Diagnostic tools and CFTR functional assays in cystic fibrosis: utility and availability in Switzerland
DOI: https://doi.org/10.4414/smw.2021.20496
Clara
Fernandez Elviroa, Sylvain
Blanchona, Sylke
Hoehnelb, Urs
Schumacherc, Alain
Sautyd, Nathalie
Brandenbergb, Nicolas
Regameyc
a Department Woman-Mother-Child, Service of Paediatrics, Paediatric Pulmonology and Cystic Fibrosis Unit, University Hospital of Lausanne and Faculty of Biology and Medicine, University of Lausanne, Switzerland
b Institute of Bioengineering, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
c Division of Respiratory Medicine, Children’s Hospital Lucerne, Switzerland
d Division of Respiratory Medicine, Hospital of Neuchâtel, Switzerland
Summary
Cystic fibrosis (CF) is a genetic disease caused by a bi-allelic mutation of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. When the diagnosis cannot be confirmed by a positive sweat test or/and the identification of two CF-causing variants, international guidelines recommend the use of CFTR functional assays. These tests assess whether CFTR activity is normal or diminished/absent through measurement of CFTR-mediated chloride secretion/absorption.
CFTR functional assays are not only useful for diagnostic purposes but can also serve as a surrogate outcome for clinical trials of CFTR modulators, which are emerging therapeutic agents designed to correct the malfunctioning protein. In the near future they could also be used as precision-medicine techniques, to help guidance and optimisation of treatment.
Until now, sweat testing has been the only CFTR functional assay available in Switzerland. Since 2020, the Centre Hospitalier Universitaire Vaudois (CHUV) at Lausanne and the Lucerne Children’s Hospital perform nasal potential difference measurement. Moreover, The Ecole Polytechnique Fédérale de Lausanne (EPFL) established a reliable procedure to generate adult intestinal organoids, i.e., stem cell-derived in-vitro grown mini tissues, extracted from rectal biopsies, which can be used to assess CFTR function in vitro.
This narrative review describes the most popular CFTR functional assays, as well as their indications, limitations and availability in Switzerland.
Abbreviations
- CFTR
-
cystic fibrosis transmembrane conductance regulator
- CF
-
cystic fibrosis
- ENaC
-
epithelial sodium channel
- NPD
-
nasal potential difference measurements
- ICM
-
intestinal current measurements
- Isc
-
transepithelial short-circuit current
Introduction
A variant in both alleles of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, located on chromosome seven (7q31.2), is responsible for the genetic disease called cystic fibrosis (CF), which is the most common genetic disorder in Caucasians and has an autosomal recessive inheritance. The CFTR gene encodes for a protein located on the apical membrane of cells that acts as an anion channel. The CFTR protein can be found in several epithelial tissues, including airways, gastrointestinal and reproductive tracts, as well as sweat and salivary glands. The CFTR channel is responsible for the transport of chloride and bicarbonate, and has an indirect influence on sodium transport via the down-regulation of the apical epithelial sodium channel (ENaC) [1, 2].
In the majority of cases, the diagnosis of CF is unequivocal. It requires a positive newborn screening, a family history of CF in a sibling or a clinical feature suggestive of CF (e.g., chronic, recurrent sinopulmonary disease, exocrine pancreatic insufficiency, salt loss syndrome, obstructive azoospermia in males). The diagnosis of CF is confirmed in the presence of a sweat test in the diagnostic range (chloride ≥60 mmol/l) and/or a bi-allelic CF-causing variant in the CFTR gene [3, 4].
CF diagnosis is, however, not always straightforward. So far, a large number of CFTR variants (>2000) have been described, but less than 10% of these variants are disease-causing and the clinical relevance of many of them is not fully understood (www.cftr2.org). Moreover, there is a large variability in clinical CF phenotype not solely explained by the CFTR genotype [5]. A small but significant proportion of patients have a normal sweat test (chloride ≤29 mmol/l) or a sweat test in the intermediate range (30–59 mmol/l) and only one or no evident pathogenic CFTR variant. In these cases, the diagnosis of CF can neither be confirmed nor excluded, with potentially dramatic clinical, psychological and economic implications for patients. For such patients, international guidelines recommend the use of CFTR functional assays to help establish a correct definitive diagnosis [3, 4, 6].
Herein, we provide a summary of the most popular CFTR functional assays, as well as their indications, limitations and availability in Switzerland. Previous reviews in the field have addressed the reliability, validity and responsiveness of CFTR biomarkers as clinical endpoints in trials of CFTR modulators [7] and their importance for CF drug development [8]. Forskolin-induced organoid swelling assays have been described in detail elsewhere [9].
CFTR functional assays
CFTR activity can be measured with a variety of techniques called “CFTR functional assays”. These assays can determinate if CFTR-mediated chloride secretion/absorption is normal or diminished/absent. Several of them are validated tests, recognised in diagnostic guidelines, such as sweat testing, nasal potential difference measurement (NPD) and intestinal current measurement (ICM). Other assays are under development such as organoids and β-adrenergic sweat-rate assays. Limitations, advantages and clinical applicability of each technique are summarised in table 1.
Table 1 Description of the advantages and limitation of each technique, their clinical applicability, their availability in Switzerland, their cost and the age of the patient from which on the test can be done.
|
Modality
|
Advantages
|
Limitations
|
Clinical applicability
|
Availability in Switzerland
|
Price (CHF)
|
Age
|
| Sweat test |
Gold standard for CF diagnosis
Noninvasive
Good reproducibility
Easy to perform (painless)
Economical |
Delayed results (hours)
Minimum weight of the patient 2 kg (neonates)
False positive/negative tests in patients with malnutrition, hydration imbalance, metabolic and endocrine disease, skin problems and some medications |
Gold standard for CF diagnosis
Biomarker of CFTR function to test the efficacy of novel CFTR modulators (only biomarker required to continue a modulator) |
8 CF centres*
|
130 |
From 3 days and 36 weeks corrected gestational age |
| NPD |
Measurement in vivo of CFTR and ENaC activities
Little invasive
Immediate results
International standardisation
Validated as endpoint in clinical trials and for CF diagnosis by international guidelines |
Sophisticated set-up, need of a pharmacy producing perfusion solutions
Susceptible to alterations of nasal mucosa (nasal polyps, prior sinus surgery, infection, inflammation)
High within-subject variability
Patient cooperation needed (patient has to stay still for 30 min) |
Helps to clarify diagnosis in patients with questionable presentation
In vivo biomarker of CFTR function to test the efficacy of novel CFTR modulators (phase II proof of concept studies) |
CHUV (Lausanne) / Lucerne Children’s Hospital |
600–800 |
From 6 years |
| ICM |
Little invasive
More reproducible and sensitive than NPD (not influenced by mucosa inflammation)
Patient’s cooperation not needed
Detect CFTR selective mutations affecting HCO3 transport
Validated as endpoint in clinical trials and for diagnosis by international guidelines |
Ex vivo measurement
Delayed results (days)
Very little availability
Not standardised, various protocols
Intact rectal mucosa needed
Contraindicated in bleeding disorders |
Helps to clarify diagnosis in patients with questionable presentation
Ex vivo biomarker of CFTR function to test the efficacy of novel CFTR modulators (phase II proof of concept studies) |
No |
? |
Every age |
| β-adrenergic tests |
Measurement in vivo of CFTR activity
Little invasive
Immediate results |
Very little availability
Mostly experimental, not standardised, various protocols
Patient cooperation needed |
Research purpose |
No |
? |
From 8 years (new technique that do not require patient’s cooperation) |
| Organoids |
Little invasive
Fast follow-up tests possible on biobanked material, e.g., when new treatments become available
Automatable
Highly reproducible, reduced human error-driven variability |
In vitro measurement
Delayed results (weeks)
Mostly experimental, not standardised
Requires highly specialised laboratories
High cost |
Research purpose
In vitro biomarker of CFTR function to test the efficacy of novel CFTR modulators (phase I proof of concept studies, www.hitcf.org) |
EPFL |
2000–5000 (can decrease at higher scale) |
Every age |
CFTR functional assays can be used to resolve cases of unclear CF diagnosis [2, 4, 6]. A typical example would be the case of a patient with clinical features compatible with CF (recurrent sinopulmonary disease) showing intermediate sweat chloride values (30–59 mmol/l) and two CFTR variants of unclear clinical significance (www.cftr2.org). Other examples would be when a diagnosis of CF cannot be confirmed in an infant with positive CF newborn screening, a situation called “cystic fibrosis screen positive, inconclusive diagnosis” (CFSPID), or when a diagnosis of CF cannot be confirmed in a patient (most commonly adult) with CFTR-related single organ disease, a situation called “CFTR-related disorder” (CFTR-RD) [10–12].
Another potential application of CFTR functional assays is to optimise CFTR modulator treatments, which are emerging therapeutics designed to correct the malfunctioning protein. Indeed, given the variability of spectrum, severity and progression of CF disease not always predictable based on CFTR genotype and the high price of CFTR modulators, drug response should ideally be assessed before their administration to a patient [13]. CFTR functional assays have the potential to provide useful information on the therapeutic efficacy of a given drug on the CFTR function of an individual person. They could thus serve as surrogate outcomes for clinical trials of CFTR protein modulators and be used in precision medicine to select the optimal treatment needed by a given individual [13–15].
The sweat test
The sweat test measures chloride concentration in sweat collected after stimulation of the sweat gland. An elevated chloride concentration reflects defective chloride reabsorption in the sweat ducts of patients with an alteration in CFTR protein function. The analysis is performed on sweat collected after 30 minutes of stimulation of the sweat gland with pilocarpine applied by iontophoresis (fig. 1) [16].

Figure 1 Sweat testing. A. Sweating is induced by pilocarpine iontophoresis using pilocarpine loaded disc gels. B. Sweat is collected using the Macroduct® Sweat Collection System. A blue dye allows easy visualisation of the progression of sweat collection in the collector tube.
The normal values of sweat chloride concentrations are ≤29 mmol/l, intermediate values are between 30 and 59 mmol/l and abnormal values are ≥60 mmol/l, at any age.
To date, the sweat test done with the Macroduct® collection system remains the gold standard for CF diagnosis. It is also the cheapest test. Sweat testing should be performed by experienced personnel, following national or international guidelines [3, 4, 17]. The principal limitations of the test are the age and weight of the patient. Sweat tests should not be performed in infants before the age of 3 days, <36 weeks corrected gestational age or a weight <2 kg [17]. Further, malnutrition, hydration imbalance, metabolic and endocrine diseases, skin problems and some medications can lead to false-positive or false-negative sweat tests.
Nasal potential difference measurement (NPD)
The measurement of NPD can be used to assess ionic transport across the airway epithelium. Indeed, 2–3 cm distal to the nostril the nasal epithelium becomes a ciliated pseudostratified columnar epithelium, similar to the one found in the distal airways. The airway epithelium is formed of contiguous cells separated by tight junctions and organised as a polarised structure, with a mucosal apical side (to lumen) and a serous basolateral side (to blood). In this kind of tissue, the CFTR protein is highly expressed on the apical membranes of cells [18, 19]. The epithelium is the site of ion exchange between the two compartments. This results in a polarisation of the membrane (negative on the intracellular side and positive on the extracellular side), creating a transmembrane electric potential [20, 21].
In NPD testing, the membrane electric potential is measured using a high impedance voltmeter between two electrodes: one placed in the mucosa of the airway epithelium in the nostril (exploring electrode, outside the cells) and the other placed in the subcutaneous compartment in the forearm (the reference electrode, inside the cells) (fig. 2). The technique has been standardised and standard operating procedure (SOP) protocols are available and are used by the European Cystic Fibrosis Society-Clinical Trials Network [22].
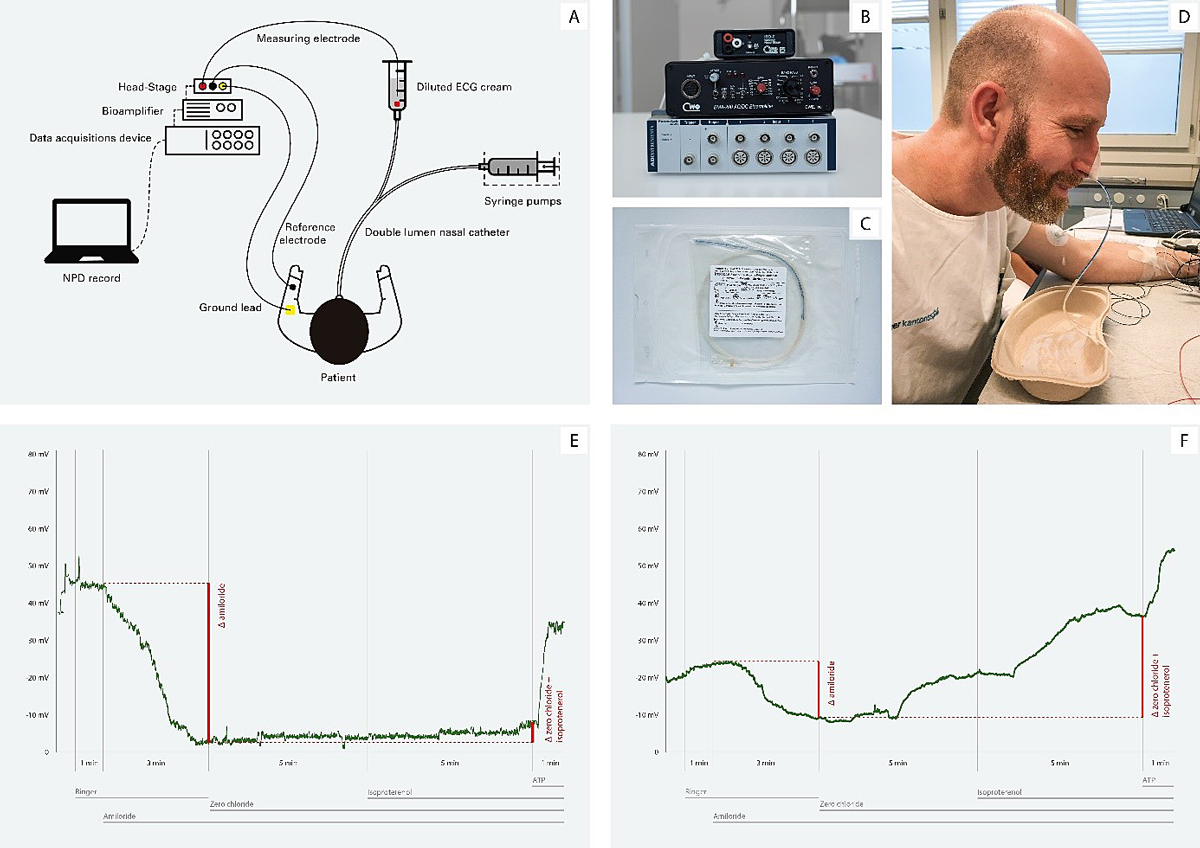
Figure 2 Nasal potential difference (NPD) measurement. A. Schematic illustration of the setup. B. Measurement instruments: A/D converter PowerLab 4/26, Bioamplifier BMA-200, ISO-Z Headstage. C. Custom made nasal catheter, produced by Marquat®. D. View showing the placement and attachment of the catheter. E. NPD tracing in a cystic fibrosis (CF) patient. F. NPD tracing in a healthy control subject. NPD baseline is more negative in CF than in controls; the change in NPD towards positive values is more pronounced in CF than in controls after blocking sodium channels with amiloride; the NPD in CF changes barely or not at all in CF after stimulation of cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels with zero-chloride solution and isoprenaline, but changes markedly toward negative values in controls; NPD changes to more negative values in both CF and controls after stimulation of alternative chloride channels with ATP.
First, basal potential is measured. As sodium absorption is the primary ion transport activity in normal epithelium, basal potential is usually negative, with values between −15 and −25 mV in healthy controls. In CF patients, basal potential is much more negative (−35 to −50 mV) due to the absence of inhibition of the activity of the ENaC by CFTR, leading to excessive sodium absorption. Compounds that activate or inhibit the different ion channel transporters are then sequentially perfused in the airway mucosa while the induced depolarisation is measured. Amiloride, which inhibits the ENaC, is perfused first. As a result, sodium is not absorbed by the cells, leading to a depolarisation of the membrane near to 0 mV. In CF patients, the depolarisation is greater due to a more negative basal potential. A zero-chloride solution that creates a chemical gradient for chloride secretion via chloride channels, including the CFTR, is then perfused. In order to equilibrate the membrane electric potential, chloride is secreted out of the cell through the CFTR channel, causing a large repolarisation of the potential. By adding isoprenaline, CFTR is directly stimulated by the cAMP pathway, which increase the repolarisation. In CF patients, there is a lack of response to the perfusion of the zero-chloride solution or to the stimulation of the CFTR channel by isoprenaline and thus no repolarisation is observed. Finally, a solution containing ATP is perfused, which increases the repolarisation in both CF and non-CF epithelia through CFTR-independent ion channels. This last is a control step that confirms that the epithelium is responsive to stimuli.
Besides the basal potential difference, key outcome parameters in NPD measurement include the response to amiloride perfusion (Δamiloride PD), the response to zero-chloride and isoprenaline perfusion (Δ-Cl-free + isoprenaline PD) and composite scores such as the Sermet and the Wilschanski score, which have been shown to adequately differentiate CF from non-CF patients [23–26].
The advantages of NPD testing are that it measures ionic transport of the airway epithelium in vivo, and that CFTR and ENaC activity can be differentially assessed. Its drawbacks lie in the need of a sophisticated infrastructure and a pharmacy able to produce the perfusion solutions. The need of an intact nasal mucosa to obtain adequate measurements and the difficulties in performing measurements in subjects with pulmonary exacerbations also limit its widespread use [7, 27].The large intrasubject variability, which has been estimated to be at +7.2 mV of measurement error for the Δ-Cl-free + isoprenaline PD, is an additional limitation of the technique, which can be overcome by averaging measurements in both nostrils [28]. Because the patient’s cooperation is needed, NPD measurement is usually feasible from the age of 6 years, although some groups have successfully performed NPD in toddlers.
Intestinal current measurement (ICM)
Intestinal current measurement uses rectal biopsies to study ex vivo CFTR activity in a tissue with high CFTR expression that remains unaltered by disease progression. There are numerous protocols and different equipment used in different centres [29]. In 2011, the European Cystic Fibrosis Society-Clinical Trials Network developed a SOP to harmonise the technique as an outcome parameter for clinical trials [23, 30]. The protocol used in the SOP is described hereafter and is available upon request at the ECFS (ECFS-CTN@uzleuven.be).
The first step in ICM is to obtain a rectal biopsy. This procedure is little invasive and can be quickly done at all ages, without requiring anaesthesia or preparation. The fresh biopsy has to be immediately mounted and introduced into an Ussing chamber, in which the measurement is made.
In contrast to NPD testing, which measures a potential difference, ICM directly measures electrical currents (transepithelial short-circuit current: Isc) generated in response to a variety of chloride secretory agents. Isc represents a direct measure of the net movement of ions across the epithelium. The first compound to be added is indomethacin, which reduces cAMP formation and thus CFTR-dependent chloride secretion to baseline. Then amiloride is added, as in NPD, which will reduce the intestinal Isc, inhibiting ENaC. The tissue is subsequently treated with forskolin and isobutyl-1-methylxanthine, which raise intracellular cAMP to activate CFTR channels. Genistein is then added, directly stimulating CFTR channels, which increases Isc. Carbachol is added next, stimulating basolateral potassium channels and driving chloride exit across CFTR chloride channels, thus hyperpolarising the cell even more. After, 4,49-diisothiocyanostilbene-2,29-disulphonic acid is added, which inhibits non‐CFTR chloride channels, allowing a more selective measurement of CFTR response. Finally, histamine, whose effect is similar to those of carbachol, is added [30].
The sum of the Isc response after stimulation with forskolin, carbachol and histamine is established as the ICM parameter that allows best differentiation between “CF unlikely” and “possible CF”, with a cut-off value of 34 mA/cm2 [31].
Compared with NPD measurement, ICM has the advantage of not being influenced by alterations of the nasal/respiratory mucosa that result from chronic inflammation and infection. Furthermore, it is especially interesting as it allows recognition of CFTR variants with residual chloride secretion [30, 32]. ICM measurement requires sophisticated equipment and can only be performed in research facilities in close proximity to a hospital setting. The procedure is time-consuming and expensive.
Beta-adrenergic sweat-rate assays
Human eccrine sweat glands produce sweat via two independent neural pathways: the β-adrenergic pathway and the cholinergic pathway [33]. The β-adrenergic secretion pathway is dependent on CFTR activity and is altered in CF patients whereas the cholinergic pathway is independent of CFTR activity [34]. Sweat glands highly express CFTR and are easily accessible to be tested and little affected by CF disease progression [33, 35].
Different methods have been developed to measure in vivo the secretory function of the sweat gland coil [35]. One method is the evaporimeter, which consists of two probes placed on the skin surface to measure the quantity of transpiration (or vapour, calculated from the humidity and temperature). Another method is bubble imaging, where an oil-filled reservoir placed on the skin surface captures by digital imaging the development of spherical sweat bubbles as they materialise. A third method and the last to be described, uses a pixilated image-sensor positioned on the forearm that detects changes in the images taken with the different sweat rates [36].
By comparing the β-adrenergic and cholinergic sweat rate, after stimulation of the sweat gland with a variety of compounds, CF, CF carriers and healthy subjects can be discriminated. The stimulation of the β-adrenergic and cholinergic pathways is done sequentially by intradermal injections and/or iontophoresis [36, 37].
Beta-adrenergic sweat-rate assays have not been standardised yet and show great variability between scientific groups in gland stimulation and measurement techniques. Nevertheless, the concept and feasibility of these test are promising, so that effort is being made to improve them for wider use in diagnosis and clinical trials.
Forskolin-induced organoid swelling assays
Under specific culture conditions, stem cells obtained from an intestinal tissue biopsy can multiply, differentiate and spontaneously assemble into hollow three-dimensional structures called “intestinal organoids”. Stem cell-derived organoids are self-renewing and self-organising in vitro cell culture systems that closely resemble native tissues in terms of cellular composition, architecture and key aspects of physiology [38]. Such intestinal organoids are genetically and phenotypically stable, can be expanded in vitro over long periods of time, bio-banked, thawed, re-expanded and re-tested if needed. Recently, organoids of the gastrointestinal tract have been established and used to assess drug-induced restoration of CFTR function in vitro [39, 40]. CFTR expression in rectal tissue is higher than in airway epithelium, making the study of intestinal organoids particularly interesting.
The first step to generate organoids is to sample rectal biopsies and extract the rectal stem-cell containing crypts from the collected tissue. The isolated crypts are then embedded in a three-dimensional in vitro culture system and the stem cells expanded until sufficient amount of material is generated for banking and testing (typically 1 month). Once ready, the organoids are grown into microtitre plates and the trans-epithelial fluid transport is evaluated by exposure to a forskolin stimulus for 1 hour. Forskolin increases intracellular cAMP and activates CFTR channels. As a consequence, chloride and water secretion into the lumen of the organoid occurs and induces swelling of the organoid. Traditionally, forskolin-induced swelling is quantified using microscopy and expressed as the maximum surface area increase compared with the non-swollen baseline, or as area under the curve, taking into account stimulation with increasing forskolin concentrations. Image-based methods using fluorescent labels are also used to trace the liquid intake within the organoid lumen. The EPFL has recently developed a label-free high-precision tracing technique to measure these fluxes, thus indirectly measuring CFTR activity (fig. 3).
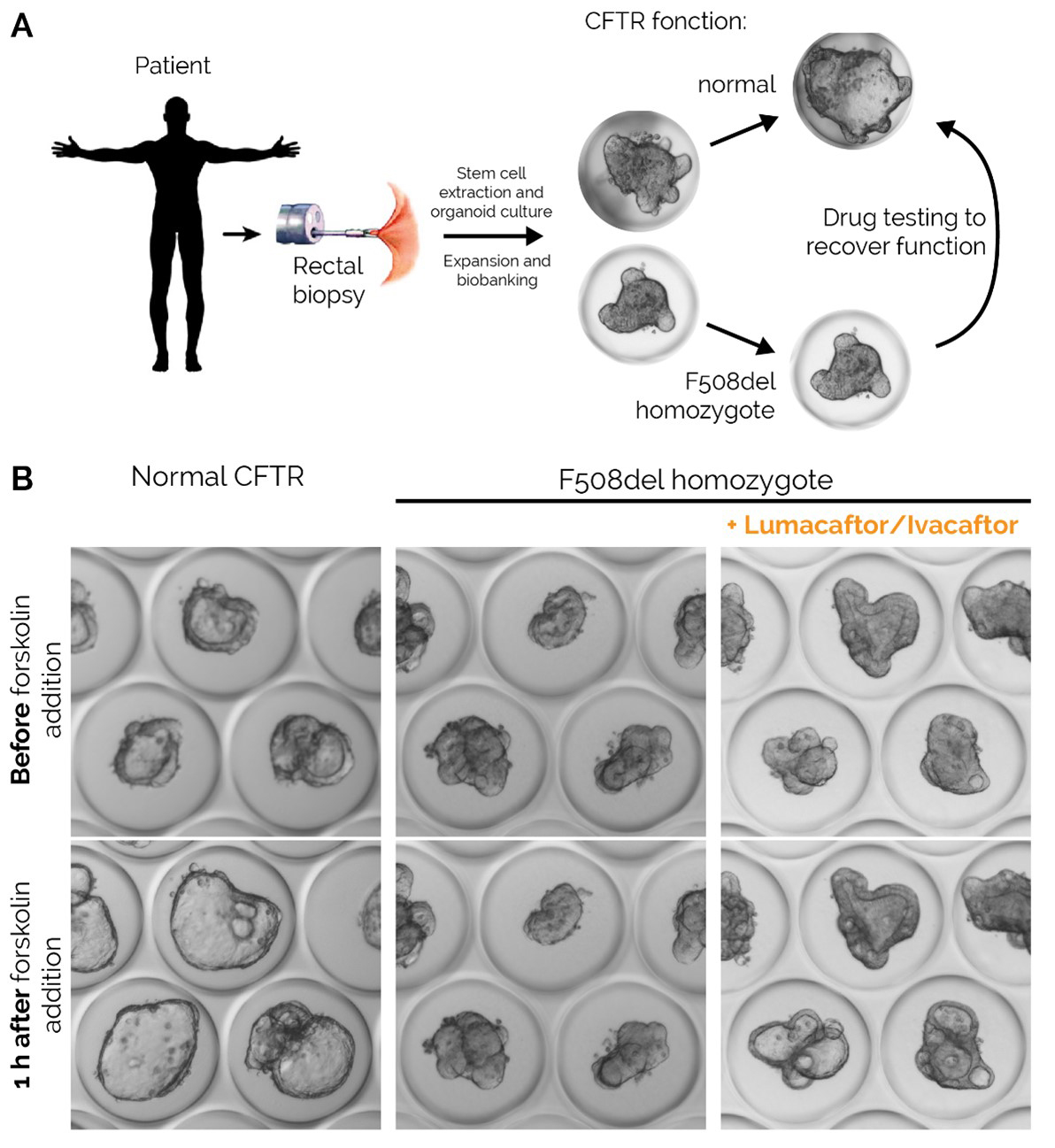
Figure 3 Rectal organoids of cystic fibrosis (CF) and non-CF control patients. A. Schematic description of the rectal organoid functional assay. B. Representative example of trans-epithelial fluid transport through healthy organoids and F508del homozygote organoids from a CF patient with or without exposure to the drug combination lumacaftor/ivacaftor. CFTR = cystic fibrosis transmembrane conductance regulator.
While retaining high expression of CFTR, rectal organoids provide an appropriate measure of the fluid transport through the rectal epithelium in contrast to NPD or ICM, which measure the airways membrane electrical potential and a net movement of ions across the rectal epithelium, respectively.
Organoids provide unique advantages in terms of scalability and automation, thus significantly decreasing human error-driven variability of CFTR functional assays. In addition, only a small amount of biopsied tissue is needed to generate material for the lifetime of a patient and the test appears to reliably measure CFTR function at the epithelial membrane level (fig. 3B). When used in precision medicine to assess the effect of CFTR modulators in vitro, organoids have the potential to identify the optimal drug or drug combination for each patient with sufficient throughput and reliability. However, the generation of organoids requires access to specialised infrastructure (cell culture laboratories), which limits its widespread establishment. Moreover, the components needed to grow rectal stem cells into organoids are costly at low scale and obtaining results can take weeks. It is an in vitro, not yet standardised measurement that is still mostly experimental but has been considerably improved in the last years.
A recent study including 21 patients failed to show any correlation between swelling in organoids, ICM, NPD and clinical parameters (including lung function) before and after treatment with the CFTR modulator lumacaftor/ivacaftor, questioning the suitability of these biomarkers of CFTR function to predict clinical efficacy of CFTR modulators [41]. However, these findings should be generalised with caution owing to the small sample size of the study, its short duration (16 weeks) and the absence of improvement in any of the other parameters known to improve upon CFTR modulator treatment. It is unknown whether similar observations would have been made in a larger study using a highly effective CFTR modulator such as elexacaftor/tezacaftor/ivacaftor.
Other techniques
In the last few years, numerous techniques such as the use of human nasal epithelial cells spheroids [42], human bronchial epithelial cell lines such as the CFBE410 cell line [43] and primary human nasal epithelial cells cultures [44] have been developed in order to better understand CFTR activity [45] and the mechanism of action of CFTR modulators [46]. All of these techniques use easily accessible nasal epithelial or tracheo-bronchial cells to create an in vitro model of the CF airway epithelium, with the final aim to create a differentiated epithelium with morphological and clinical features similar to the human airway epithelium. Most of these techniques are exclusively used for research purpose in specialised laboratories and have currently little clinical applicability. However, they could be important tools for precision medicine, especially in patients with rare CF-causing variants in the future.
Situation and availability in Switzerland
The diagnostic pathway of CF in Switzerland has changed greatly since the implementation of CF newborn screening in 2011, and the majority of CF cases are now diagnosed shortly after birth. A positive newborn screening test is defined by an elevation of immunoreactive trypsinogen levels in a heel prick test (Guthrie card) combined with the identification of a CFTR variant. If no variant is found, the confirmation of an elevated immunoreactive trypsinogen in a second heel prick test defines a positive test. Infants with positive CF newborn screening are referred to a CF centre for sweat testing. In the event of a positive sweat test, CF diagnosis is confirmed.
In 2019, Swiss investigators reported that the addition of the Nanoduct®, a newer sweat conductivity analysis system specially developed for newborns because it requires only 3–5 μl of sweat and measures conductivity in situ, might speed up the diagnostic process and therefore help to reduce the stressful time of uncertainty for parents and help to start appropriate treatment earlier [47].
Since 2020, the paediatric pulmonology and cystic fibrosis unit of the CHUV at Lausanne and the paediatric cystic fibrosis centre of Lucerne Children’s Hospital perform NPD measurement on paediatric and adult patients, with the aim of providing additional testing of CFTR function to patients with an unclear diagnosis despite sweat testing and extended genetic analysis. In the near future, NPD measurement could also be used as a surrogate endpoint in clinical trials, allowing easy and early assessment of drug efficacy without having to wait for months or even years for hard clinical endpoints such as body mass index or spirometry changes. In addition, the technique could be used to evaluate the functional restoration of CFTR by new modulators, which increase CFTR protein production, stability and activity [15, 48–51].
Furthermore, the EPFL has established a robust SOP to generate, expand and biobank rectal organoids. The use of these organoids to evaluate the benefits of specific CFTR modulators on CFTR function in a patient-specific fashion prior to prescription is of great interest [14]. The use of rectal organoids for CF diagnosis in Switzerland using the novel approach developed by EPFL is currently being evaluated.
Hopefully, in the near future, the techniques described above will enable precision medicine for CF patients to be practiced in Switzerland. We might be able to test the efficacy of new CFTR modulators in vitro using organoids derived from patients, with direct measurement of the improvement of CFTR activity. This would allow selective treatment of responders, thus avoiding unnecessary individual trials, side effects and wastage of resources. Short-term assessment of CFTR activity in vivo by sweat testing and NPD measurement would further confirm or refute the clinical efficacy of the selected CFTR modulator for the patient under treatment.
Conclusion
In the last few decades CFTR functional assays have been developed to clarify diagnosis in patients in whom CF cannot be confirmed or refuted using classic diagnostic tools such as sweat testing and genetic analysis. Additionally, these functional assays are increasingly being used as outcome parameters for clinical trials of new CFTR modulators. Finally, they may become invaluable tools in precision medicine aimed at selecting the best drug or drug combination for restoring CFTR function in a given CF patient. Thus, given their large field of application, the availability of these techniques in Switzerland seems of utmost importance. Efforts should be done to encourage their implementation, so that CF diagnosis and treatment can be optimised.
References
1
Harris
A
. Cystic fibrosis gene. Br Med Bull. 1992;48(4):738–53. doi:.https://doi.org/10.1093/oxfordjournals.bmb.a072575
2
Saint-Criq
V
,
Gray
MA
. Role of CFTR in epithelial physiology. Cell Mol Life Sci. 2017;74(1):93–115. doi:.https://doi.org/10.1007/s00018-016-2391-y
3
Farrell
PM
,
White
TB
,
Ren
CL
,
Hempstead
SE
,
Accurso
F
,
Derichs
N
, et al.
Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181:S4–15, 15.e1. doi:.https://doi.org/10.1016/j.jpeds.2016.09.064
4
Castellani
C
,
Duff
AJA
,
Bell
SC
,
Heijerman
HGM
,
Munck
A
,
Ratjen
F
, et al.
ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17(2):153–78. doi:.https://doi.org/10.1016/j.jcf.2018.02.006
5
Ratjen
F
,
Döring
G
. Cystic fibrosis. Lancet. 2003;361(9358):681–9. doi:.https://doi.org/10.1016/S0140-6736(03)12567-6
6
Bombieri
C
,
Claustres
M
,
De Boeck
K
,
Derichs
N
,
Dodge
J
,
Girodon
E
, et al.
Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10(Suppl 2):S86–102. doi:.https://doi.org/10.1016/S1569-1993(11)60014-3
7
De Boeck
K
,
Kent
L
,
Davies
J
,
Derichs
N
,
Amaral
M
,
Rowe
SM
, et al.; European Cystic Fibrosis Society Clinical Trial Network Standardisation Committee. CFTR biomarkers: time for promotion to surrogate end-point. Eur Respir J. 2013;41(1):203–16. doi:.https://doi.org/10.1183/09031936.00057512
8
Muhlebach
MS
,
Clancy
JP
,
Heltshe
SL
,
Ziady
A
,
Kelley
T
,
Accurso
F
, et al.
Biomarkers for cystic fibrosis drug development. J Cyst Fibros. 2016;15(6):714–23. doi:.https://doi.org/10.1016/j.jcf.2016.10.009
9
van Mourik
P
,
Beekman
JM
,
van der Ent
CK
. Intestinal organoids to model cystic fibrosis. Eur Respir J. 2019;54(1):1802379. doi:.https://doi.org/10.1183/13993003.02379-2018
10
Sermet-Gaudelus
I
,
Brouard
J
,
Audrézet
MP
,
Couderc Kohen
L
,
Weiss
L
,
Wizla
N
, et al.
Guidelines for the clinical management and follow-up of infants with inconclusive cystic fibrosis diagnosis through newborn screening. Arch Pediatr. 2017;24(12):e1–14. doi:.https://doi.org/10.1016/j.arcped.2017.07.015
11
Simmonds
NJ
. Is it cystic fibrosis? The challenges of diagnosing cystic fibrosis. Paediatr Respir Rev. 2019;31:6–8.
12
Southern
KW
,
Barben
J
,
Gartner
S
,
Munck
A
,
Castellani
C
,
Mayell
SJ
, et al.
Inconclusive diagnosis after a positive newborn bloodspot screening result for cystic fibrosis; clarification of the harmonised international definition. J Cyst Fibros. 2019;18(6):778–80. doi:.https://doi.org/10.1016/j.jcf.2019.04.010
13
Marson
FAL
,
Bertuzzo
CS
,
Ribeiro
JD
. Personalized or Precision Medicine? The Example of Cystic Fibrosis. Front Pharmacol. 2017;8:390. doi:.https://doi.org/10.3389/fphar.2017.00390
14
Boj
SF
,
Vonk
AM
,
Statia
M
,
Su
J
,
Vries
RR
,
Beekman
JM
, et al.
Forskolin-induced Swelling in Intestinal Organoids: An In Vitro Assay for Assessing Drug Response in Cystic Fibrosis Patients. J Vis Exp. 2017;120:e5519. doi:.https://doi.org/10.3791/55159
15
Graeber
SY
,
Dopfer
C
,
Naehrlich
L
,
Gyulumyan
L
,
Scheuermann
H
,
Hirtz
S
, et al.
Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2018;197(11):1433–42. doi:.https://doi.org/10.1164/rccm.201710-1983OC
16
Collie
JT
,
Massie
RJ
,
Jones
OA
,
LeGrys
VA
,
Greaves
RF
. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol. 2014;49(2):106–17. doi:.https://doi.org/10.1002/ppul.22945
17
LeGrys
VA
,
Yankaskas
JR
,
Quittell
LM
,
Marshall
BC
,
Mogayzel
PJ, Jr
; Cystic Fibrosis Foundation. Diagnostic sweat testing: the Cystic Fibrosis Foundation guidelines. J Pediatr. 2007;151(1):85–9. doi:.https://doi.org/10.1016/j.jpeds.2007.03.002
18
Puchelle
E
,
Gaillard
D
,
Ploton
D
,
Hinnrasky
J
,
Fuchey
C
,
Boutterin
MC
, et al.
Differential localization of the cystic fibrosis transmembrane conductance regulator in normal and cystic fibrosis airway epithelium. Am J Respir Cell Mol Biol. 1992;7(5):485–91. doi:.https://doi.org/10.1165/ajrcmb/7.5.485
19
Gaillard
D
,
Ruocco
S
,
Lallemand
A
,
Dalemans
W
,
Hinnrasky
J
,
Puchelle
E
. Immunohistochemical localization of cystic fibrosis transmembrane conductance regulator in human fetal airway and digestive mucosa. Pediatr Res. 1994;36(2):137–43. doi:.https://doi.org/10.1203/00006450-199408000-00002
20
Boucher
RC
. Molecular insights into the physiology of the ‘thin film’ of airway surface liquid. J Physiol. 1999;516(Pt 3):631–8. doi:.https://doi.org/10.1111/j.1469-7793.1999.0631u.x
21Lodish HBA, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. The Action Potential and Conduction of Electric Impulses. In: Molecular Cell Biology 4th edition. New York: WH Freeman; 2000.
22
Solomon
GM
,
Bronsveld
I
,
Hayes
K
,
Wilschanski
M
,
Melotti
P
,
Rowe
SM
, et al.
Standardized Measurement of Nasal Membrane Transepithelial Potential Difference (NPD). J Vis Exp. 2018;139:e57006. doi:.https://doi.org/10.3791/57006
23
De Boeck
K
,
Derichs
N
,
Fajac
I
,
de Jonge
HR
,
Bronsveld
I
,
Sermet
I
, et al.; ECFS Diagnostic Network Working Group; EuroCareCF WP3 Group on CF diagnosis. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros. 2011;10(Suppl 2):S53–66. doi:.https://doi.org/10.1016/S1569-1993(11)60009-X
24
Tridello
G
,
Menin
L
,
Pintani
E
,
Bergamini
G
,
Assael
BM
,
Melotti
P
. Nasal potential difference outcomes support diagnostic decisions in cystic fibrosis. J Cyst Fibros. 2016;15(5):579–82. doi:.https://doi.org/10.1016/j.jcf.2016.06.009
25
Wilschanski
M
,
Famini
H
,
Strauss-Liviatan
N
,
Rivlin
J
,
Blau
H
,
Bibi
H
, et al.
Nasal potential difference measurements in patients with atypical cystic fibrosis. Eur Respir J. 2001;17(6):1208–15. doi:.https://doi.org/10.1183/09031936.01.00092501
26
Sermet-Gaudelus
I
,
Girodon
E
,
Sands
D
,
Stremmler
N
,
Vavrova
V
,
Deneuville
E
, et al.
Clinical phenotype and genotype of children with borderline sweat test and abnormal nasal epithelial chloride transport. Am J Respir Crit Care Med. 2010;182(7):929–36. doi:.https://doi.org/10.1164/rccm.201003-0382OC
27
Yaakov
Y
,
Kerem
E
,
Yahav
Y
,
Rivlin
J
,
Blau
H
,
Bentur
L
, et al.
Reproducibility of nasal potential difference measurements in cystic fibrosis. Chest. 2007;132(4):1219–26. doi:.https://doi.org/10.1378/chest.06-2975
28
Kyrilli
S
,
Henry
T
,
Wilschanski
M
,
Fajac
I
,
Davies
JC
,
Jais
JP
, et al.
Insights into the variability of nasal potential difference, a biomarker of CFTR activity. J Cyst Fibros. 2020;19(4):620–6. doi:.https://doi.org/10.1016/j.jcf.2019.09.015
29
Beekman
JM
,
Sermet-Gaudelus
I
,
de Boeck
K
,
Gonska
T
,
Derichs
N
,
Mall
MA
, et al.
CFTR functional measurements in human models for diagnosis, prognosis and personalized therapy: Report on the pre-conference meeting to the 11th ECFS Basic Science Conference, Malta, 26-29 March 2014. J Cyst Fibros. 2014;13(4):363–72. doi:.https://doi.org/10.1016/j.jcf.2014.05.007
30
Clancy
JP
,
Szczesniak
RD
,
Ashlock
MA
,
Ernst
SE
,
Fan
L
,
Hornick
DB
, et al.
Multicenter intestinal current measurements in rectal biopsies from CF and non-CF subjects to monitor CFTR function. PLoS One. 2013;8(9):e73905. doi:.https://doi.org/10.1371/journal.pone.0073905
31
Derichs
N
,
Sanz
J
,
Von Kanel
T
,
Stolpe
C
,
Zapf
A
,
Tümmler
B
, et al.
Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax. 2010;65(7):594–9. doi:.https://doi.org/10.1136/thx.2009.125088
32
Sousa
M
,
Servidoni
MF
,
Vinagre
AM
,
Ramalho
AS
,
Bonadia
LC
,
Felício
V
, et al.
Measurements of CFTR-mediated Cl- secretion in human rectal biopsies constitute a robust biomarker for Cystic Fibrosis diagnosis and prognosis. PLoS One. 2012;7(10):e47708. doi:.https://doi.org/10.1371/journal.pone.0047708
33
Quinton
PM
. Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda). 2007;22(3):212–25. doi:.https://doi.org/10.1152/physiol.00041.2006
34
Sato
K
,
Sato
F
. Defective beta adrenergic response of cystic fibrosis sweat glands in vivo and in vitro. J Clin Invest. 1984;73(6):1763–71. doi:.https://doi.org/10.1172/JCI111385
35
Kim
J
,
Farahmand
M
,
Dunn
C
,
Davies
Z
,
Frisbee
E
,
Milla
C
, et al.
Evaporimeter and Bubble-Imaging Measures of Sweat Gland Secretion Rates. PLoS One. 2016;11(10):e0165254. doi:.https://doi.org/10.1371/journal.pone.0165254
36
Salinas
DB
,
Peng
YH
,
Horwich
B
,
Wee
CP
,
Frisbee
E
,
Maarek
JM
. Image-based β-adrenergic sweat rate assay captures minimal cystic fibrosis transmembrane conductance regulator function. Pediatr Res. 2020;87(1):137–45. doi:.https://doi.org/10.1038/s41390-019-0503-8
37
Quinton
P
,
Molyneux
L
,
Ip
W
,
Dupuis
A
,
Avolio
J
,
Tullis
E
, et al.
β-adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am J Respir Crit Care Med. 2012;186(8):732–9. doi:.https://doi.org/10.1164/rccm.201205-0922OC
38
Sato
T
,
Vries
RG
,
Snippert
HJ
,
van de Wetering
M
,
Barker
N
,
Stange
DE
, et al.
Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–5. doi:.https://doi.org/10.1038/nature07935
39
Dekkers
JF
,
Wiegerinck
CL
,
de Jonge
HR
,
Bronsveld
I
,
Janssens
HM
,
de Winter-de Groot
KM
, et al.
A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–45. doi:.https://doi.org/10.1038/nm.3201
40
Dekkers
JF
,
Berkers
G
,
Kruisselbrink
E
,
Vonk
A
,
de Jonge
HR
,
Janssens
HM
, et al.
Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8(344):344ra84. doi:.https://doi.org/10.1126/scitranslmed.aad8278
41
Graeber
SY
,
van Mourik
P
,
Vonk
AM
,
Kruisselbrink
E
,
Hirtz
S
,
van der Ent
CK
, et al.
Comparison of Organoid Swelling and In Vivo Biomarkers of CFTR Function to Determine Effects of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation. Am J Respir Crit Care Med. 2020;202(11):1589–92. doi:.https://doi.org/10.1164/rccm.202004-1200LE
42
Brewington
JJ
,
Filbrandt
ET
,
LaRosa
FJ, 3rd
,
Moncivaiz
JD
,
Ostmann
AJ
,
Strecker
LM
, et al.
Generation of Human Nasal Epithelial Cell Spheroids for Individualized Cystic Fibrosis Transmembrane Conductance Regulator Study. J Vis Exp. 2018;134:e57492. doi:.https://doi.org/10.3791/57492
43
Ehrhardt
C
,
Collnot
EM
,
Baldes
C
,
Becker
U
,
Laue
M
,
Kim
KJ
, et al.
Towards an in vitro model of cystic fibrosis small airway epithelium: characterisation of the human bronchial epithelial cell line CFBE41o-. Cell Tissue Res. 2006;323(3):405–15. doi:.https://doi.org/10.1007/s00441-005-0062-7
44
Müller
L
,
Brighton
LE
,
Carson
JL
,
Fischer
WA, 2nd
,
Jaspers
I
. Culturing of human nasal epithelial cells at the air liquid interface. J Vis Exp. 2013;80:e50646. doi:.https://doi.org/10.3791/50646
45
Veit
G
,
Bossard
F
,
Goepp
J
,
Verkman
AS
,
Galietta
LJ
,
Hanrahan
JW
, et al.
Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol Biol Cell. 2012;23(21):4188–202. doi:.https://doi.org/10.1091/mbc.e12-06-0424
46
Veit
G
,
Roldan
A
,
Hancock
MA
,
Da Fonte
DF
,
Xu
H
,
Hussein
M
, et al.
Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight. 2020;5(18):e139983. doi:.https://doi.org/10.1172/jci.insight.139983
47
Rueegg
CS
,
Kuehni
CE
,
Gallati
S
,
Jurca
M
,
Jung
A
,
Casaulta
C
, et al.; Swiss Cystic Fibrosis Screening Group. Comparison of two sweat test systems for the diagnosis of cystic fibrosis in newborns. Pediatr Pulmonol. 2019;54(3):264–72. doi:.https://doi.org/10.1002/ppul.24227
48
Masson
A
,
Schneider-Futschik
EK
,
Baatallah
N
,
Nguyen-Khoa
T
,
Girodon
E
,
Hatton
A
, et al.
Predictive factors for lumacaftor/ivacaftor clinical response. J Cyst Fibros. 2019;18(3):368–74. doi:.https://doi.org/10.1016/j.jcf.2018.12.011
49
Accurso
FJ
,
Rowe
SM
,
Clancy
JP
,
Boyle
MP
,
Dunitz
JM
,
Durie
PR
, et al.
Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi:.https://doi.org/10.1056/NEJMoa0909825
50
Accurso
FJ
,
Van Goor
F
,
Zha
J
,
Stone
AJ
,
Dong
Q
,
Ordonez
CL
, et al.
Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J Cyst Fibros. 2014;13(2):139–47. doi:.https://doi.org/10.1016/j.jcf.2013.09.007
51
Mesbahi
M
,
Shteinberg
M
,
Wilschanski
M
,
Hatton
A
,
Nguyen-Khoa
T
,
Friedman
H
, et al.
Changes of CFTR functional measurements and clinical improvements in cystic fibrosis patients with non p.Gly551Asp gating mutations treated with ivacaftor. J Cyst Fibros. 2017;16(1):45–8. doi:.https://doi.org/10.1016/j.jcf.2016.08.006