Research projects in human genetics in Switzerland: analysis of research protocols submitted to cantonal ethics committees in 2018
DOI: https://doi.org/10.4414/smw.2021.20403
Susanne
Driessen, Pietro
Gervasoni
swissethics, Haus der Akademien, Bern, Switzerland
Summary
AIM OF THE STUDY
This analysis provides a full national overview of genetic research dossiers pertaining to clinical and nonclinical trials, and to further-use research projects submitted for approval to ethics committees in Switzerland in 2018. It addresses the research type, medical field, number of individuals or datasets involved, diagnostic laboratories and data privacy, as well as the procedures foreseen for obtaining consent, communicating results, and dealing with excess data and incidental findings. The analysis results should constitute a basis for future discussions surrounding regulatory and ethical procedures that govern genetic investigations in biomedical research in Switzerland.
METHODS
All research dossiers approved by ethics committees in 2018 were screened for genetic investigations. A sample of 122 dossiers were analysed in depth, with regards to the frequency of genetic investigations, overall purpose and number of human beings or datasets included, in addition to the diagnostic categories and methodologies that were employed. The number of genes, biosample storage conditions and laboratory types concerned were also recorded. The processes for obtaining informed consent, communicating the results to the participants and predetermined principles for handling incidental findings were analysed.
RESULTS
Genetic investigations were retrieved from 9% of all research applications. The focus of most clinical trials was pharmacogenetics, whereas research projects of further use of data and/or biological material were mostly investigator-initiated and focused on basic genetic research and multiple gene analysis. Overall, big datasets (i.e., more than 100 or even 1000 sets) were included, especially in further-use research projects. Nongermline somatic genetic investigations were a large research field in oncology (56%), whereas genetic germline testing was mostly performed in neurology or psychiatry. In most cases, numerous genes were analysed. Modern sequencing techniques were employed, rendering excess genetic information nearly inevitable. Information regarding the storage of genetic data was mostly lacking, whereas information regarding the biosample storage was mostly provided. Data protection and informed consent procedures aligned with legal, regulatory and ethical standards. Procedures for communicating genetic analysis results and incidental findings to research participants were not predetermined in most research protocols, and they were handled differently from informed consent and general consent forms.
CONCLUSIONS
This study overviewed the key dimensions of regulatory and ethical assessments pertaining to genetic investigations that are performed on human beings as part of research projects in Switzerland. The data’s potential impact to shape the Federal Act on Human Genetic Testing and the Human Research Act in the future is also discussed. A direct transfer of standards for quality, consultation and communication of genetic testing within clinical genetic routines to genetic testing of human beings in the research context is neither required nor appropriate. It would bear a high risk of excluding patients and the Swiss health system from seminal innovations in medicine and life-science research.
Introduction
Genetic analysis pertaining to human beings is now routinely carried out for diagnostic purposes in almost all medical specialties. The outcome may inform treatment allocation and genetic management. The amount of genetic data thereby collected has rapidly increased over time, partially through the widespread use of massive sequencing methods, which are modern, fast and cheap [1]. International efforts such as the Human Genome Project and the HapMap Project, combined with genome-wide association and sequencing studies, have helped to identify more than 60,000 genetic associations across thousands of human diseases and traits [2]. In addition, knowledge of the causality between drug effects, drug safety profiles and genetic predisposition (pharmacogenomics) is still evolving, thus becoming increasingly useful to predict therapeutic responses [3].
Generally, the acquisition of genetic data, both in clinical routine and research projects, is a sensitive topic that involves ethical, legal and social issues (ELSI), all of which could have a political impact. The results of genetic analysis in clinical routine are verified by accredited laboratories, which exhibit the highest possible standards of quality. In addition, genetic counselling is mandatory and legally predetermined [4]. This ensures that clinical results based on genetic analysis are valid and placed in the proper context for each individual person. This legal aspect also raises fundamental questions, such as whether genetic data can still meet the requirements for confidentiality and data protection, and whether anonymisation is still possible when dealing with genetic data [5].
Outside of clinical routine, large amounts of genetic data are being collected in biomedical research, with far-reaching insights expected. All these research-derived data adhere to the well-defined procedures of regulatory and ethical approval [6]. The Swiss Federal Act on Human Genetic Testing (HGTA) [4] was recently revised to address public protection issues, such as avoiding direct-to-customer genetic testing [7] and providing a revised definition of genetic data, among other topics. The conduct of genetic analysis in research projects involving humans currently falls under the Human Research Act (HRA), not under the HGTA. It is currently being discussed whether the HGTA rules should also apply to genetic investigations performed in research involving humans, in the same way as genetic analysis conducted in clinical routine.
The current regulatory research framework does not include the fundamental standards of, for example, data quality, genetic counselling and informed consent procedures by geneticists, which are mandatory when genetic germline testing is performed in clinical routine. Whether this is relevant to only very few cases or individuals undergoing genetic investigations in research, or whether this represents a critical imbalance in the protection of individuals who undergo genetic testing is largely unknown, because the confidentiality of research protocols precludes researchers, regulators, societies and politicians likewise to obtain a broad overview of the amount and type of genetic research investigations performed. The type of genetic testing performed, the methodology used for data acquisition and the level of coding used, as well as the informed consent procedure involved, are largely protected by confidentiality contracts. According to the World Health Organization (WHO) [8], about 50% of clinical trials go unreported, leaving an incomplete and potentially misleading picture of the research conducted in the public domain.
Here, we provide a comprehensive, national-level overview of the type, amount, methodology and procedures applied to genetic investigations in human research projects. This research outcome may serve as a basis for the rational assessment of the current ethical, regulatory and legal framework; moreover, it may guide discussions about potential adaptations in this vibrant field, which concerns humans and their sensitive personal data.
Methods
Terminology concerning genetic analysis, genetic investigation and genetic testing
In accordance with article 3 of the Federal Act on Human Genetic Testing (HGTA), genetic analysis in clinical routine refers to cytogenetic and molecular genetic analyses aimed to determine hereditary characteristics of human genetic material or those that are acquired during the embryonic phase. This also includes all other laboratory tests whose primary purpose is to provide such information about genetic material. The term “genetic test”, which includes the research context, also encompasses genetic tests that are carried out in a biomedical context, without being subject to the HGTA. This includes, for example, the genetic investigation of a tumour for which underlying somatic mutations are neither hereditary nor acquired during the embryonic phase, but which have occurred during the subject’s life [9]. However, there is a grey area in these definitions because novel sequencing techniques also facilitate the discovery of inherited traits in tumour samples.
In this article, “genetic investigations” and “genetic testing” are employed as synonyms. However, the term “genetic analysis” refers to the genetics of clinical routine within the HGTA context. The development of specific genetic tests, if applicable, is discussed separately. The term “research dossier” includes all the documents that were submitted for ethical approval, including the protocol, patient information sheet and contract.
Data source and endpoints investigated
Since 1 January 2016, it has been mandatory for sponsors and project leaders of Swiss research projects that fall under the HRA to submit all applications to Swiss ethics committees. This is executed through the centralised electronic submission portal, the Business Administration System for Ethical Committees (BASEC). swissethics is the national umbrella organisation of the seven cantonal ethics committees and operator of BASEC. Thus, on 11 June 2019, swissethics was mandated by the Federal Office of Public Health (FOPH) to search BASEC in order to extract and analyse research projects including genetic research that were submitted to and approved by the ethics committees in 2018. This mandate authorises swissethics to publish the key results of this survey in a scientific journal. All sponsors of and applicants to these research projects have provided agreement to this review by submitting their applications through BASEC. None of the applicants made use of their right to object.
As per the HRA, the applications can be differentiated into three categories:
- Clinical trials
- These are research projects in which persons are prospectively assigned to a health-related intervention in order to investigate its effects on health or structure and function of the human body. Clinical trials are governed by the Clinical Trials Ordinance (ClinO).
- Research projects involving persons
- These are research projects in which biological material is prospectively sampled or health-related personal data are collected from a human being. These are known as
- Research projects involving the further use (FU) of data and/or biological material that have already been collected for research purposes or clinical routine in the past.
- These research projects are governed by chapter 3 of the HRO. This category further distinguishes between projects in which participants’ informed consent or general consent is obtained prior to conducting the research project and those in which such informed consent cannot be obtained for specific reasons. In the latter case, the ethics committee decides whether certain specific legal conditions that are established by article 34 of the HRA are met, and whether the ethics committee can approve the project, if the requirements are actually fulfilled. In this manuscript, this entire category is referred to as further-use projects.
Research applications were analysed with regards to the general frequency of genetic investigations, overall purpose, number of research participants or data sets that were included, and the diagnostic categories and methods that were employed. We also recorded the number of genes that were studied, the conditions for storage and retention of the biosamples and genetic data, and the type of laboratories or third parties that were involved. The processes involved in the informed consent procedure, result communication and handling of potential surplus information, i.e., incidental findings, were also analysed.
Study sample
Of the 2369 applications submitted to BASEC in 2018, all were included in the analysis for the present study. Of these, 537 applications were clinical trials. For research projects involving human being and further-use projects involving data and biological material, the BASEC submission procedure requires the applicant to directly state in the submission document whether the project contains genetic research. As a result, projects containing genetic research could be directly extracted from the database. The 537 clinical trial protocols were screened for predefined keywords that referred to genetic research or methodology. These keywords included DNA, RNA, gene, geno-, marker, code, transcription, translation, replication, and chromosome. Most of the protocols were in English; when protocols were in French or German, the respective keywords from the appropriate language were employed. There were no protocols in Italian. Positive hits were then manually analysed to determine whether genetic research involving human beings was planned in each clinical trial. This process identified overall 128 clinical trials containing genetic investigations. Among these, the first 45 clinical trials with genetic investigations, i.e., a sample of 35%, were selected according to their submission date, and an in-depth protocol analysis of the qualitative above-defined endpoints was then performed on this sample.
In addition, the full analysis comprised all nonclinical trials involving human beings (according to chapter 2, HRO), nonclinical trials containing genetic investigations (with 17 out of 844 projects submitted from this category), and all further-use research projects involving data and/or biological material containing genetic investigations (according to chapter 3, HRO, with 60 out of 987 submitted in this category).
Overall, 122 research applications were thus analysed in detail, including 45 clinical trials, 17 research projects involving human beings, and 60 research projects involving further use of data and/or biological material.
Results
Frequency, characteristics and number of participants/data sets
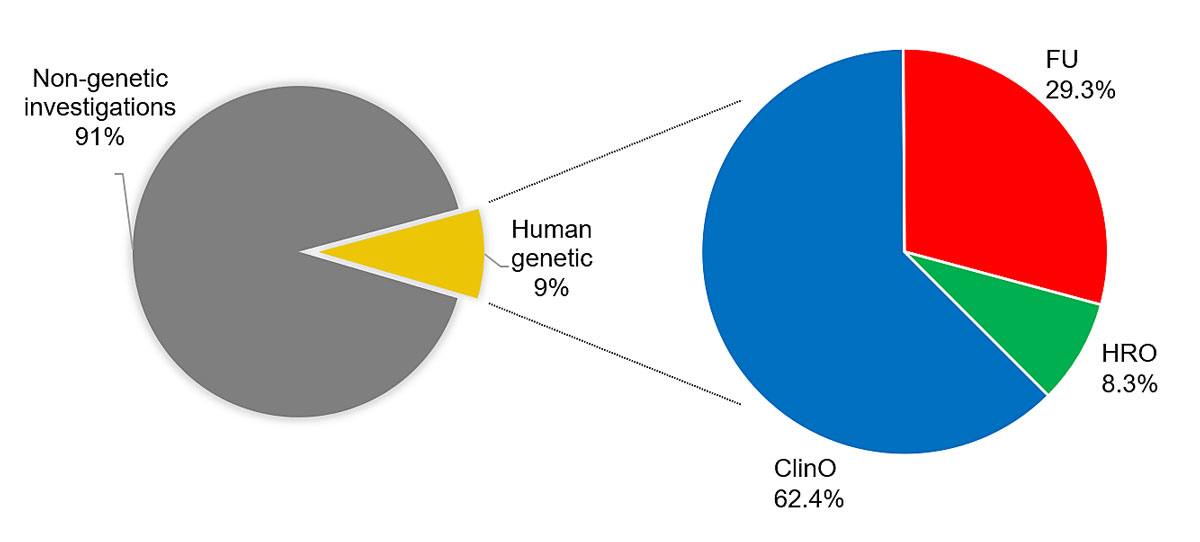
Genetic investigations involving human beings were retrieved from 9% of all applications submitted to Swiss ethics committees in 2018 (figure 1). Genetic investigations were mostly carried out in clinical trials: 128 applications out of 537 clinical trials, corresponding to 24%, included genetic research. This was followed by further-use research projects, with 60 applications out of 987 projects, corresponding to 6.1%. Finally, there were 13 nonclinical trials out of 844 projects overall, corresponding to only 1.5%. Genetic research within clinical trials was performed in phase I (11%), phase I/II (8%), phase II (31%), phase III (39%) and phase IV (11%) trials (data not shown).
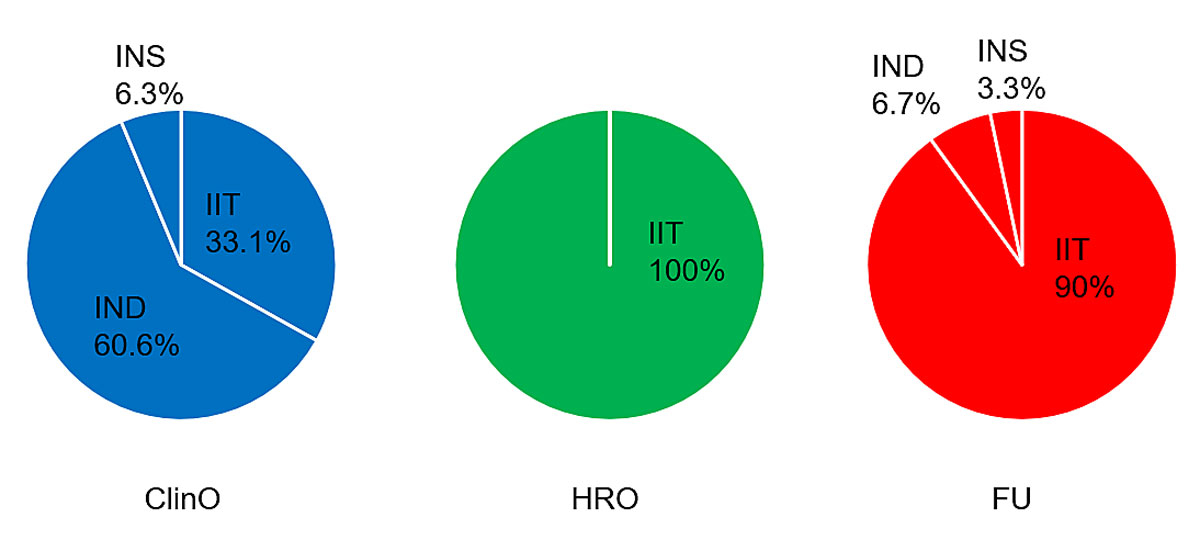
Genetic investigations almost exclusively focused on the nature or specific genetic aspects of the diseases investigated. Of all the clinical trials that included genetic research, the regulatory sponsor was the pharmaceutical industry in 63% of trials, principal investigators in 31% and academic institutions or foundations in 6%. Conversely, in nonclinical trials according to chapter 2, HRO, and in further-use research projects, principal investigators / project leaders almost exclusively acted as sponsors or project leaders in the regulatory sense (100% in observational research projects and 90% in further-use research projects; figure 2).
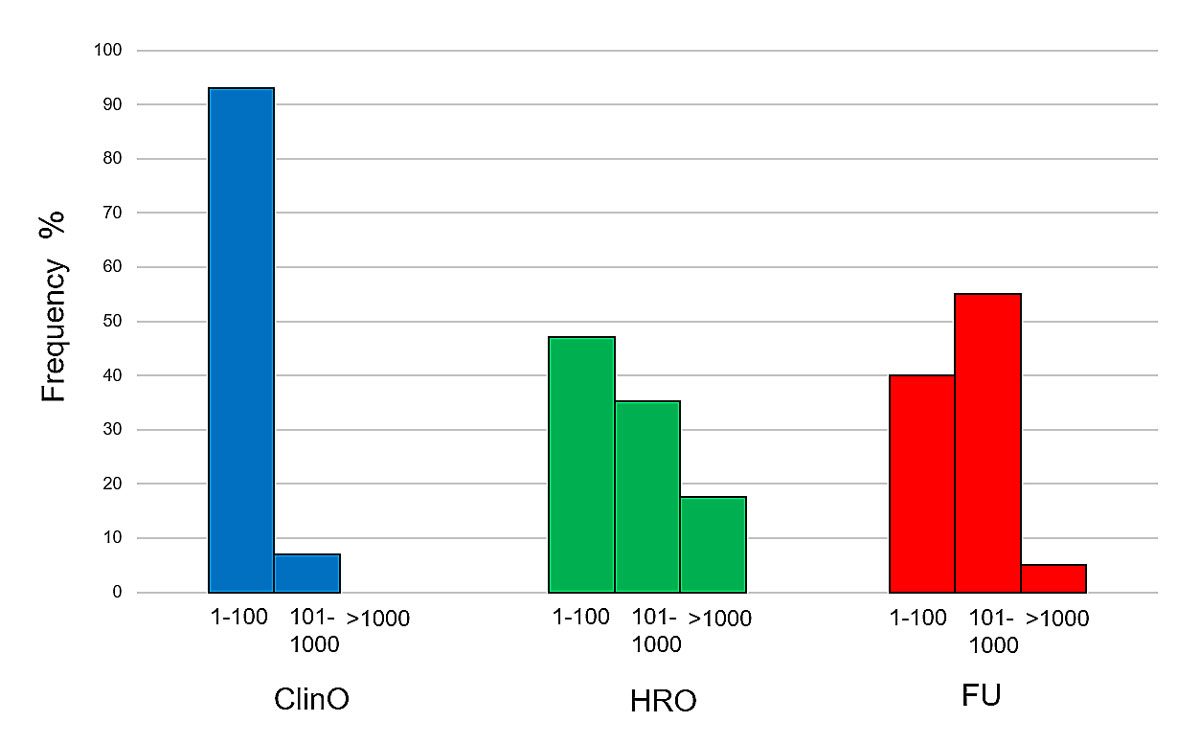
For participant/dataset analysis, patients and datasets that were subject to genetic investigations were grouped into three groups according to their number and size. These were research projects involving 1–100, 101–1000 and more than 1000 human beings or datasets. According to the numbers provided in the trial protocols and BASEC application forms, around 44,000 participants/datasets were intended for analysis. Most often, clinical trials included 1–100 human beings (93% of protocols). In contrast, nonclinical trials with persons or further-use projects mainly included larger cohorts/datasets. For example, for further-use projects, 55% of protocols included 101–1000 datasets, and 5% even more than 1000 datasets (figure 3). It should be noted that these numbers of datasets intended for analysis were provided by the applicant during the submission process. The actual number of participants enrolled or datasets analysed in these investigations cannot be correctly derived from these data. In 13% of applications, genetic investigations were conducted on children and adolescents, whereas no adults lacking capacity were included in the research projects.
The purpose of genetic research
Based on its overall purpose, genetic analysis was classified into four groups: (1) basic genetic research; (2) search for known or unknown genetic biomarkers; (3) genetic screening for already predetermined genes; (4) development of genetic tests. In clinical trials, genetic research primarily aimed to assess potential genetic biomarkers, particularly in pharmacogenetics, which accounted for 54% of trials with genetic investigations. This was followed by basic research (28%, data not shown). For further-use research projects, basic research was the primary purpose, with 85% of projects including genetic investigations, followed by the development or assessment of genetic biomarkers (13%). Overall, only 8% of all applications were focused on genetic screening of well-known genes. Among all the research protocols evaluated, none employed genetic investigations with the aim of developing a specific genetic test.
Diagnostic categories and methodology
Genetic research was mainly carried out in oncology (56%). Somatic genetic testing, i.e., testing to investigate genetic mutations that occurred after birth such as in tumour cells, and germline genetic testing, i.e., testing to examine inherited mutations, differ significantly and provide different information. Whereas germline mutations are generally transferred to the offspring, somatic tumour mutations provide information on tumour growth and aggressiveness. However, the comparison between tumour gene mutations and germline gene mutations may help better understanding of both tumour development and progression. In oncology, almost all projects thus included two analysis referring to somatic and germline tissue mutations. As shown in table 1, genetic cancer-related research was mainly carried out in the setting of haematological and gynaecological malignancies, the latter primarily concerning breast and ovarian cancers. Thoracic tumours were the next most common, followed by urogenital and gastrointestinal malignancies. Other carcinomas were accounted for 23% of overall oncological research. The proportion of genetic research in benign tumours amounted to only 3% of the sample assessed.
Table 1 Diagnostic categories of oncological versus nononcological diseases.
|
Diagnosis
|
ClinO (%)
n = 45
|
HRO (%)
n = 17
|
FU (%)
n = 60
|
Total (%)
n = 122
|
|
Malignant oncological
|
60.9
|
36.8
|
59.1
|
56.5
|
| Haematology |
14.3 |
0.0 |
25.6 |
18.9 |
| Gynaecology |
14.3 |
28.6 |
12.8 |
14.9 |
| Thoracic |
17.9 |
0.0 |
12.8 |
13.5 |
| Gastroenterology |
14.3 |
0.0 |
10.3 |
10.8 |
| Urogenital |
3.6 |
28.6 |
10.3 |
9.5 |
| Neurology |
14.3 |
0.0 |
2.6 |
6.8 |
| Dermatology |
3.6 |
14.3 |
0.0 |
2.7 |
| Other carcinomas |
17.9 |
28.6 |
25.6 |
23.0 |
|
Benign oncological
|
0.0
|
0.0
|
6.1
|
3.1
|
|
Nononcological
|
39.1
|
63.2
|
34.8
|
40.5
|
| Neurology/psychiatry |
16.7 |
16.7 |
21.7 |
18.9 |
| Healthy volunteers |
11.1 |
8.3 |
8.7 |
9.4 |
| Pulmonology |
16.7 |
8.3 |
0.0 |
7.5 |
| Dermatology |
11.1 |
8.3 |
4.3 |
7.5 |
| Cardiology |
0.0 |
16.7 |
0.0 |
3.8 |
| Infectious diseases |
0.0 |
8.3 |
4.3 |
3.8 |
| Haematology |
0.0 |
8.3 |
0.0 |
1.9 |
| Others, including rare diseases |
44.4 |
25.0 |
60.9 |
47.2 |
|
Total
|
100.0
|
100.0
|
100.0
|
100.0
|
Genomic studies were exclusively focused on hereditary germline genetic material and carried out in the nononcological area (40% in total), with genetic research primarily related to neurology and psychiatry (19% of nongermline research; table 1). Next followed genetic research on either healthy individuals or subjects affected by diseases pertaining to pulmonology, dermatology and cardiology. The group of other diseases was extremely heterogeneous and also included very rare diseases. This group accounted for a large proportion of the sample (47% of total germline genetic projects). The considerable heterogeneity of this group meant that further subgroups could not be analysed.
Conventional cytogenetics are no longer used in oncological or nononcological research. Instead, the methods of choice are molecular cytogenetics and sequencing methods for detecting mutations, deletions, inversions and others. Classic Sanger sequencing was only mentioned in two individual research protocols, since next-generation whole exome or whole genome sequencing methods have become the standard.
The methodologies employed differed considerably between research projects. Commercially available kits that cover a defined gene spectrum were most frequently used to determine somatic tumour aberrations in oncological research projects. For hereditary genetic testing, whole exome and whole genome sequencing with standard equipment were usually employed in a potentially nonvalidated manner.
The range of genes analysed and retention of biological material
The number of genes examined in the research protocols was classified according to three groups: few genes (1–10), several genes (11–100) and many genes (over 100). In total, 17% of protocols fell within the category of 1–10 genes, 21% fell within the category of 11–100 genes and 16% investigated more than 100 genes (data not shown). Interestingly, 36% of the research protocols involving genetic research failed to specify the number of genes or amount of genetic information that was analysed.
Overall, 62% of genetic investigations in the analysed research projects were carried out in dedicated research laboratories focusing on research and not certified by the FOPH [10]. Overall, 17% of genetic research testing is conducted in pathology institutes, i.e., academic hospitals. Only 5% of the research genetic testing is carried out in specialised genetic laboratories that are certified by the FOPH [10]. The latter are probably “hybrid laboratories”, performing genetic analysis as a diagnostic tool for clinical routine and in a research setting as well. In 16% of projects, no information was provided regarding the type of diagnostic laboratory involved.
The retention and storage of biosamples used for these genetic investigations followed different patterns in the clinical trials, observational studies or further-use research projects. Most biological material that was retrieved within clinical trials was stored abroad in central biobanks of international pharmaceutical sponsors (54%), whereas biological material from observational projects and further-use projects was stored in Switzerland in academic research institutions (65% and 48%, respectively) or pathology institutions (36% for further use).
Coding, data analysis and data storage
The data analysis of genetic research involving humans was almost exclusively performed in a coded form (97%). Only one research protocol specified that data had to be anonymised before analysis. No genetic investigations planned to use un-coded information. Extremely large amounts of data were generated in individual research projects, including a project that created 1 terabyte of data per tumour sample. Genetic investigations were directly carried out by research staff from the associated institution in 65% of cases, whereas genetic investigations were to be performed by external commercial providers in 29% of applications. The vast majority of research projects lacked specific information regarding the ways in which data would be stored after completing the research project (88% of projects). Likewise, details about the format of genetic data storage were not specified in almost all protocols, including information related to raw data, base pair sequences, sequence variants and individual gene results.
Informed consent procedure
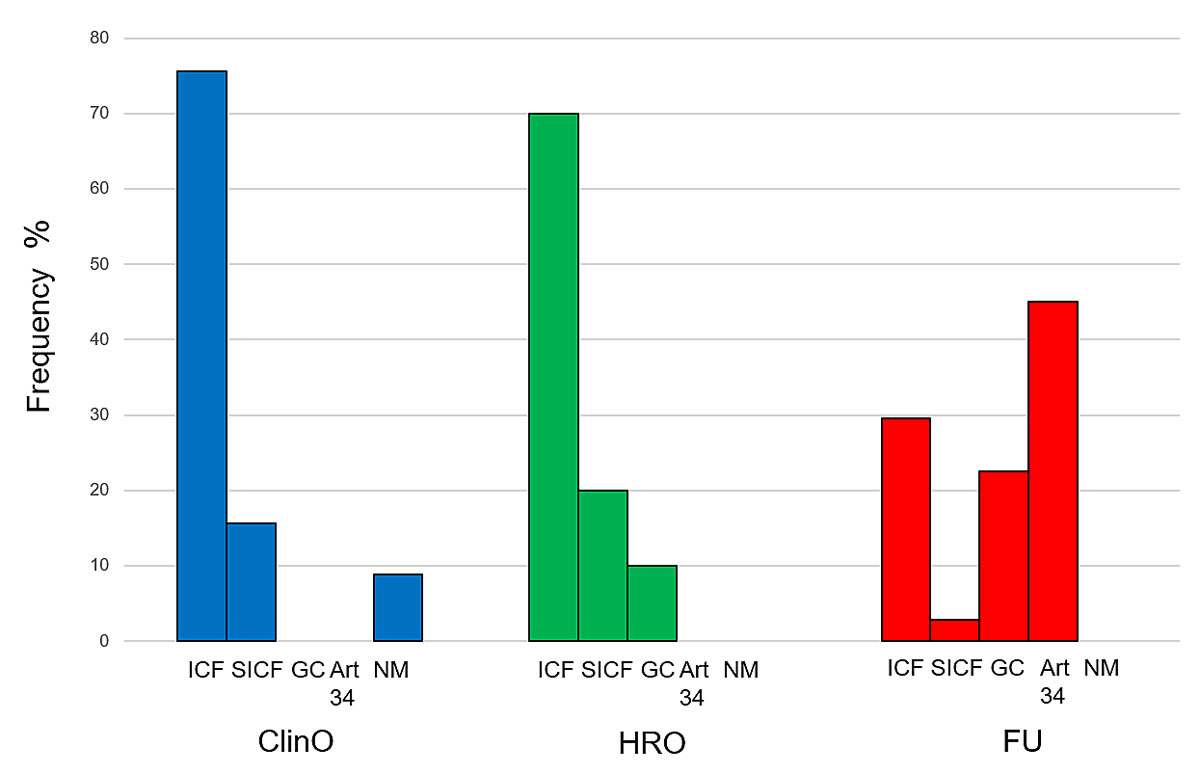
Research participants were informed about genetic investigations in different ways. In most cases, participants were informed about the genetic investigations being conducted on their biosamples in the project’s informed consent form (ICF) or in an additional specific informed consent form (SICF), which acts as an addendum to the project’s ICF (figure 4). In Switzerland, using project-unspecific general consent is permitted for further-use research projects that involve coded genetic data and/or biological material. General consent was employed in 27% of further-use projects.
In a large fraction of the further-use projects analysed (42%), the ethics committee approved using health-related personal data and/or biological material with only partial or a total absence of explicit participant consent. Many projects had a mix of consent and no consent, which were counted as projects without consent. Consequently, it is not possible to precisely ascertain the number of genetic investigations that were conducted without consent, according to the provisions of article 34 of the HRA.
In four approved clinical trials, no specific information about the planned conduct of genetic investigations was given to participants, according to the information provided in research applications. Genetic counselling by a medical genetic specialist prior to the genetic analysis, which is a standard procedure in clinical routine, was documented in only four research protocols (3%).
Communicating results and incidental findings
For 55% of genetic investigations, there was an intention to communicate the genetic research results to participants (“you will be informed”), according to the information provided in the ICF and the research protocol. Interestingly, in clinical trials and nonclinical trials that involved persons, results from the genetic investigations performed were communicated in 84% and 71% of cases, respectively. By contrast, further-use projects included plans to communicate results in 35% of cases.
The communication of potential incidental genetic findings is an essential ethical issue. In general, it was more commonly scheduled for projects to communicate than not communicate incidental findings to participants, based on the submitted dossiers. In 36% of cases, the ICF informed participants that they would be contacted in the case of potentially relevant incidental clinical findings, as based on current knowledge. In about 10% of cases, the participant could decide whether s/he wanted to be informed or not. In only 16% of cases, incidental findings were explicitly not communicated. About 20% of protocols for further-use research projects did not obtain participants’ consent; in these cases, the logistical or ethical reasons for not communicating incidental findings are not applicable, since re-contacting people was impossible, for example because research participants had already passed away. Overall, 8% of projects lacked information in the ICF and research protocol, whereas incidental findings were excluded for methodological reasons in 10% of cases, such as only a low number of specific genes being analysed by methods that did not generate a relevant amount of excess genetic information.
Discussion
Genetic research generates essential knowledge for science and society, exerting a major impact on the future advancement of biomedicine, personalised health and life science. Ethical and legal issues such as consent procedures and data security are of critical importance. Genetic analysis in research differs fundamentally from diagnostic genetic testing in clinical routine. Because of this diversity, different definitions of genetic data in the HRA research context on one hand, and clinical standard diagnostic procedures under the HGTA on the other hand, are not only acceptable but rather necessary. Genetic investigation within clinical trials and genetics in further-use projects are very heterogenic and characterised by different patterns in project leader, number of genes analysed, purpose of the project and handling of incidental findings. In industry-sponsored clinical trials, genetic investigation usually comprises a targeted analysis of a small number of well-defined potential biomarkers for pharmacological interventions. In contrast, further-use projects that are initiated by researchers or academic institutions explore genomic mechanisms or investigate genetic variants based on a very large number of genes and participants.
The current results indicate that genetic data protection within research projects are usually compliant with the applicable provisions of national law and international guidance documents, based on the dossier-provided information. However, these documents do not specifically regulate details of genetic data storage, i.e., whether genetic data are stored per base pair sequence, gene variants or genes, or other. It remains unclear whether the data protection level for genetic investigation-derived data in other countries, such as the United States (US) or Asia, is comparable to that applied in Europe and Switzerland, or whether it fulfils the Taipei Declaration criteria that focus on the ethical principles pertaining to health databases and biobanks [11]. Considering the amplification of individual genetic data that are currently available, as well as data storage and related risks, adequate data protection rules should be applied worldwide in the near future.
Today, the high safety standards for clinical trial participants, which have been achieved via regulatory efforts over past decades, must be balanced against the additional regulatory burden, with a negative impact on biomedical research speed and effectiveness. At present, the regulatory framework is already associated with increased drug development times and decreased patient benefits, particularly in medical fields such as oncology [12]. The current national discussion designed to adapt the regulatory requirements for somatic genetic investigations to the regulatory level of germline research should therefore be revisited. Most innovative drug developments, particularly those pertaining to oncology and neurology research, currently include genetic patient testing in clinical trials. The rationale behind this is the effort to design and develop innovative medicinal products able to target those patients who are most likely to benefit from them. Further increasing the regulatory burden of such trials may result in a situation where the proportion of Swiss patients within such global drug development projects is meant to further decrease. Swiss patients are then more likely to be excluded from having access to experimental next-generation drug treatment options, while, similarly, Swiss clinical centres and researchers are not permitted to participate in major medical progress, especially regarding personalised “tailored” drugs. The current results demonstrate that all germline and nongermline research projects submitted to Swiss ethical committees in 2018 would fall under such a strict genetic research regulation. As a result, these research projects would be required to meet the particularly strict rules pertaining to quality, informed consent, consultation and communication that are currently applied to genetic diagnostic procedures performed outside of the research context.
Genetic investigations in research are mainly not carried out in laboratories accredited by the FOPH, with their results thus not validated. They are therefore exploratory in nature and hypothesis-generating, as regards their experimental nature. In the clinical setting, genetic results are confirmed by means of a second independent method [13], the latter generating a verified, definitive result. In addition, the diagnostic laboratories involved regularly undergo internal and external quality control, resulting in certification and regular recertification [14–16]. This gold standard of routine clinical genetic analysis is not applied to any of the evaluated research projects per protocol, but the research protocols are not especially focused on the quality expertise of research genetic laboratories. Indeed, laboratories involved in genetic analysis in the research context may have comparable quality standards – or even superior methodology and experience in the field – although they are not certified by the FOPH.
Regarding communication of genetic results and incidental research findings, the main foci of researchers, participants and other parties are quite distinct [17]. Significant differences in communicating incidental findings to participants were observed, ranging from active communication to no such communication, or leaving the decision to the research participant. All these variants display limitations with respect to an ethically adequate handling of the right to either know or not to know [18, 19]. The American College of Medical Genetics and Genomics (ACMG) published a list of genes that are actionable; if one or more variants of these genes are found incidentally (revised 2016 and 2019) [20, 21], this should thus be communicated to the participant. These actionable variants according to the ACMG list will not be detected accidentally in any further-use project if the researchers are not actively looking for these variants. Additionally, the vast majority of variants identified in research projects are variants of unknown significance (VOUS), especially in the absence of good phenotypic information, family history and segregation, as is the case in the research setting compared to clinical routine. The significance of VOUS regarding the impact on individual health is, by definition, unknown. Some gene variants are reclassified at a later time with regard to their pathogenicity [22], rendering the context even more complex. In summary, incidental findings in further-use projects should often not be communicated to participants, owing to their explorative nature and lack of a clear outcome. These incidental findings are not validated and confirmed in a way that would be mandatory in clinical routine. Therefore, they may not be considered as real results with corresponding validity.
In exceptional cases, such as when researchers explicitly look for actionable genes, the social and ethical impact of these findings may prove to be significant, not only for the individuals themselves, but also for their relatives and offspring. The ELSI (ethical, legal and social issues) group of the Swiss Personalised Health Network therefore suggested including a multidisciplinary Expert Panel Board that takes part in the difficult decision-making process with respect to communication or non-communication [23]. Although this approach appears extremely useful, this board should only be involved when actionable incidental findings are expected owing to the specific nature of the research project.
Concerning the informed consent procedure in genetic research, it remains unclear how this is managed in daily practice, and especially whether and what essential verbal information is additionally provided. This significantly differs from the clinical setting, where careful genetic consultation has been implemented as a procedure ensuring legal and ethical standards [16]. The complexity of the written and mutual informed consent process cannot simply be addressed by changing the HRA, revising the informed consent documents or providing general recommendations. Every single personal situation is unique for the individual and should be handled in its particular context. From the authors’ point of view, oral information and counselling conducted by an experienced genetic researcher should be considered in the research context when actionable mutations are expected.
At present, the HRA and HGTA provide the current legal framework for genetic investigations in a research setting and for genetic analysis in the medical setting. The current different definitions of genetic data in both Acts and the associated different practices in research and routine should be maintained. Further, there are practical aspects aimed to avoid further regulatory burdens. Indeed, double quality checks on genetic research data in FOPH-accredited laboratories would hinder most current projects, given that the additional benefit is unknown, and resources are lacking.
In summary, ethicists play a fundamental role ensuring that the three HRA pillars, including the protection of dignity, privacy and research participants’ health, are guaranteed at all times, and that the participants’ risks are adequately balanced. At the same time, the legal aspects in this very dynamic and changing field must be continuously assessed and adapted to medical progress [24]. Transparency with respect to research participants is probably the most essential step to build further trust in future biomedical genetic research. The protection of the individual is the uppermost priority, but it should not preclude the important progress through research involving human beings.
Acknowledgment
swissethics wishes to thank the FOPH, Division of Biomedicine, Human Research Section, Bern, for financing and supporting this project. The authors thank Michael Tüller for the BASEC data export and CremerConsulting for linguistic support. The authors are also grateful to all the researchers who submitted a wealth of interesting research protocols and dossiers in their research fields.
References
1
van El
CG
,
Cornel
MC
,
Borry
P
,
Hastings
RJ
,
Fellmann
F
,
Hodgson
SV
, et al.; ESHG Public and Professional Policy Committee. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013;21(6):580–4. doi:.https://doi.org/10.1038/ejhg.2013.46
2
Claussnitzer
M
,
Cho
JH
,
Collins
R
,
Cox
NJ
,
Dermitzakis
ET
,
Hurles
ME
, et al.
A brief history of human disease genetics. Nature. 2020;577(7789):179–89. Published online January 8, 2020. doi:.https://doi.org/10.1038/s41586-019-1879-7
3
Roden
DM
,
McLeod
HL
,
Relling
MV
,
Williams
MS
,
Mensah
GA
,
Peterson
JF
, et al.
Pharmacogenomics. Lancet. 2019;394(10197):521–32. Published online August 5, 2019. doi:.https://doi.org/10.1016/S0140-6736(19)31276-0
4Federal Act on Human Genetic Testing (HGTA) of 8 October 2004 (Status as of 1 January 2014). The Federal Assembly of the Swiss Confederation (810.12).
5
Vokinger
KN
. Gesundheitsdaten im digitalen Zeitalter. Jusletter. 2020;27. doi:.https://doi.org/10.38023/c41ccdd8-8614-4ede-9926-a4331d4d6622
6Federal Act on Research involving Human Beings (Human Research Act, HRA) of 30 September 2011 (Status as of 1 January 2020). The Federal Assembly of the Swiss Confederation (810.30).
7
Horton
R
,
Crawford
G
,
Freeman
L
,
Fenwick
A
,
Wright
CF
,
Lucassen
A
. Direct-to-consumer genetic testing. BMJ. 2019;367:l5688. doi:.https://doi.org/10.1136/bmj.l5688
8WHO. Public Disclosure of Clinical Trial Results, Reporting of Findings of Clinical Trials. https://www.who.int/ictrp/results/en/. last accessed 2020 June 8..
9Swissmedic: Merkblatt genetic tests: MU500_00_003e_MB - Merkblatt_AW - Anweisung / V2.0 / ans / wam / 01.01.2017. https://www.swissmedic.ch/dam/swissmedic/en/dokumente/medizinprodukte/mu500_00_003d_mbgentests.pdf.download.pdf/mu500_00_003e_mbgenetictests.pdf
10
https://www.bag.admin.ch/bag/de/home/gesetze-und-bewilligungen/gesuche-bewilligungen/gesuche-bewilligungen-bereich-genetische-untersuchungen/zyto-molekulargenetische-untersuchungen.html
11WMA Declaration of Taipei on Ethical Considerations regarding Health Databases and Biobanks. 67th WMA General Assembly, Taipei, Taiwan, October 2016. https://www.wma.net/policies-post/wma-declaration-of-taipei-on-ethical-considerations-regarding-health-databases-and-biobanks/
12
Stewart
DJ
,
Whitney
SN
,
Kurzrock
R
. Equipoise lost: ethics, costs, and the regulation of cancer clinical research. J Clin Oncol. 2010;28(17):2925–35. doi:.https://doi.org/10.1200/JCO.2009.27.5404
13
Schweizerische Gesellschaft für Medizinische Genetik. SGHG. https://sgmg.ch
14Verordnung über genetische Untersuchungen beim Menschen (GUMV) of 14 February 2007 (status as of 1 February 2019). The Swiss Federal Council (810.122.1), in public consultation May 2020.
15
Schweizerische Kommission für Qualitätssicherung im Medizinischen Labor (QUALAB). http://www.qualab.ch
16
Schweizerische Akkreditierungsstelle (SAS). https://www.sas.admin.ch
17
Jamal
L
,
Schupmann
W
,
Berkman
BE
. An Ethical Framework for Genetic Counseling in the Genomic Era. J Genet Couns. 2019. doi: .https://doi.org/10.1002/jgc4.1207
18
Appelbaum
PS
,
Waldman
CR
,
Fyer
A
,
Klitzman
R
,
Parens
E
,
Martinez
J
, et al.
Informed consent for return of incidental findings in genomic research. Genet Med. 2014;16(5):367–73. Published online October 24, 2013. doi:.https://doi.org/10.1038/gim.2013.145
19
Castellanos
A
,
Phimister
EG
,
Stefánsson
K
,
Clayton
EW
. Disclosure of Genetic Risk Revealed in a Research Study. N Engl J Med. 2020;382(8):763–5. doi:.https://doi.org/10.1056/NEJMclde1915107
20
Kalia
SS
,
Adelman
K
,
Bale
SJ
,
Chung
WK
,
Eng
C
,
Evans
JP
, et al.
Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55. doi:.https://doi.org/10.1038/gim.2016.190
21
ACMG Board of Directors. The use of ACMG secondary findings recommendations for general population screening: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21(7):1467–8. doi:.https://doi.org/10.1038/s41436-019-0502-5
22
Bombard
Y
,
Brothers
KB
,
Fitzgerald-Butt
S
,
Garrison
NA
,
Jamal
L
,
James
CA
, et al.
The Responsibility to Recontact Research Participants after Reinterpretation of Genetic and Genomic Research Results. Am J Hum Genet. 2019;104(4):578–95. doi:.https://doi.org/10.1016/j.ajhg.2019.02.025
23Recommendations developed by the Swiss Personalised Health Network, ELSI advisory group: Reporting actionable genetic findings to research participants. February 2020. https://sphn.ch/2020/05/19/reporting-actionable-genetic-findings-to-research-participants
24Federal Office Public Health. Human Research Act (HRA): Results of the evaluation and further action. 6 December 2019 (available in German and French). https://www.bag.admin.ch/bag/en/home/medizin-und-forschung/forschung-am-menschen/evaluation-humanforschungsgesetz.html