 2 or higher) can substitute tissue biopsy for diagnosing ATTRv amyloidosis with cardiac involvement in the absence of concomitant monoclonal gammopathy, but false negative results can occur [I, B].
Staining of biopsy samples with Congo red and visualisation of atypical colours (green, yellow or orange birefringence) under polarised light is indicative of the presence of amyloid fibrils. Once Congo red-positive material is detected, the subtype of amyloid has to be determined by immunohistochemistry, and if immunohistochemistry is inconclusive, by mass spectrometry or immunoelectron microscopy [6, 27, 45–47].
Amyloid infiltration can be demonstrated in tissue samples taken from a surrogate site (e.g., abdominal fat pad, salivary glands or rectum), or from a clinically involved organ. Accessory salivary gland biopsy has a sensitivity of 75% for amyloid detection in patients with ATTRv amyloidosis when performed in an experienced centre [48]. Sural nerve biopsy is performed only occasionally, mostly to rule out differential diagnoses. It is always recommended to ask for past biopsies (most often gastrointestinal), when available, to be tested for amyloid deposits.
Detection of amyloid deposits can be challenging, and negative biopsy results should not rule out the diagnosis. Repeated tissue biopsy may be required (see section 3.1) [49].
In one study, ATTR amyloid was detectable in 27.3% of abdominal fat aspirates. ATTR amyloid deposits were detected in 35.4% of bone marrow biopsies and 33.3% of gastrointestinal biopsies when abdominal fat aspiration was not able to provide evidence of amyloid [50]. When ATTR amyloid was detectable in abdominal fat aspirates, it was also detectable in 66.6% of bone marrow and 68.2% of gastrointestinal biopsies. The sensitivity of screening biopsies is lower in patients with ATTR amyloidosis than in those with AL amyloidosis [50].
A blood test should include at least complete blood count, creatinine, eGFR, liver values including alkaline phosphatase, N-terminal pro-B-type natriuretic peptide (NT-proBNP), troponin T, serum protein, albumin, prealbumin, albuminuria, proteinuria, serum free light chains, and serum and urine protein electrophoresis with immunofixation. Further tests such as thyroid stimulating hormone (TSH) / free thyroxine (fT4), iron status, vitamin B12 and screening for diabetes mellitus should be performed to rule out concomitant diagnoses affecting the nervous system.
SAN recommends genetic counselling for all patients with hereditary amyloidosis [I, C]. Genetic testing should be offered to all symptomatic relatives, and discussed individually with asymptomatic relatives, starting with the patient’s siblings. Genetic counselling should be coordinated by amyloidosis specialists together with a certified genetic counselling centre [I, C].
Pre-symptomatic testing should be considered [ IIa, C]. Since the application on 1 April 2007 of the Law on Human Genetic Analysis (GUMG /LAGH), genetic counselling and informed consent are formally required before any genetic testing and should be performed by dedicated centres. A list of Swiss genetic counselling centres can be found on the homepage of the Swiss Society of Medical Genetics (www.sgmg.ch). Cost coverage of genetic testing for symptomatic patients by the healthcare insurance is either under the provisions for amyloidotic polyneuropathy, or in the case of non-neurological manifestations under the orphan disease provisions. For the latter, a request for coverage has to be obtained from the health insurance beforehand, which we also recommend for the amyloidotic polyneuropathy provisions, although not mandatory there.
Considering the short length of the TTR gene, and the limited number of coding exons (3 exons), genetic investigation of pathological mutations associated with hereditary forms of systemic amyloidosis can be performed by Sanger sequencing. An updated list of all amyloidogenic mutations identified so far is available on www.amyloidosismutations.com [51]. There are few non-amyloidogenic mutations.
The increasing availability of effective and early treatments led to the promotion of pre-symptomatic testing for hereditary TTR amyloidosis, with differing counselling protocols among countries [52, 53].
Especially in endemic areas for early-onset ATTRv amyloidosis, harbouring a TTR mutation might entail decisions about reproduction (including the choice of prenatal or pre-implant diagnosis, depending on the respective legal premises), with a psychological impact that warrants adequate support [54, 55]. Overall, pre-symptomatic testing for ATTRv amyloidosis appears to be feasible when performed within a multidisciplinary team. Family investigation sometimes contributes to identifying affected family members.
Given the increasing availability of therapeutic options, how best to monitor mutation carriers and when to initiate treatment is currently a debated topic (see section 4.1.).
A comprehensive neurological assessment for ATTRv amyloidosis requires time and resources. It includes standardised questionnaires, a complete personal and family history, and a systematic clinical work-up for sensory, autonomic and motor neuropathy. Table 6 summarises the recommended initial neurological work-up for specialised neurological centres caring for patients with ATTRv amyloidosis. Standardised baseline assessments and follow-up visits not only guarantee quality of patient care, but are needed for regulatory reasons in patients receiving disease-modifying drugs. Recommendations for initial and periodic assessments are summarised in tables 5 and 6.
Table 5Recommendations for baseline and follow-up assessment of patients with ATTRv amyloidosis.
| Tool | Baseline | 3–6 monthly | 12 monthly | |
| General health assessment | Patient history, physical examination, standard laboratory testing | X | X | X |
| Weight | X | X | X | |
| Conventional BMI | X | X | X | |
| Modified BMI | X | X | X | |
| Albumin | X | X | X | |
| Prealbumin | X | X | X | |
| Medication | Check for completeness, interactions, harmful prescriptions | X | X | X |
| Neurology | Norfolk QOL | X | X | X |
| Compass 31 or CADT | X | X | X | |
| SFN S1Q | X | X | X | |
| RODS | X | X | X | |
| Schellong | X | X | X | |
| Composite nerve conduction score | X | - | X | |
| Sudoscan | X | - | X | |
| Grip test | X | - | X | |
| NIS LL and UL | X | X | X | |
| PND and FAP stage | X | X | X | |
| Cardiology | NT-proBNP | X | X | X |
| ECG | X | X | X | |
| 24-hour ECG | X | X for cardiac ATTR | X for cardiac ATTR | |
| TTE | X | X for cardiac ATTR | X for cardiac ATTR | |
| CMR | X | - | (consider repeating at 2 years) | |
| 6-min walking test | X | X | X | |
| Scintigraphy using bone-avid radiotracers | (X) discuss case-by-case | - | - | |
| NYHA class | X | X | X | |
| Nephrology | Serum creatinine/eGFR | X | X | X |
| Urine creatinine/protein/albumin | X | |||
| Sonography | X | - | - | |
| Ophthalmology | Visual acuity, check for amyloid deposits | X | - | X |
| Gastroenterology | Ask for early satiety, postprandial fullness, nausea, vomiting and weight loss | Endoscopy, oesophageal and intestinal manometry, and gastric emptying studies if symptomatic | ||
| Ask about constipation, diarrhoea, incontinence | Endoscopy, search for bacterial overgrowth, bile acid and fat malabsorption if symptomatic | |||
| Alkaline phosphatase | X | X | X | |
| Abdominal sonography | X | - | - | |
| Psychology/social service | Check if support needed | X | X | X |
ATTRv: variant transthyretin amyloidosis; BMI: body mass index; CADT: compound autonomic dysfunction test; CMR: cardiac MRI; COMPASS score: composite autonomic symptom score; ECG: electrocardiography; NYHA: New York Heart Association; eGFR: estimated glomerular filtration rate; FAP: familial amyloid polyneuropathy; NIS-LL and -UL: neuropathy impairment score in the lower limbs and upper limbs; Nt.ProBNP: N-terminal pro-B-type natriuretic peptide; PND: polyneuropathy disability score; QOL: quality of life; RODS questionnaire: Rasch‐built overall disability scale questionnaire; SNF SIQ: small fibre neuropathy symptom inventory questionnaire.
Table 6Neurological assessment in ATTRv neuropathy.
| Evaluation | Purpose | |
| Sensory motor neuropathy | SFN SIQ questionnaire | Small fibre loss |
| Nerve conduction study | Composite nerve conduction score | |
| NIS-LL and -UL | Small and large fibre loss | |
| Examination for pain and thermal sensory loss | Small fibre loss | |
| Disability | RODS questionnaire | Overall disability |
| Locomotion | PND Score | Autonomy to walk |
| 6-min walking test | Endurance | |
| Autonomic neuropathy | COMPASS-31 questionnaire | Overall dysfunction |
| Sudoscan | Sweat gland denervation | |
| Schellong Test | Orthostatic dysfunction | |
| Quality of life | Norfolk QOL | Overall QOL |
ATTRv: variant transthyretin amyloidosis; COMPASS score: composite autonomic symptom score; NIS-LL and -UL: neuropathy impairment score in the lower limbs and upper limbs; PND score: polyneuropathy disability score; QOL: quality of life; RODS questionnaire: Rasch‐built overall disability scale questionnaire; SNF SIQ: small fibre neuropathy symptom inventory questionnaire.
Nerve conduction study
A nerve conduction study is mandatory to evaluate peripheral neuropathy, usually demonstrating symmetrical axonal sensorimotor polyneuropathy in patients with ATTRv amyloidosis [56, 57]. Of note, if done early in the disease course, the nerve conduction study can be normal since up to 5% of patients with ATTR amyloidosis have involvement of only unmyelinated C-fibres [58]. The nerve conduction study can also show demyelinating features with slowing of conduction velocity and prolonged distal motor latencies leading to a misdiagnosis of chronic inflammatory demyelinating polyneuropathy. The composite nerve conduction score should be calculated at baseline and follow-up [59]. Autonomic function can be assessed with sympathetic skin response and R-R interval.
Small fibre assessment
Detection of small fibre involvement starts with specific questionnaires (SFN S1Q, see table 5) and clinical examination, looking for pain and loss of sensation of temperature. Clinical examination can be completed with quantitative tests (Sudoscan® for sudomotor function, quantitative sensory testing), when available. The gold standard for diagnosing small fibre neuropathy is quantification of intra-epidermal nerve fibres in a skin biopsy (70–90% sensitivity and specificity) [60, 61]. However, skin biopsy is usually not necessary to diagnose small fibre neuropathy in patients with ATTR amyloidosis, unless it is also used for amyloid detection and characterisation (this approach requires expertise and is only possible in few centres).
A minimal cardiac baseline assessment should include blood pressure, Schellong test, electrocardiography (ECG), 24-hour ECG, exercise test and/or 6-minute walking test, transthoracic echocardiography (TTE) including speckle-tracking echocardiography based left ventricular deformation analysis (global longitudinal strain), and/or cardiac magnetic resonance imaging (CMR) [62, 63]. The indication for bone scintigraphy is discussed in detail in section 3.1. In general, ATTR amyloidosis with cardiac involvement can be assessed without the need of biopsies, with scintigraphy using bone-avid radiotracers. Evidence-based data demonstrate that bone scintigraphy using technetium-labelled radiotracers provides very high diagnostic accuracy in the non-invasive assessment of cardiac ATTR [64]. However, the sensitivity of bone scintigraphy may be lower in ATTRv amyloidosis than in ATTRwt amyloidosis, depending on the type of mutation [65–68]. As recently reported and for reasons still unknown, patients with early-onset p.V50M and the p.F84L (also known as “F64L”) variant do not show cardiac tracer uptake [69, 70].
SAN recommends cardiac and neurological staging of all patients with ATTRv amyloidosis [I, B].
Cardiac biomarkers, estimated glomerular filtration rate and functional class according to the New York Heart Association (NYHA) have been extensively studied to define prognosis in cardiac ATTR amyloidosis [71–74] (tables 7 and 8).
Table 7Clinical stages of cardiac ATTR.
| eGFR (ml/min/1.73 m 2 ) | NT-pro BNP (ng/l) | |
| Stage I | ≥45 | <3000 |
| Stage II | ≥45 | >3000 |
| <45 | <3000 | |
| Stage III | <45 | >3000 |
ATTR: transthyretin amyloidosis; eGFR: estimated glomerular filtration rate: NT-pro BNP: N-terminal pro-B-type natriuretic peptide
Table 8New York Heart Association (NYHA) functional classification.
| Stage | Symptoms |
| I | No limitations of physical activity. Normal physical activity does not cause symptoms of heart failure. |
| II | Slight limitations of physical activity. Comfortable at rest, but normal physical activity leads to symptoms of heart failure. |
| III | Moderate limitations of physical activity. Comfortable at rest, but less than normal physical activity leads to symptoms or heart failure. |
| IV | Unable to carry out any physical activity without symptoms of heart failure, or symptoms of heart failure at rest. |
The neurological stage should be determined according to the transthyretin familial amyloid polyneuropathy (TTR-FAP; Coutinho) staging system and the peripheral neuropathy disability (PND) score (table 9) [75, 76].
Table 9TTR-FAP stage and peripheral neuropathy disability (PND) score.
| TTR-FAP stage | Symptoms | PND |
| Stage 0 | Asymptomatic | - |
| Stage I | Mild, ambulatory, symptoms at lower limbs limited | I sensory disturbance (LL/UL), preserved walking capacity |
| II walking difficulties but no need for walking aid | ||
| Stage II | Moderate, further neuropathic deterioration, ambulatory but requires assistance | IIIa One stick or one crutch required for walking |
| IIIb Two sticks or two crutches required for walking | ||
| Stage III | Severe, bedridden, bound to wheelchair, generalized weakness | IV confined to wheelchair or bed |
LL: lower limbs; TTR-FAP: transthyretin familial amyloid polyneuropathy; UL: upper limbs
SAN recommends disease-modifying treatment according to current drug approval status in Switzerland [I, B].
ATTRv amyloidosis with neurological manifestations: Patisiran (Onpattro ® ) monotherapy has received marketing authorisation, and is approved for the treatment of symptomatic, primarily neurological patients with hereditary transthyretin-mediated amyloidosis (ATTRv) with a PND stage ≥I and ≤IIIb, or a TTR-FAP stage >0 and ≤2, and with an initial NIS-LL between 5 and 130. One of the two designated Swiss ATTRv treatment centres (CHUV, Lausanne and University Hospital Zurich) must apply for reimbursement, initiate and follow up treatment for all Swiss patients. Patients are required to have a life expectancy of >2 years and NYHA functional class II or lower. Patients have to be included in a designated registry. Reimbursement of patisiran for treatment initiation and continuation is regulated in detail by the Federal Office of Public Health (FOPH, list of specialties).
Exclusively cardiac ATTRv amyloidosis (not qualifying for patisiran): Tafamidis (Vyndaqel ® ) has received marketing authorisation in Switzerland for the treatment of transthyretin amyloidosis in adult patients with wild-type or hereditary cardiomyopathy, but is not yet reimbursed. A reimbursement request has to be approved by the healthcare insurance prior to treatment initiation. The requests are currently evaluated according to article 71b KVV.
Liver transplantation should be considered as a second-line treatment option for patients with early-onset ATTRv amyloidosis with a primarily neurological phenotype and RNAi (patisiran, Onpattro ® ) treatment failure or intolerance [IIa, C].
Heart transplantation should be considered as an option for younger patients with predominant and advanced cardiac involvement not responding to disease-modifying drugs, or for whom disease-modifying drugs are not available or unlikely to be effective [IIa, C].
The aim of a disease-modifying treatment is to halt protein aggregation and amyloid formation and to prevent further tissue damage (reducing the concentration of the circulating precursor protein, stabilising its native structure or interfering with the aggregation of misfolded prefibrillar forms) (fig. 3). Reabsorption of existing amyloid deposits is another potential therapeutic approach [77]. Immunotherapy has been investigated in the past decade using an anti-SAP (anti-serum amyloid P) antibody that targets a common constituent of all amyloid deposits. However, despite encouraging preliminary results, this approach has not completed phase III clinical development [78]. Doxycycline is currently the only investigational drug potentially active on different types of amyloidosis based on its in vitro and in vivo efficacy to disrupt fibrils of various type [79, 80].
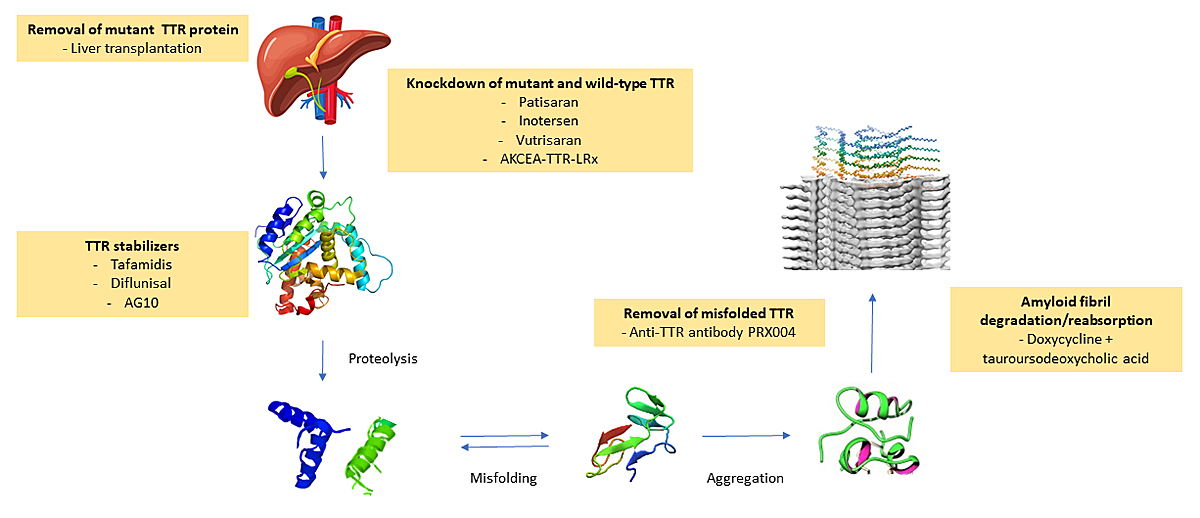
Figure 3 Current and investigational therapeutic strategies for transthyretin amyloidosis. Therapeutic interventions target different steps in the process of transthyretin amyloid formation.
During the last decade, different phase III randomised trials reported the efficacy and safety of treatments preventing or delaying ATTR amyloidosis progression. As a result, patients’ quality of life and survival have significantly improved [81–85].
Treatments targeting different steps of the amyloidogenic cascade have been developed (fig. 3). The gene-silencing agents patisiran and inotersen suppress the synthesis of hepatic TTR [81, 82], whereas small-molecule ligands, such as tafamidis, stabilise the TTR tetramer, preventing dissociation into amyloidogenic monomeric forms [84]. Although not registered for this indication, the clinical efficacy of diflunisal as a TTR stabilszer has been demonstrated in a phase III study [83].
Liver transplantation was the first effective treatment for ATTRv amyloidosis. It halts the production of variant TTR, with production of wild-type TTR by the donor liver. Liver transplantation was demonstrated to stop progression of the neuropathy after a mean follow-up of 4 years in patients with p.V50M at an early stage [86]. Worldwide, more than 2000 patients with ATTRv amyloidosis underwent liver transplantation [87]. Long-term follow-up data reported doubling of the survival time for patients with early-onset ATTR p.V50M amyloidosis at initial stages; the impact of on survival in late-onset p.V50M or other variants is not clear, even when combined liver-heart transplantation is considered [88].
Nevertheless, ATTR amyloidosis can progress after liver transplantation, as wild-type TTR tends to accumulate on pre-existing amyloid deposits [89, 90], and because the production of mutant TTR in the choroid plexus ependymal cells of the brain and retinal epithelium in the eye persists. Long-term manifestations in transplanted patients include: progressive ocular damage leading to glaucoma and visual loss and central nervous system manifestations typically consisting of transient ischaemic attack (TIA)-like episodes, stroke or seizures due to extensive cerebral amyloid angiopathy [91]. Patients with ATTRv amyloidosis and ultimately die from progressive neuropathy, cardiac amyloidosis or stroke [92].
Tafamidis
Tafamidis meglumine, an analogue of thyroxine, can stabilise both wild-type and fibrillogenic variants of TTR, inhibiting tetramer dissociation, which is the rate-limiting step in the amyloidogenic process [84]. This effect slows ATTR amyloidosis progression. Tafamidis is administered orally. Its safety and efficacy in slowing polyneuropathy progression in early-stage p.V50M were demonstrated in an 18-month randomised, placebo-controlled trial followed by two open-label extension studies [84]. Based on these results, the drug was approved in Europe, Japan, Israel and several countries in Latin America, but not in Switzerland, for patients with isolated ATTR polyneuropathy. The results of long-term treatment indicate a sustained delay in neurological deterioration, with earlier initiation associated with better outcomes [84, 93, 94]. Real-world data support a survival benefit in both early-onset and late-onset p.V50M compared with historical controls and liver transplantation patients [33].
Patients with cardiac ATTR amyloidosis were studied in the randomised controlled ATTR-ACT trial, with 75% of patients having ATTRwt amyloidosis and 25% having ATTRv amyloidosis [95]. In this trial tafamidis 80 mg, tafamidis 20 mg or placebo were studied in a 2:1:2 ratio for 30 months. The safety profile of tafamidis was similar to placebo. The study was not powered to detect a possible treatment effect in the subgroup of patients with ATTRv amyloidosis, for whom an inferior outcome has been reported [96]. Overall, the ATTR-ACT showed a benefit for tafamidis-treated vs placebo-treated patients regarding a hierarchically assessed primary endpoint of all-cause mortality, followed by frequency of cardiovascular-related hospitalisations [95]. Based on these results, tafamidis free acid was recently approved by the US Food and Drug Administration (FDA), and the European Medicines Agency (EMA) for the treatment of cardiac ATTRwt amyloidosis and cardiac ATTRv amyloidosis, at the dose 61 mg/day, according to bioequivalence with tafamidis meglumine 80 mg [97]. Tafamidis has received marketing authorisation for cardiac ATTR amyloidosis, but is not yet reimbursed, in Switzerland.
TTR gene silencing
The gene silencing strategy halts the hepatic production of TTR either by means of RNA interference (RNAi) or with antisense oligonucleotides.
Patisiran, an RNAi agent, lowers the production of mutant and wild-type TTR by targeting the 3′ untranslated region of TTR mRNA. In the APOLLO trial, 225 patients with polyneuropathy (stage I or II) were randomised to receive patisiran at 0.3 mg/kg i.v. every 3 weeks or placebo for 18 months [81]. The mean modified neuropathy impairment score (NIS) + 7 (mNIS+7), improved in patisiran-treated patients compared with baseline by –6 points versus progression by +28 points in the placebo arm. Up to 56% of patients improved their mNIS+7 score compared with baseline and 51% their QoL [98]. Improvement in total COMPASS-31 score and in individual domains orthostatic intolerance and gastrointestinal symptoms was observed [99]. Most frequent AEs were infusion related reactions or oedema, with an overall good safety profile. In the prespecified cardiac subpopulation, patisiran decreased mean left ventricular wall thickness, global longitudinal strain and NT-proBNP levels compared with placebo, suggesting a beneficial effect on cardiac involvement [100].
Inotersen is a second-generation antisense oligonucleotide that degrades both mutant and wild-type TTR mRNA, decreasing median serum TTR concentration by ̴ ~75%. Its safety and efficacy were demonstrated in a multicentre, randomised trial, in which patients with ATTRv polyneuropathy (stage I and II) received either inotersen 300 mg/week s.c. injection or placebo for 15 months [82]. Inotersen significantly improved the two co-primary endpoints, progression of mNIS+7 and Norfolk-QOL, compared with placebo at 15 months [101]. A rare risk for thrombocytopenia and glomerulonephritis was reported.
Both inotersen and patisiran obtained marketing authorisation by the EMA and FDA in ATTRv amyloidosis with polyneuropathy stage I and II. Only patisiran obtained marketing authorisation by Swissmedic and is reimbursed based on detailed criteria defined by the Swiss FOPH. Vitamin A supplementation is needed to prevent potential risks of eye damage due to drug-related reduced levels of the vitamin.
Diflunisal
Diflunisal is a nonsteroidal anti-inflammatory drug which binds within the two thyroxine binding sites of TTR [102]. In a phase III, randomised, placebo-controlled trial of patients with ATTRv polyneuropathy resulting from a diverse number of mutations, diflunisal (250 mg orally twice daily) was well tolerated [83]. Diflunisal is not available in Switzerland, and the Cantonal Pharmacy has to be informed prior to its use. Patients who agree to a "compassionate use" of diflunisal, can order the drug via an international pharmacy.
Doxycycline
The combination of doxycycline and tauroursodeoxycholic acid was evaluated in a phase II trial enrolling 20 patients (3 patients having ATTRwt amyloidosis). No progression of cardiac or neurological involvement was noted [103, 104].
Gene editing and/or seeding inhibitors
CRISPR/Cas9 technology is in pre-clinical development to silence expression of TTR, as are TTR fibril capping agents [105].
Supportive treatment is extremely important in the management of all patients with amyloidosis and is discussed in more detail in the chapter on ATTRwt amyloidosis (see section 3.4.2.).
Clinical and biological follow-up in specialised centres every 3–6 months is recommended, including a comprehensive follow-up with disability and QOL questionnaires, and a standardised cardiological and neurological work-up (table 5) [I, C]. A standardised ophthalmological assessment should be performed every 1–2 years [I, C]. SAN recommends that the lead for patient follow-up should be cardiological for patients with predominantly cardiac manifestations and neurological for patients with predominantly neurological manifestations [I, C]. In Switzerland, for regulatory reasons, patients treated with patisiran must have a yearly assessment in a reference centre for amyloidosis (CHUV, Lausanne / University Hospital Zurich) to evaluate treatment continuation.
Patients with confirmed diagnoses should be regularly monitored for disease progression by an interdisciplinary team. Self-administered questionnaires are important outcome measures. Assessments should be scheduled every 3–6 months in patients with advanced-stage cardiac or neurological disease, and every 6 months in patients with early-stage disease.
ATTRwt amyloidosis is a disease of the elderly, with a median age at diagnosis of 74 years. It is rarely diagnosed in the 40s–60s [106, 107]. Cardiac involvement by ATTRwt amyloidosis is mainly responsible for the clinical presentation, but soft tissue involvement is also common, manifesting as bilateral carpal tunnel syndrome, stenosis of the lumbar spine, and/or spontaneous biceps tendon rupture [24, 105, 108–110]. Neuropathy can be detected in up to 10% of patients, though few cases with significant peripheral and/or autonomic neuropathy are reported [107].
Endomyocardial biopsy is the gold standard for cardiac ATTR amyloidosis diagnosis and is nearly 100% sensitive and specific if biopsy specimens are collected from multiple sites (at least four are recommended) and tested for amyloid deposits by Congo red staining [111]. Methods for the identification of the precursor protein are immunohistochemistry (in experienced pathology laboratories) or laser dissection, and tandem mass spectrometry analysis [112]. Endomyocardial biopsy specimens are usually obtained via a femoral or internal jugular venous approach from the right ventricular septum, which has the advantage of posing a low risk of cardiac perforation. The overall risk of major complications is relatively low in experienced hands (~1%) [113].
In the absence of monoclonal gammopathy, scintigraphy using bone-avid radiotracers can accurately diagnose ATTR amyloidosis with cardiac involvement, without the need for biopsies. Three technetium-labeled radiotracers have been evaluated for ATTR amyloidosis identification. These include 99mTc-pyrophosphate (available in the United States), 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) or 99mTc hydroxymethylenediphosphonate (HMDP), [113], the latter two being available in Europe.
The mechanisms of myocardial retention of these bone-avid tracers are not fully understood, but have been linked to the presence of microcalcifications that are more common in ATTR amyloidosis than in AL amyloidosis [114, 115]. The results of an international collaboration with a large cohort of endomyocardial biopsy-proven cases of ATTRwt amyloidosis with cardiac involvement report a 100% specificity for ATTRwt amyloidosis when semi-quantitative Perugini grade 2 or 3 uptake of these tracers (fig. 4) was seen in the absence of a monoclonal gammopathy by serum and urine testing in patients with heart failure and echocardiographic or magnetic resonance findings typical of amyloidosis [116].
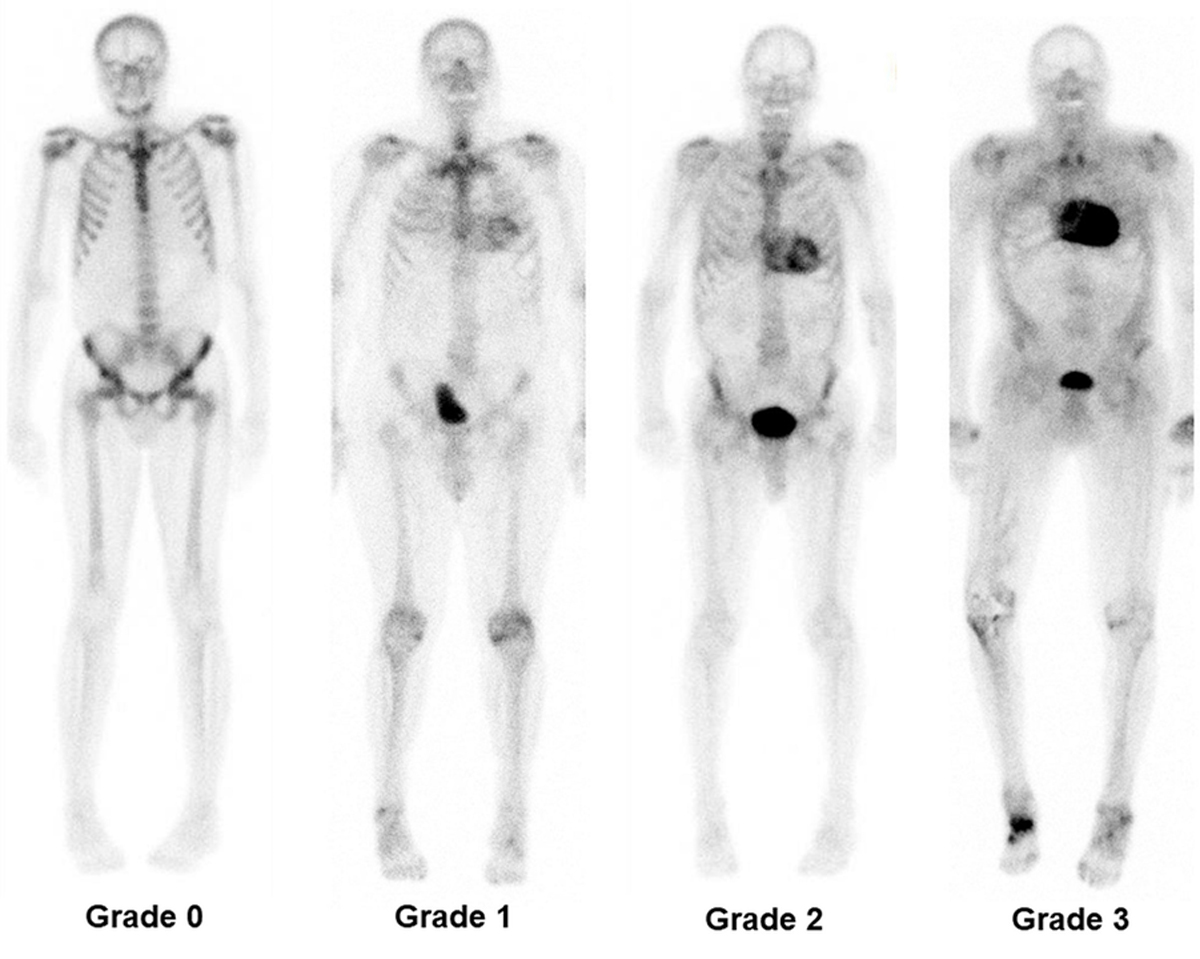
Figure 4 Tc-DPD myocardial scintigraphy whole-body planar images. From left to right grade 0, grade 1, grade 2 and grade 3 myocardial uptake are shown. (Image courtesy PD Dr. Aju P. Pazhenkottil, Cardiac Imaging, Department of Nuclear Medicine, University Hospital of Zurich, Switzerland).
A non-biopsy diagnostic approach is recognised as part of the diagnostic algorithm for patients with suspicted of ATTRwt amyloidosis. Scintigraphy alone cannot ensure a diagnosis with 100% specificity in patients with a concurrent monoclonal gammopathy (monoclonal immunoglobulin and/or free monoclonal immunoglobulin light chain) (fig. 5) [117].
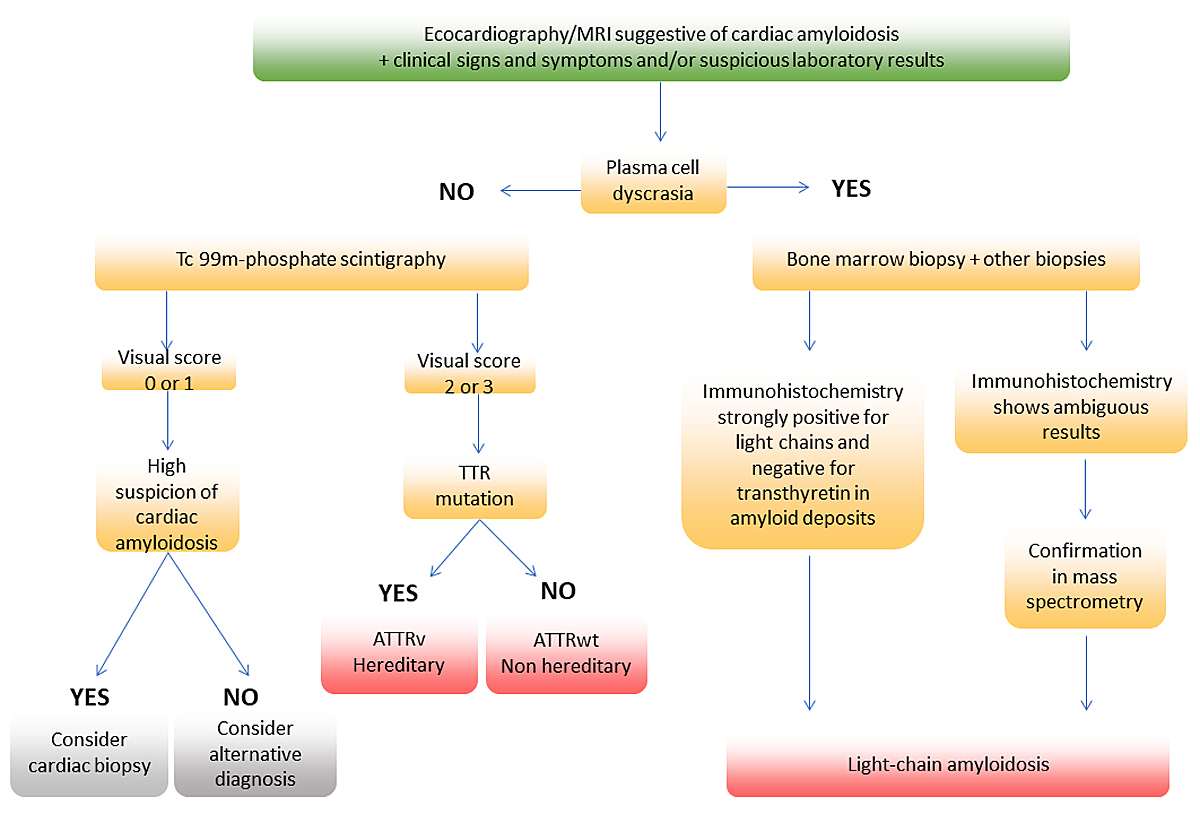
Figure 5 Simplified diagnostic algorithm for patients with suspected cardiac amyloidosis.
The diagnosis of ATTRwt amyloidosis should be suspected in older persons with HFpEF, or in patients with otherwise unexplained elevated troponin levels and/or high levels of NT-proBNP. ATTRwt amyloidosis should also be excluded in patients with left ventricular hypertrophy and a concomitant atrioventricular block [6]. Personal history of previous lumbar spinal stenosis, carpal tunnel syndrome and spontaneous biceps tendon rupture may also raise suspicions of ATTRwt amyloidosis (see above).
In contrast, a family history of progressive neuropathy and heart failure at early stage should raise suspicion of ATTRv amyloidosis.
Interdisciplinary discussion of the results obtained during the screening for monoclonal gammopathy with specialists in haemato-oncology should be considered [ IIa, C].
The following screening panel for monoclonal gammopathy should be considered: serum protein electrophoresis, serum immunofixation, serum free light chain measurement and urine immunofixation [IIa, B].
Elevation of serum troponin and NT-proBNP levels are often seen in ATTRwt amyloidosis, even in the absence of a severe cardiomyopathy or symptoms of heart failure [118]. An initial blood test should include complete blood count, creatinine, creatinine clearance, eGFR, liver values including alkaline phosphatase, NT-proBNP, troponin T, microalbuminuria, proteinuria, serum free light chains, and serum and urine protein electrophoresis with immunofixation. The evaluation of the presence of a monoclonal protein should be done by physicians with experience in the field.
If a monoclonal protein is detected, assessments should include amyloid typing on a tissue biopsy from a clinically affected organ endomyocardial biopsy if the heart is clinically affected], or the abdominal fat pad, the rectum and the salivary gland as surrogate organs.
TTR genetic testing should be considered in all patients with cardiac ATTR amyloidosis regardless of their age [IIa, C]. The results have significant implications for family members at risk (see section 2.2.3).
A visit to a neurologist may be considered at baseline for patients with ATTRwt amyloidosis and neuropathy [IIb, C].
A cardiological work-up is recommended at baseline, including ECG, 24-hour ECG, blood pressure and a TTE. The need for bone scintigraphy, cardiac MRI and myocardial biopsy should be discussed by an interdisciplinary team on a case-to-case basis [I, B].
Electrocardiography
A discrepancy between left ventricular wall thickness and QRS voltages (low voltage) on a 12-lead ECG is commonly seen in cardiac amyloidosis [119], progressing to low-voltage and a pseudo-infarction pattern at later disease stages [120]. Atrioventricular block is another suggestive feature of ATTRwt amyloidosis and may be seen in up to 20% of patients with cardiac amyloidosis [121]. Cardiac conduction abnormalities may be the first manifestation of ATTRwt amyloidosis, and amyloid infiltration of the sinus and atrioventricular nodes may prompt pacemaker implantation.
Echocardiography
Echocardiography is the most important tool for the diagnosis of cardiac amyloidosis. Amyloid deposits lead to a progressive thickening of the left- and right ventricular walls. This thickening typically presents with an increased echogenicity and a granular sparkling appearance [122]. Amyloid deposits may also be seen as a thickening of the atrioventricular valves and of the inter-atrial septum. During early stages of disease, systolic function is preserved in most patients, but progressive diastolic dysfunction is observed. At later stages, severe diastolic dysfunction with a restrictive filling pattern is typically associated with dilated atria due to the chronically elevated left-sided filling pressures [122]. Systolic dysfunction is a sign of advanced disease.
Characteristic of amyloidosis is an impaired global longitudinal peak strain in speckle tracking analysis, with a gradient from the apex to the base – with almost normal function at the apex (apical sparing) [123]. Pericardial effusion is present in most cases.
Endomyocardial biopsy
Endomyocardial biopsy is the gold standard for diagnosing cardiac amyloidosis but is an invasive procedure and not always required to establish the diagnosis. See figure 5 for a non-biopsy diagnosis approach. The biopsy must be assessed by a pathologist with experience in the field.
Cardiac MRI
CMR is highly sensitive and specific for the assessment of cardiac amyloidosis. CMR offers the gold standard for structural and functional assessment combined with tissue characterisation using T1 mapping and late gadolinium enhancement.The sensitivity (80–100%) and specificity (8094%) are high for the detection of amyloidosis in patients with suspected infiltrative disease [62, 124]. Late gadolinium enhancement and quantitative parametric mapping techniques such as prolonged T1 mapping times and extracellular volume measurements are used for diagnosis and prognosis assessment [125127].
Bone scintigraphy
Radionuclide imaging plays a distinctive role in the non-invasive diagnosis of cardiac amyloidosis. High sensitivity and specificity for ATTR amyloid deposits has been proven for 99mTc-DPD and 99mTc-HMDP tracers. A Perugini score of 2 or more, indicating a cardiac uptake equal to or greater than rib uptake, is a widely accepted criterion for defining the presence of cardiac TTR amyloid deposits, in the absence of a monoclonal gammopathy (see above) [68, 114, 128]. Cardiac scintigraphy may be helpful for early identification of affected individuals before the occurrence of cardiac involvement assessed by other diagnostic techniques (increased wall thickness) [129–132].
This technique is currently extensively applied; however, important limitations have to be taken into account [133].
Cardiac positron emission tomography
Cardiac poisitron emission tomography (PET) using the amyloid tracers 18F-florbetapir, 18F-florbetaben, or 11C-Pittsburgh B compound, is accurate in diagnosing cardiac involvement with a target to background (left ventricular myocardium to blood pool) ratio >1.5 or a retention index >0.030/min being typical features [134].
The cost of using of amyloid PET tracers for the diagnosis of cardiac amyloidosis is not currently covered by health insurance in Switzerland and a general diagnostic strategy using these is therefore not recommended by the SAN consensus.
The abdominal fat pad aspirate has a low sensitivity for ATTRwt amyloidosis (12–15%) [50, 135]. If biopsy proof of ATTR is required, but endomyocardial biopsy is not feasible, biopsies from surrogate sites such as the rectum or the salivary glands should be obtained.
Initial staging of all patients with ATTRwt amyloidosis should be considered [IIa, B].
Gilmore et al. proposed a staging system that is applicable to both ATTRwt and ATTRv cardiac amyloidosis, using two simple and universally measured serum biomarkers: NT-pro BNP and eGFR (see table 7) [71]. The staging system discriminates between the ~20% patients with cardiac ATTR amyloidosis who have a median survival of approximately 2 years, the ~40% who have a median survival of about 4 years, and the ~40% who have a median survival of around 6 years [71]. Initial risk stratification should also include functional NYHA class (table 8), which seems to be predictive for treatment response [95]. Two other commonly used staging systems are based on the cardiac markers NT-proBNP and troponin T, or diuretic dose and NYHA functional class, respectively [73, 136].
SAN recommends treatment guided by experienced centres, using a multidisciplinary approach (cardiology, haemato-oncology, neurology, nephrology, gastroenterology) [I, C].
Treatment with tafamidis 61 mg should be considered for patients with cardiac ATTRwt amyloidosis and dyspnoea NYHA class I–III [IIa, B].
Case-by-case discussions should be considered prior to tafamidis initiation for patients with significant comorbidities that interfere with intermediate-term survival [IIa, C].
Case-by-case discussions should be considered for patients with complex cardiac situations (e.g., concomitant aortic stenosis, concomitant severe coronary artery disease), patients with a formal indication for implantation of an internal cardioverter defibrillator, and patients with typical angina due to microvascular disease [IIa, C].
The optimal timing to start treatment for ATTRwt amyloidosis remains unclear, though it is known that there is a long latency period before organ damage manifests. Patients treated at earlier stages of the disease are reported to have a greater survival benefit [95].
Cardiac disease monitoring relies on NYHA class, laboratory testing (cardiac biomarkers NT-proBNP, BNP and troponin), functional testing (6-minute walking test or stress test) and cardiac imaging (echocardiography, CMR).
Tafamidis
The ATTR-ACT phaseIII clinical trial assessed tafamidis 20 mg versus tafamidis 80 mg versus placebo in wild-type and hereditary ATTR cardiomyopathy [95]. A total of 441 patients were enrolled for 30 months. Primary outcome measurement was a combined endpoint of all-cause mortality and cardiovascular-related hospitalisations [95]. Tafamidis was associated with lower all-cause mortality and a lower rate of cardiovascular-related hospitalisations, when compared with placebo. As anticipated above, this benefit was observed mainly in the subgroup of patients with NYHA class I and II, but not in the subgroup of patients with NYHA class III. The safety profile of tafamidis was similar to placebo. Based on these results, tafamidis free acid was approved by the FDA and EMA for the treatment of cardiac ATTRwt amyloidosis and cardiac ATTRv amyloidosis at the dose 61 mg/day, according to bioequivalence with tafamidis meglumine 80 mg [97]. Tafamidis free acid has received marketing authorisation in Switzerland for the treatment of transthyretin amyloidosis in adult patients with wild-type or hereditary cardiomyopathy, but is not yet reimbursed. A reimbursement request has to be approved by the healthcare insurance prior to treatment initiation. The requests are currently evaluated according to article 71b KVV.
TTR gene silencing
Patisiran and inotersen have not yet been extensively studied in patients with ATTRwt amyloidosis. The effect of patisiran in patients with cardiac ATTR amyloidosis including both wild-type and variant subtypes is being evaluated in an ongoing phase III trial (APOLLO-B, NCT03997383). Accrual has been completed and first results are expected in mid-2022. Vutrisiran, a subcutaneously administered investigational RNAi therapeutic, is currently being studied in a large phase III trial (HELIOS-B, NCT04153149) [137].
Doxycycline
The combination of doxycycline and tauroursodeoxycholic acid was evaluated in a phase II trial enrolling 20 patients (3 patients having ATTRwt amyloidosis) [103, 104]. No progression of cardiac or neurological involvement was reported. The combination of doxycycline and tauroursodeoxycholic acid is currently being studied in a single-centre phase III trial (NCT03481972).
Diflunisal
The experience with diflunisal in ATTR cardiomyopathy has been limited to open-label, single-centre studies. Diflunisal (250 mg orally twice daily) was generally well tolerated, with adverse effects in a minority of subjects, including thrombocytopenia and renal dysfunction. Diflunisal was associated with a survival benefit similar to tafamidis in one nonrandomised ATTRwt amyloidosis study [138]. Despite its application in patients with concomitant use of oral anticoagulants, significant bleeding has not been reported, but patients were highly selected in this study. The lower toxicity could be related to the reduced dosage of diflunisal administered for TTR stabilisation, which is lower than the dose indicated as an anti-inflammatory drug [139]. Diflunisal can be considered for selected patients with ATTRwt amyloidosis without severe renal dysfunction (eGFR >45 ml/min/1.73 m2), with normal platelet function and count, no volume overload and without recent haemodynamic instability. A proton pump inhibitor might be added and kidney function should be regularly monitored. Diflunisal is not available in Switzerland, and the Cantonal Pharmacy has to be informed prior to its use. Patients who agree to a "compassionate use" of diflunisal can order the drug via an international pharmacy.
The key objective of ATTRwt amyloidosis supportive management is to maintain a stable fluid balance. To achieve euvolaemia, sodium and fluid restriction and personalised diuretic therapy are central for patients with symptomatic HFpEF. On the other hand, excessive diuretic treatment can cause symptomatic hypotension and worsening of renal perfusion. Due to their bioavailability, loop diuretics (torasemide and furosemide) are favoured, and may be combined with aldosterone receptor antagonists or thiazide diuretics. Serum potassium levels should be monitored regularly [140).
When adjusting antihypertensive treatment, clinicians should be aware that calcium channel blockers (verapamil type only) can cause high degree heart block and beta-blockers may not be well tolerated (because of a fixed stroke volume – cardiac output depends solely on heart rate) [141].
Compression stockings and midodrine may be useful in advanced stages with arterial hypotension, when cardiorenal syndrome can also occur [141].
In atrial fibrillation, tachycardia and bradycardia may not be well tolerated. Data on the outcome of catheter ablation in ATTR amyloidosis are limited. Antiarrhythmic treatment (amiodarone) and cardioversion can be offered once intracardiac thrombi have been excluded, independent of the presence of an active and therapeutic anticoagulation [141, 142]. In the case of atrial fibrillation, long-term anticoagulation is recommended regardless of the CHADs-VASc score [141].
Patients with syncope, pre-syncope or palpitations should undergo a 24-hour ECG assessment. A 24-hour ECG should be recorded every 6–12 months to screen for atrial fibrillation and conduction disturbances. Indications for permanent pacing should follow the European Society of Cardiology guidelines [143].
For gastric and bowel disease, symptomatic treatment should follow that of a gastroparesis of other causes (hygienic measures such as smoking cessation, avoidance of alcohol and sparkling drinks, meal splitting). Metoclopramide, domperidone and erythromycin can be administered in the case of no response. To limit constipation, a fibre-rich diet and osmotic laxatives can be prescribed [144, 145].
Physiotherapy can be beneficial for patients with cardiac ATTR amyloidosis, especially for patients with concomitant gait disorder (stenosis of the lumbar spine) or patients at high risk for falls.
Although heart transplantation is not a viable option for most patients with ATTR cardiopathy because of advanced age, younger patients with predominantly cardiac involvement should be referred to a transplant centre in good time. In the current International Society of Heart/Lung Transplantation guidelines, heart transplant in cardiac amyloidosis may be considered on an individual basis after multidisciplinary assessment [146, 147]. Outcomes in carefully selected patients are similar to those of patients transplanted for other causes of heart failure [148].
Cardiological follow-up should be considered every 3–6 months, including clinical and laboratory evaluation (NT-proBNP, troponin T, creatinine, proteinuria, albuminuria), and a comprehensive evaluation including ECG, 24-hour ECG, TTE every 6–12 months, ergometry, depending on disease severity and treatment [IIa, C]. Regular blood pressure and body weight home monitoring may be considered in order to adapt diuretic dose targeting euvolaemia [IIb, C].
The increasing availability of effective and early treatments led to the promotion of pre-symptomatic testing for hereditary TTR amyloidosis [52–55] (see section 2.2.3). The screening of asymptomatic carriers of amyloidogenic mutations should be individualised based on patient preferences and the predicted age at disease onset. Based on the specific TTR mutation and the onset age in other affected family members, the predicted age at disease onset can be estimated. Systemic monitoring of asymptomatic carriers should be considered at least 10 years prior to the predicted age at disease onset [149, 150] [IIa, C].
For patients who received an organ from a liver-transplant donor with ATTRv amyloidosis, a baseline evaluation of cardiac function (ECG, TTE), renal function screening (eGFR, proteinuria), and a neurological assessment including laboratory testing for peripheral neuropathy and a nerve conduction study are mandatory. A 6-monthly or yearly follow-up for signs of neuropathy (including BMI, autonomic dysfunction) should be considered [IIa, B]. Prospective data on the development of de novo neuropathy in domino liver recipients related to mutant TTR report a rate of 25% [151]. In this setting, polyneuropathy can develop within years after liver tarnsplantation and is a rapidly progressive disease. Advanced age of the recipient is a known risk factor.
No guidelines are available regarding follow-up in this setting. A baseline evaluation for patients in whom ATTR has been detected in biopsies obtained from the yellow ligament, from muscle tendons or from the carpal tunnel, is recommended. A cardiac work-up including ECG, TTE (including speckle-tracking echocardiography based left ventricular strain analysis) and genetic testing for ATTRv amyloidosis should be considered [IIa, C]. The necessity for further investigations with bone scintigraphy and CMR should be discussed on a case-to-case basis. An ongoing Swiss study (NCT03966105), is investigating the prevalence of ATTR amyloidosis in patients undergoing surgery for carpal tunnel syndrome or stenosis of the lumbar spine.
All authors have completed and submitted the International Committee of Medical Journal Editors form for disclosure of potential conflicts of interest.
AC: unrestricted Pfizer research grant (ID#53938109); AJF: fees from Alnylam and Pfizer relevant for this article, fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Fresenius, Imedos Systems, Mundipharma, Novartis, Orion Pharma, Roche, Schwabe Pharma, Vifor, and Zoll, unrelated to this article; BG: fees from Alnylam, Pfizer and SOBI relevant for this article; HHJ: advisory boards of Alnylam pharmaceuticals; MT: Pfizer grant for hATTR research (ID#54435979), served on advisory boards for Alnylam, received speaker honoraria and travel grants from Alnylam; GST: consulting fees and travel grants from Alnylam; RS: unrestricted research grant from Pfizer and fees from Alnylam, Pfizer related to this article as well as fees from Mundipharma, Janssen, Takeda not relevant for this article; SD: Pfizer research grant on behalf of the Institition.
1. Benson MD , Buxbaum JN , Eisenberg DS , Merlini G , Saraiva MJ , Sekijima Y , et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020 Dec;27(4):217–22. https://doi.org/10.1080/13506129.2020.1835263
2. Merlini G , Dispenzieri A , Sanchorawala V , Schönland SO , Palladini G , Hawkins PN , et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018 Oct;4(1):38. https://doi.org/10.1038/s41572-018-0034-3
3. Adams D , Koike H , Slama M , Coelho T . Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019 Jul;15(7):387–404. https://doi.org/10.1038/s41582-019-0210-4
4. Mohamed-Salem L , Santos-Mateo JJ , Sanchez-Serna J , Hernández-Vicente Á , Reyes-Marle R , Castellón Sánchez MI , et al. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int J Cardiol. 2018 Nov;270:192–6. https://doi.org/10.1016/j.ijcard.2018.06.006
5. Longhi S , Guidalotti PL , Quarta CC , Gagliardi C , Milandri A , Lorenzini M , et al. Identification of TTR-related subclinical amyloidosis with 99mTc-DPD scintigraphy. JACC Cardiovasc Imaging. 2014 May;7(5):531–2. https://doi.org/10.1016/j.jcmg.2014.03.004
6. Maurer MS , Bokhari S , Damy T , Dorbala S , Drachman BM , Fontana M , et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019 Sep;12(9):e006075. https://doi.org/10.1161/CIRCHEARTFAILURE.119.006075
7. Winburn I , Ishii T , Sumikawa T , Togo K , Yasunaga H . Estimating the Prevalence of Transthyretin Amyloid Cardiomyopathy in a Large In-Hospital Database in Japan. Cardiol Ther. 2019 Dec;8(2):297–316. https://doi.org/10.1007/s40119-019-0142-5
8. Gilstrap LG , Dominici F , Wang Y , El-Sady MS , Singh A , Di Carli MF , et al. Epidemiology of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ Heart Fail. 2019 Jun;12(6):e005407. https://doi.org/10.1161/CIRCHEARTFAILURE.118.005407
9. Buxbaum JN . Transthyretin and the Transthyretin Amyloidoses. Protein Misfolding, Aggregation, and Conformational Diseases. Protein Reviews2007. p. 259-83.
10. Mangrolia P , Murphy RM . Retinol-Binding Protein Interferes with Transthyretin-Mediated β-Amyloid Aggregation Inhibition. Biochemistry. 2018 Aug;57(33):5029–40. https://doi.org/10.1021/acs.biochem.8b00517
11. Yee AW , Aldeghi M , Blakeley MP , Ostermann A , Mas PJ , Moulin M , et al. A molecular mechanism for transthyretin amyloidogenesis. Nat Commun. 2019 Feb;10(1):925. https://doi.org/10.1038/s41467-019-08609-z
12. Marcoux J , Mangione PP , Porcari R , Degiacomi MT , Verona G , Taylor GW , et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015 Oct;7(10):1337–49. https://doi.org/10.15252/emmm.201505357
13. Pepys MB . The Pentraxins 1975-2018: Serendipity, Diagnostics and Drugs. Front Immunol. 2018 Oct;9:2382. https://doi.org/10.3389/fimmu.2018.02382
14. Kisilevsky R . Theme and variations on a string of amyloid. Neurobiol Aging. 1989 Sep-Oct;10(5):499–500. https://doi.org/10.1016/0197-4580(89)90109-7
15. Bellotti V , Nuvolone M , Giorgetti S , Obici L , Palladini G , Russo P , et al. The workings of the amyloid diseases. Ann Med. 2007;39(3):200–7. https://doi.org/10.1080/07853890701206887
16. Schmidt HH , Waddington-Cruz M , Botteman MF , Carter JA , Chopra AS , Hopps M , et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018 May;57(5):829–37. https://doi.org/10.1002/mus.26034
17. Waddington-Cruz M , Schmidt H , Botteman MF , Carter JA , Stewart M , Hopps M , et al. Epidemiological and clinical characteristics of symptomatic hereditary transthyretin amyloid polyneuropathy: a global case series. Orphanet J Rare Dis. 2019 Feb;14(1):34. https://doi.org/10.1186/s13023-019-1000-1
18. Ueda M , Horibata Y , Shono M , Misumi Y , Oshima T , Su Y , et al. Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol. 2011 Dec;24(12):1533–44. https://doi.org/10.1038/modpathol.2011.117
19. Tanskanen M , Peuralinna T , Polvikoski T , Notkola IL , Sulkava R , Hardy J , et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–9. https://doi.org/10.1080/07853890701842988
20. Menter T , Haslbauer JD , Nienhold R , Savic S , Hopfer H , Deigendesch N , et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020 Aug;77(2):198–209. https://doi.org/10.1111/his.14134
21. Castaño A , Narotsky DL , Hamid N , Khalique OK , Morgenstern R , DeLuca A , et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017 Oct;38(38):2879–87. https://doi.org/10.1093/eurheartj/ehx350
22. González-López E , Gallego-Delgado M , Guzzo-Merello G , de Haro-Del Moral FJ , Cobo-Marcos M , Robles C , et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015 Oct;36(38):2585–94. https://doi.org/10.1093/eurheartj/ehv338
23. Nitsche C , Scully PR , Patel KP , Kammerlander AA , Koschutnik M , Dona C , et al. Prevalence and Outcomes of Concomitant Aortic Stenosis and Cardiac Amyloidosis. J Am Coll Cardiol. 2021 Jan;77(2):128–39. https://doi.org/10.1016/j.jacc.2020.11.006
24. Aus dem Siepen F , Hein S , Prestel S , Baumgärtner C , Schönland S , Hegenbart U , et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019 Dec;108(12):1324–30. https://doi.org/10.1007/s00392-019-01467-1
25. Sperry BW , Reyes BA , Ikram A , Donnelly JP , Phelan D , Jaber WA , et al. Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J Am Coll Cardiol. 2018 Oct;72(17):2040–50. https://doi.org/10.1016/j.jacc.2018.07.092
26. Eldhagen P , Berg S , Lund LH , Sorensson P , Suhr OB , Westermark P . Transthyretin amyloid deposits in lumbar spinal stenosis and assessment of signs of systemic amyloidosis. J Intern Med. 2020.
27. Adams D , Ando Y , Beirao JM , Coelho T , Gertz MA , Gillmore JD , et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2020. https://doi.org/10.1007/s00415-019-09688-0
28. Quarta CC , Buxbaum JN , Shah AM , Falk RH , Claggett B , Kitzman DW , et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015 Jan;372(1):21–9. https://doi.org/10.1056/NEJMoa1404852
29. Koike H , Misu K , Ikeda S , Ando Y , Nakazato M , Ando E , et al.; Study Group for Hereditary Neuropathy in Japan . Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002 Nov;59(11):1771–6. https://doi.org/10.1001/archneur.59.11.1771
30. Hörnsten R , Pennlert J , Wiklund U , Lindqvist P , Jensen SM , Suhr OB . Heart complications in familial transthyretin amyloidosis: impact of age and gender. Amyloid. 2010 Jun;17(2):63–8. https://doi.org/10.3109/13506129.2010.483114
31. Olsson M , Norgren N , Obayashi K , Plante-Bordeneuve V , Suhr OB , Cederquist K , et al. A possible role for miRNA silencing in disease phenotype variation in Swedish transthyretin V30M carriers. BMC Med Genet. 2010 Sep;11(1):130. https://doi.org/10.1186/1471-2350-11-130
32. Yamashita T , Ueda M , Misumi Y , Masuda T , Nomura T , Tasaki M , et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: experience from a single-referral center in Japan. J Neurol. 2018 Jan;265(1):134–40. https://doi.org/10.1007/s00415-017-8640-7
33. Coelho T , Inês M , Conceição I , Soares M , de Carvalho M , Costa J . Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018 Nov;91(21):e1999–2009. https://doi.org/10.1212/WNL.0000000000006543
34. Parman Y , Adams D , Obici L , Galán L , Guergueltcheva V , Suhr OB , et al.; European Network for TTR-FAP (ATTReuNET) . Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016 Feb;29 Suppl 1:S3–13. https://doi.org/10.1097/WCO.0000000000000288
35. Théaudin M , Lozeron P , Algalarrondo V , Lacroix C , Cauquil C , Labeyrie C , et al.; French FAP Network (CORNAMYL) Study Group . Upper limb onset of hereditary transthyretin amyloidosis is common in non-endemic areas. Eur J Neurol. 2019 Mar;26(3):497–e36. https://doi.org/10.1111/ene.13845
36. Lozeron P , Lacroix C , Theaudin M , Richer A , Gugenheim M , Adams D , et al. An amyotrophic lateral sclerosis-like syndrome revealing an amyloid polyneuropathy associated with a novel transthyretin mutation. Amyloid. 2013 Sep;20(3):188–92. https://doi.org/10.3109/13506129.2013.818535
37. Adams D , Lozeron P , Theaudin M , Mincheva Z , Cauquil C , Adam C , et al.; French Network for FAP . Regional difference and similarity of familial amyloidosis with polyneuropathy in France. Amyloid. 2012 Jun;19(sup1 Suppl 1):61–4. https://doi.org/10.3109/13506129.2012.685665
38. Mariani LL , Lozeron P , Théaudin M , Mincheva Z , Signate A , Ducot B , et al.; French Familial Amyloid Polyneuropathies Network (CORNAMYL) Study Group . Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015 Dec;78(6):901–16. https://doi.org/10.1002/ana.24519
39. Adams D , Coelho T , Obici L , Merlini G , Mincheva Z , Suanprasert N , et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015 Aug;85(8):675–82. https://doi.org/10.1212/WNL.0000000000001870
40. Cortese A , Vegezzi E , Lozza A , Alfonsi E , Montini A , Moglia A , et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017 May;88(5):457–8. https://doi.org/10.1136/jnnp-2016-315262
41. Lozeron P , Mariani LL , Dodet P , Beaudonnet G , Théaudin M , Adam C , et al. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology. 2018 Jul;91(2):e143–52. https://doi.org/10.1212/WNL.0000000000005777
42. Koike H , Katsuno M . Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019 Feb;7(1):E11. https://doi.org/10.3390/biomedicines7010011
43. Adams D , Théaudin M , Cauquil C , Algalarrondo V , Slama M . FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014 Mar;14(3):435. https://doi.org/10.1007/s11910-013-0435-3
44. Briani C , Cavallaro T , Ferrari S , Taioli F , Calamelli S , Verga L , et al. Sporadic transthyretin amyloidosis with a novel TTR gene mutation misdiagnosed as primary amyloidosis. J Neurol. 2012 Oct;259(10):2226–8. https://doi.org/10.1007/s00415-012-6529-z
45. Yakupova EI , Bobyleva LG , Vikhlyantsev IM , Bobylev AG . Congo Red and amyloids: history and relationship. Biosci Rep. 2019 Jan;39(1):BSR20181415. https://doi.org/10.1042/BSR20181415
46. El-Meanawy A , Mueller C , Iczkowski KA . Improving sensitivity of amyloid detection by Congo red stain by using polarizing microscope and avoiding pitfalls. Diagn Pathol. 2019 Jun;14(1):57. https://doi.org/10.1186/s13000-019-0822-4
47. Schwotzer R , Flammer AJ , Gerull S , Pabst T , Arosio P , Averaimo M , et al. Expert recommendation from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med Wkly. 2020 Dec;150:w20364.
48. de Paula Eduardo F , de Mello Bezinelli L , de Carvalho DL , Della-Guardia B , de Almeida MD , Marins LV , et al. Minor salivary gland biopsy for the diagnosis of familial amyloid polyneuropathy. Neurol Sci. 2017 Feb;38(2):311–8. https://doi.org/10.1007/s10072-016-2760-1
49. Gillmore JD , Maurer MS , Falk RH , Merlini G , Damy T , Dispenzieri A , et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016 Jun;133(24):2404–12. https://doi.org/10.1161/CIRCULATIONAHA.116.021612
50. Cohen OC , Sharpley F , Gilbertson JA , Wechalekar AD , Sachchithanantham S , Mahmood S , et al. The value of screening biopsies in light-chain (AL) and transthyretin (ATTR) amyloidosis. Eur J Haematol. 2020 Sep;105(3):352–6. https://doi.org/10.1111/ejh.13458
51. Rowczenio DM , Noor I , Gillmore JD , Lachmann HJ , Whelan C , Hawkins PN , et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014 Sep;35(9):E2403–12. https://doi.org/10.1002/humu.22619
52. Schmidt HH , Barroso F , González-Duarte A , Conceição I , Obici L , Keohane D , et al. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2016 Sep;54(3):353–60. https://doi.org/10.1002/mus.25210
53. Paneque M , Félix J , Mendes Á , Lemos C , Lêdo S , Silva J , et al. Twenty Years of a Pre-Symptomatic Testing Protocol for Late-Onset Neurological Diseases in Portugal. Acta Med Port. 2019 Apr;32(4):295–304. https://doi.org/10.20344/amp.10526
54. Leite  , Dinis MA , Sequeiros J , Paúl C . Illness representations, knowledge and motivation to perform presymptomatic testing for late-onset genetic diseases. Psychol Health Med. 2017 Feb;22(2):244–9. https://doi.org/10.1080/13548506.2016.1159704
55. Obici L , Kuks JB , Buades J , Adams D , Suhr OB , Coelho T , et al.; European Network for TTR-FAP (ATTReuNET) . Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016 Feb;29 Suppl 1:S27–35. https://doi.org/10.1097/WCO.0000000000000290
56. Castro J , Miranda B , Castro I , de Carvalho M , Conceição I . The diagnostic accuracy of Sudoscan in transthyretin familial amyloid polyneuropathy. Clin Neurophysiol. 2016 May;127(5):2222–7. https://doi.org/10.1016/j.clinph.2016.02.013
57. Cazzato D , Lauria G . Small fibre neuropathy. Curr Opin Neurol. 2017 Oct;30(5):490–9. https://doi.org/10.1097/WCO.0000000000000472
58. Brouwer BA , de Greef BT , Hoeijmakers JG , Geerts M , van Kleef M , Merkies IS , et al. Neuropathic Pain due to Small Fiber Neuropathy in Aging: Current Management and Future Prospects. Drugs Aging. 2015 Aug;32(8):611–21. https://doi.org/10.1007/s40266-015-0283-8
59. Suanprasert N , Berk JL , Benson MD , Dyck PJ , Klein CJ , Gollob JA , et al. Retrospective study of a TTR FAP cohort to modify NIS+7 for therapeutic trials. J Neurol Sci. 2014 Sep;344(1-2):121–8. https://doi.org/10.1016/j.jns.2014.06.041
60. Devigili G , Rinaldo S , Lombardi R , Cazzato D , Marchi M , Salvi E , et al. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain. 2019 Dec;142(12):3728–36. https://doi.org/10.1093/brain/awz333
61. Haroutounian S , Todorovic MS , Leinders M , Campagnolo M , Gewandter JS , Dworkin RH , et al. Diagnostic criteria for idiopathic small fiber neuropathy: A systematic review. Muscle Nerve. 2021 Feb;63(2):170–7. https://doi.org/10.1002/mus.27070
62. Maceira AM , Joshi J , Prasad SK , Moon JC , Perugini E , Harding I , et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005 Jan;111(2):186–93. https://doi.org/10.1161/01.CIR.0000152819.97857.9D
63. Martinez-Naharro A , Kotecha T , Norrington K , Boldrini M , Rezk T , Quarta C , et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc Imaging. 2019 May;12(5):810–9. https://doi.org/10.1016/j.jcmg.2018.02.006
64. Treglia G , Glaudemans AW , Bertagna F , Hazenberg BP , Erba PA , Giubbini R , et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging. 2018 Oct;45(11):1945–55. https://doi.org/10.1007/s00259-018-4013-4
65. Perugini E , Guidalotti PL , Salvi F , Cooke RM , Pettinato C , Riva L , et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005 Sep;46(6):1076–84. https://doi.org/10.1016/j.jacc.2005.05.073
66. Galat A , Rosso J , Guellich A , Van Der Gucht A , Rappeneau S , Bodez D , et al. Usefulness of (99m)Tc-HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid. 2015;22(4):210–20. https://doi.org/10.3109/13506129.2015.1072089
67. Rapezzi C , Quarta CC , Guidalotti PL , Pettinato C , Fanti S , Leone O , et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011 Jun;4(6):659–70. https://doi.org/10.1016/j.jcmg.2011.03.016
68. Perugini E , Guidalotti PL , Salvi F , Cooke RM , Pettinato C , Riva L , et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005 Sep;46(6):1076–84. https://doi.org/10.1016/j.jacc.2005.05.073
69. Musumeci MB , Cappelli F , Russo D , Tini G , Canepa M , Milandri A , et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020 Jun;13(6):1314–21. https://doi.org/10.1016/j.jcmg.2019.10.015
70. Suhr OB , Lundgren E , Westermark P . One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017 Apr;281(4):337–47. https://doi.org/10.1111/joim.12585
71. Gillmore JD , Damy T , Fontana M , Hutchinson M , Lachmann HJ , Martinez-Naharro A , et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018 Aug;39(30):2799–806. https://doi.org/10.1093/eurheartj/ehx589
72. Merlini G , Lousada I , Ando Y , Dispenzieri A , Gertz MA , Grogan M , et al. Rationale, application and clinical qualification for NT-proBNP as a surrogate end point in pivotal clinical trials in patients with AL amyloidosis. Leukemia. 2016 Oct;30(10):1979–86. https://doi.org/10.1038/leu.2016.191
73. Grogan M , Scott CG , Kyle RA , Zeldenrust SR , Gertz MA , Lin G , et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol. 2016 Sep;68(10):1014–20. https://doi.org/10.1016/j.jacc.2016.06.033
74. Lavatelli F , Albertini R , Di Fonzo A , Palladini G , Merlini G . Biochemical markers in early diagnosis and management of systemic amyloidoses. Clin Chem Lab Med. 2014 Nov;52(11):1517–31. https://doi.org/10.1515/cclm-2014-0235
75. Coutinho P . Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: Glenner G CP, de Freitas A., editor. Amyloid and amyloidosis. Amsterdam: Excerpta Medica; 1980. p. 88-98.
76. Yamamoto S , Wilczek HE , Nowak G , Larsson M , Oksanen A , Iwata T , et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant. 2007 Nov;7(11):2597–604. https://doi.org/10.1111/j.1600-6143.2007.01969.x
77. Arosio P , Vendruscolo M , Dobson CM , Knowles TP . Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol Sci. 2014 Mar;35(3):127–35. https://doi.org/10.1016/j.tips.2013.12.005
78. Richards DB , Cookson LM , Berges AC , Barton SV , Lane T , Ritter JM , et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med. 2015 Sep;373(12):1106–14. https://doi.org/10.1056/NEJMoa1504942
79. Cardoso I , Martins D , Ribeiro T , Merlini G , Saraiva MJ . Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010 Jul;8(1):74. https://doi.org/10.1186/1479-5876-8-74
80. Giorgetti S , Raimondi S , Pagano K , Relini A , Bucciantini M , Corazza A , et al. Effect of tetracyclines on the dynamics of formation and destructuration of beta2-microglobulin amyloid fibrils. J Biol Chem. 2011 Jan;286(3):2121–31. https://doi.org/10.1074/jbc.M110.178376
81. Adams D , Gonzalez-Duarte A , O’Riordan WD , Yang CC , Ueda M , Kristen AV , et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul;379(1):11–21. https://doi.org/10.1056/NEJMoa1716153
82. Benson MD , Waddington-Cruz M , Berk JL , Polydefkis M , Dyck PJ , Wang AK , et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul;379(1):22–31. https://doi.org/10.1056/NEJMoa1716793
83. Berk JL , Suhr OB , Obici L , Sekijima Y , Zeldenrust SR , Yamashita T , et al.; Diflunisal Trial Consortium . Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013 Dec;310(24):2658–67. https://doi.org/10.1001/jama.2013.283815
84. Coelho T , Maia LF , Martins da Silva A , Waddington Cruz M , Planté-Bordeneuve V , Lozeron P , et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012 Aug;79(8):785–92. https://doi.org/10.1212/WNL.0b013e3182661eb1
85. Adams D , Suhr OB , Dyck PJ , Litchy WJ , Leahy RG , Chen J , et al. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017 Sep;17(1):181. https://doi.org/10.1186/s12883-017-0948-5
86. Adams D , Samuel D , Goulon-Goeau C , Nakazato M , Costa PM , Feray C , et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000 Jul;123(Pt 7):1495–504. https://doi.org/10.1093/brain/123.7.1495
87. Ericzon BG , Wilczek HE , Larsson M , Wijayatunga P , Stangou A , Pena JR , et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation. 2015 Sep;99(9):1847–54. https://doi.org/10.1097/TP.0000000000000574
88. Suhr OB , Larsson M , Ericzon BG , Wilczek HE ; FAPWTRʼs investigators . Survival After Transplantation in Patients With Mutations Other Than Val30Met: Extracts From the FAP World Transplant Registry. Transplantation. 2016 Feb;100(2):373–81. https://doi.org/10.1097/TP.0000000000001021
89. Yazaki M , Mitsuhashi S , Tokuda T , Kametani F , Takei YI , Koyama J , et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant. 2007 Jan;7(1):235–42. https://doi.org/10.1111/j.1600-6143.2006.01585.x
90. Liepnieks JJ , Zhang LQ , Benson MD . Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010 Jul;75(4):324–7. https://doi.org/10.1212/WNL.0b013e3181ea15d4
91. Sekijima Y , Yazaki M , Oguchi K , Ezawa N , Yoshinaga T , Yamada M , et al. Cerebral amyloid angiopathy in posttransplant patients with hereditary ATTR amyloidosis. Neurology. 2016 Aug;87(8):773–81. https://doi.org/10.1212/WNL.0000000000003001
92. Algalarrondo V , Antonini T , Théaudin M , Chemla D , Benmalek A , Castaing D , et al. Cause of death analysis and temporal trends in survival after liver transplantation for transthyretin familial amyloid polyneuropathy. Amyloid. 2018 Dec;25(4):253–60. https://doi.org/10.1080/13506129.2018.1550061
93. Waddington Cruz M , Amass L , Keohane D , Schwartz J , Li H , Gundapaneni B . Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid. 2016 Sep;23(3):178–83. https://doi.org/10.1080/13506129.2016.1207163
94. Barroso FA , Judge DP , Ebede B , Li H , Stewart M , Amass L , et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017 Sep;24(3):194–204. https://doi.org/10.1080/13506129.2017.1357545
95. Maurer MS , Schwartz JH , Gundapaneni B , Elliott PM , Merlini G , Waddington-Cruz M , et al.; ATTR-ACT Study Investigators . Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018 Sep;379(11):1007–16. https://doi.org/10.1056/NEJMoa1805689
96. Rapezzi C , Elliott P , Damy T , Nativi-Nicolau J , Berk JL , Velazquez EJ , et al. Efficacy of Tafamidis in Patients With Hereditary and Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Analyses From ATTR-ACT. JACC Heart Fail. 2021 Feb;9(2):115–23. https://doi.org/10.1016/j.jchf.2020.09.011
97. Lockwood PA , Le VH , O’Gorman MT , Patterson TA , Sultan MB , Tankisheva E , et al. The Bioequivalence of Tafamidis 61-mg Free Acid Capsules and Tafamidis Meglumine 4 × 20-mg Capsules in Healthy Volunteers. Clin Pharmacol Drug Dev. 2020 Oct;9(7):849–54. https://doi.org/10.1002/cpdd.789
98. Obici L , Berk JL , González-Duarte A , Coelho T , Gillmore J , Schmidt HH , et al. Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Amyloid. 2020 Sep;27(3):153–62. https://doi.org/10.1080/13506129.2020.1730790
99. González-Duarte A , Berk JL , Quan D , Mauermann ML , Schmidt HH , Polydefkis M , et al. Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. J Neurol. 2020 Mar;267(3):703–12. https://doi.org/10.1007/s00415-019-09602-8
100. Solomon SD , Adams D , Kristen A , Grogan M , González-Duarte A , Maurer MS , et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation. 2019 Jan;139(4):431–43. https://doi.org/10.1161/CIRCULATIONAHA.118.035831
101. Coelho T , Yarlas A , Waddington-Cruz M , White MK , Sikora Kessler A , Lovley A , et al. Inotersen preserves or improves quality of life in hereditary transthyretin amyloidosis. J Neurol. 2020 Apr;267(4):1070–9. https://doi.org/10.1007/s00415-019-09671-9
102. Miller SR , Sekijima Y , Kelly JW . Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004 May;84(5):545–52. https://doi.org/10.1038/labinvest.3700059
103. Obici L , Cortese A , Lozza A , Lucchetti J , Gobbi M , Palladini G , et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012 Jun;19(sup1 Suppl 1):34–6. https://doi.org/10.3109/13506129.2012.678508
104 Wixner J , Pilebro B , Lundgren HE , Olsson M , Anan I . Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid. 2017;24(sup1):78-9.
105. Park H , Oh J , Shim G , Cho B , Chang Y , Kim S , et al. In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer’s disease. Nat Neurosci. 2019 Apr;22(4):524–8. https://doi.org/10.1038/s41593-019-0352-0
106. Pinney JH , Whelan CJ , Petrie A , Dungu J , Banypersad SM , Sattianayagam P , et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013 Apr;2(2):e000098. https://doi.org/10.1161/JAHA.113.000098
107. Connors LH , Sam F , Skinner M , Salinaro F , Sun F , Ruberg FL , et al. Heart Failure Resulting From Age-Related Cardiac Amyloid Disease Associated With Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation. 2016 Jan;133(3):282–90. https://doi.org/10.1161/CIRCULATIONAHA.115.018852
108. Yanagisawa A , Ueda M , Sueyoshi T , Okada T , Fujimoto T , Ogi Y , et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol. 2015 Feb;28(2):201–7. https://doi.org/10.1038/modpathol.2014.102
109. Geller HI , Singh A , Alexander KM , Mirto TM , Falk RH . Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. JAMA. 2017 Sep;318(10):962–3. https://doi.org/10.1001/jama.2017.9236
110. Westermark P , Westermark GT , Suhr OB , Berg S . Transthyretin-derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci. 2014 Aug;119(3):223–8. https://doi.org/10.3109/03009734.2014.895786
111. Pellikka PA , Holmes DR Jr , Edwards WD , Nishimura RA , Tajik AJ , Kyle RA . Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med. 1988 Mar;148(3):662–6. https://doi.org/10.1001/archinte.1988.00380030168027
112. Vrana JA , Gamez JD , Madden BJ , Theis JD , Bergen HR 3rd , Dogan A . Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009 Dec;114(24):4957–9. https://doi.org/10.1182/blood-2009-07-230722
113. Sławek S , Araszkiewicz A , Gaczkowska A , Koszarska J , Celiński D , Grygier M , et al. Endomyocardial biopsy via the femoral access - still safe and valuable diagnostic tool. BMC Cardiovasc Disord. 2016 Nov;16(1):222. https://doi.org/10.1186/s12872-016-0406-0
114. Pilebro B , Suhr OB , Näslund U , Westermark P , Lindqvist P , Sundström T . (99m)Tc-DPD uptake reflects amyloid fibril composition in hereditary transthyretin amyloidosis. Ups J Med Sci. 2016;121(1):17–24. https://doi.org/10.3109/03009734.2015.1122687
115. Stats MA , Stone JR . Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: implications for utilizing nuclear medicine studies to subtype amyloidosis. Cardiovasc Pathol. 2016 Sep-Oct;25(5):413–7. https://doi.org/10.1016/j.carpath.2016.07.001
116. Gillmore JD , Maurer MS , Falk RH , Merlini G , Damy T , Dispenzieri A , et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016 Jun;133(24):2404–12. https://doi.org/10.1161/CIRCULATIONAHA.116.021612
117. Phull P , Sanchorawala V , Connors LH , Doros G , Ruberg FL , Berk JL , et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018 Mar;25(1):62–7. https://doi.org/10.1080/13506129.2018.1436048
118. Damy T , Deux JF , Moutereau S , Guendouz S , Mohty D , Rappeneau S , et al. Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid. 2013 Dec;20(4):212–20. https://doi.org/10.3109/13506129.2013.825240
119. Quarta CC , Solomon SD , Uraizee I , Kruger J , Longhi S , Ferlito M , et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014 May;129(18):1840–9. https://doi.org/10.1161/CIRCULATIONAHA.113.006242
120. González-López E , Gagliardi C , Dominguez F , Quarta CC , de Haro-Del Moral FJ , Milandri A , et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017 Jun;38(24):1895–904. https://doi.org/10.1093/eurheartj/ehx043
121. Huang J , Zhao S , Chen Z , Zhang S , Lu M . Contribution of Electrocardiogram in the Differentiation of Cardiac Amyloidosis and Nonobstructive Hypertrophic Cardiomyopathy. Int Heart J. 2015;56(5):522–6. https://doi.org/10.1536/ihj.15-005
122. Martinez-Naharro A , Baksi AJ , Hawkins PN , Fontana M . Diagnostic imaging of cardiac amyloidosis. Nat Rev Cardiol. 2020 Jul;17(7):413–26. https://doi.org/10.1038/s41569-020-0334-7
123. Rapezzi C , Fontana M . Relative Left Ventricular Apical Sparing of Longitudinal Strain in Cardiac Amyloidosis: Is it Just Amyloid Infiltration? JACC Cardiovasc Imaging. 2019 Jul;12(7 Pt 1):1174–6. https://doi.org/10.1016/j.jcmg.2018.07.007
124. Martinez-Naharro A , Treibel TA , Abdel-Gadir A , Bulluck H , Zumbo G , Knight DS , et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017 Jul;70(4):466–77. https://doi.org/10.1016/j.jacc.2017.05.053
125. Fontana M , Chung R , Hawkins PN , Moon JC . Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015 Mar;20(2):133–44. https://doi.org/10.1007/s10741-014-9470-7
126. Baggiano A , Boldrini M , Martinez-Naharro A , Kotecha T , Petrie A , Rezk T , et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020 Jan;13(1 Pt 1):69–80. https://doi.org/10.1016/j.jcmg.2019.03.026
127. Martinez-Naharro A , Kotecha T , Norrington K , Boldrini M , Rezk T , Quarta C , et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovasc Imaging. 2019 May;12(5):810–9. https://doi.org/10.1016/j.jcmg.2018.02.006
128. Galat A , Rosso J , Guellich A , Van Der Gucht A , Rappeneau S , Bodez D , et al. Usefulness of (99m)Tc-HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid. 2015;22(4):210–20. https://doi.org/10.3109/13506129.2015.1072089
129. Rapezzi C , Quarta CC , Guidalotti PL , Pettinato C , Fanti S , Leone O , et al. Role of (99m)Tc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging. 2011 Jun;4(6):659–70. https://doi.org/10.1016/j.jcmg.2011.03.016
130. Harb SC , Haq M , Flood K , Guerrieri A , Passerell W , Jaber WA , et al. National patterns in imaging utilization for diagnosis of cardiac amyloidosis: A focus on Tc99m-pyrophosphate scintigraphy. J Nucl Cardiol. 2017 Jun;24(3):1094–7. https://doi.org/10.1007/s12350-016-0478-3
131. Haq M , Pawar S , Berk JL , Miller EJ , Ruberg FL . Can 99mTc-Pyrophosphate Aid in Early Detection of Cardiac Involvement in Asymptomatic Variant TTR Amyloidosis? JACC Cardiovasc Imaging. 2017 Jun;10(6):713–4. https://doi.org/10.1016/j.jcmg.2016.06.003
132. Glaudemans AW , van Rheenen RW , van den Berg MP , Noordzij W , Koole M , Blokzijl H , et al. Bone scintigraphy with (99m)technetium-hydroxymethylene diphosphonate allows early diagnosis of cardiac involvement in patients with transthyretin-derived systemic amyloidosis. Amyloid. 2014 Mar;21(1):35–44. https://doi.org/10.3109/13506129.2013.871250
133. Hanna M , Ruberg FL , Maurer MS , Dispenzieri A , Dorbala S , Falk RH , et al. Cardiac Scintigraphy With Technetium-99m-Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2020 Jun;75(22):2851–62. https://doi.org/10.1016/j.jacc.2020.04.022
134. Dorbala S , Ando Y , Bokhari S , Dispenzieri A , Falk RH , Ferrari VA , et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 2 of 2-Diagnostic Criteria and Appropriate Utilization. J Card Fail. 2019 Nov;25(11):854–65. https://doi.org/10.1016/j.cardfail.2019.08.002
135. Quarta CC , Gonzalez-Lopez E , Gilbertson JA , Botcher N , Rowczenio D , Petrie A , et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J. 2017 Jun;38(24):1905–8. https://doi.org/10.1093/eurheartj/ehx047
136. Cheng RK , Levy WC , Vasbinder A , Teruya S , De Los Santos J , Leedy D , et al. Diuretic Dose and NYHA Functional Class Are Independent Predictors of Mortality in Patients With Transthyretin Cardiac Amyloidosis. JACC CardioOncol. 2020 Sep;2(3):414–24. https://doi.org/10.1016/j.jaccao.2020.06.007
137. Habtemariam BA , Karsten V , Attarwala H , Goel V , Melch M , Clausen VA , et al. Single-Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting N-acetylgalactosamine-Small Interfering Ribonucleic Acid Conjugate, Vutrisiran, in Healthy Subjects. Clin Pharmacol Ther. 2021 Feb;109(2):372–82. https://doi.org/10.1002/cpt.1974
138. Rosenblum H , Castano A , Alvarez J , Goldsmith J , Helmke S , Maurer MS . TTR (Transthyretin) Stabilizers Are Associated With Improved Survival in Patients With TTR Cardiac Amyloidosis. Circ Heart Fail. 2018 Apr;11(4):e004769. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004769
139. Ikram A , Donnelly JP , Sperry BW , Samaras C , Valent J , Hanna M . Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid. 2018 Sep;25(3):197–202. https://doi.org/10.1080/13506129.2018.1519507
140. Castaño A , Drachman BM , Judge D , Maurer MS . Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015 Mar;20(2):163–78. https://doi.org/10.1007/s10741-014-9462-7
141. Ruberg FL , Grogan M , Hanna M , Kelly JW , Maurer MS . Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 Jun;73(22):2872–91. https://doi.org/10.1016/j.jacc.2019.04.003
142. Mints YY , Doros G , Berk JL , Connors LH , Ruberg FL . Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Heart Fail. 2018 Oct;5(5):772–9. https://doi.org/10.1002/ehf2.12308
143. Brignole M , Auricchio A , Baron-Esquivias G , Bordachar P , Boriani G , Breithardt OA , et al.; Document Reviewers . 2013 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: the Task Force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Eur Heart J. 2013 Aug;34(29):2281–329. https://doi.org/10.1093/eurheartj/eht150
144. Cibeira MT , Ortiz-Pérez JT , Quintana LF , Fernádez de Larrea C , Tovar N , Bladé J . Supportive Care in AL Amyloidosis. Acta Haematol. 2020;143(4):335–42. https://doi.org/10.1159/000506760
145. Obici L , Suhr OB . Diagnosis and treatment of gastrointestinal dysfunction in hereditary TTR amyloidosis. Clin Auton Res. 2019 Sep;29(S1 Suppl 1):55–63. https://doi.org/10.1007/s10286-019-00628-6
146. Mehra MR , Canter CE , Hannan MM , Semigran MJ , Uber PA , Baran DA , et al.; International Society for Heart Lung Transplantation (ISHLT) Infectious Diseases, Pediatric and Heart Failure and Transplantation Councils . The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016 Jan;35(1):1–23. https://doi.org/10.1016/j.healun.2015.10.023
147. Sousa M , Monohan G , Rajagopalan N , Grigorian A , Guglin M . Heart transplantation in cardiac amyloidosis. Heart Fail Rev. 2017 May;22(3):317–27. https://doi.org/10.1007/s10741-017-9601-z
148. Barrett CD , Alexander KM , Zhao H , Haddad F , Cheng P , Liao R , et al. Outcomes in Patients With Cardiac Amyloidosis Undergoing Heart Transplantation. JACC Heart Fail. 2020 Jun;8(6):461–8. https://doi.org/10.1016/j.jchf.2019.12.013
149. Conceição I , Damy T , Romero M , Galán L , Attarian S , Luigetti M , et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019 Mar;26(1):3–9. https://doi.org/10.1080/13506129.2018.1556156
150. Dohrn MF , Auer-Grumbach M , Baron R , Birklein F , Escolano-Lozano F , Geber C , et al. Chance or challenge, spoilt for choice? New recommendations on diagnostic and therapeutic considerations in hereditary transthyretin amyloidosis with polyneuropathy: the German/Austrian position and review of the literature. J Neurol. 2020.
151. Adams D , Lacroix C , Antonini T , Lozeron P , Denier C , Kreib AM , et al. Symptomatic and proven de novo amyloid polyneuropathy in familial amyloid polyneuropathy domino liver recipients. Amyloid. 2011 Jun;18(sup1 Suppl 1):174–7. https://doi.org/10.3109/13506129.2011.574354065