Cardiovascular aspects of COVID-19
DOI: https://doi.org/10.4414/smw.2020.20417
David J.
Kurz, Franz R.
Eberli
Stadtspital Waid and Triemli, Cardiology, Zürich, Switzerland
Summary
Coronavirus disease 2019 (COVID-19) is primarily a pulmonary disease, but also affects the cardiovascular system in multiple ways. In this review, we will summarise and put into perspective findings and debates relating to the diverse aspects of cardiovascular involvement of COVID-19. We will review evidence for the role of the renin-angiotensin-aldosterone system (RAAS), the risk of pre-existing cardiovascular disease in COVID-19 susceptibility and course, and the mechanism of acute and long-term myocardial injury.
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) uses membrane-bound angiotensin converting-enzyme-2 (ACE2) as a receptor for cell entry. ACE2 is part of an important counter-regulatory circuit antagonising the harmful effects of angiotensin II on lung and heart. Modulation of ACE2 may therefore affect disease susceptibility and disease course. However, observational clinical studies and one randomised trial have so far not yielded evidence for harmful or beneficial effects of blockers of the RAAS during COVID-19. Age, gender, and multi-morbidity all increase susceptibility to SARS-CoV-2. In contrast, pre-existing cardiovascular diseases do so only minimally, but they may aggravate the disease course.
Direct SARS-CoV-2 infection of the heart tissue and myocytes is rare. Nevertheless, COVID-19 may lead to myocarditis-like acute cardiac injury, characterised by myocardial oedema, but lacking extensive myocyte loss and lymphocytic infiltration. Independent of this, increases in cardiac biomarkers (troponin, N-terminal pro-brain natriuretic peptide, D-dimer) are frequent, especially in the phase of severe systemic inflammation and acute respiratory distress syndrome, and quantitatively associated with poor outcome. The pulmonary infection may result initially in right ventricular dysfunction, but in cases with severe systemic infection hypoxia, hyperinflammation and cytokine storm heart failure may eventually ensue.
Unlike other infections and inflammatory states, COVID-19 does not appear to trigger acute coronary syndromes. In children, even mild COVID-19 can induce a multisystem inflammatory syndrome with Kawasaki-like symptoms frequently accompanied by cardiogenic shock.
Introduction
The pandemic infection known as coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), currently dominates all of our lives, of both those who work in medical institutions and the general population. To date, over 50 million people have become infected worldwide, resulting in over 1.2 million deaths. The numbers for Switzerland at the time of writing were 25,000 infections and >3000 deaths attributed to COVID-19 [1]. After a first peak in the spring of 2020 and a subsequent respite during the summer, the pandemic has returned in many European countries, with a second wave beginning in September 2020, and with Switzerland transiently suffering one of the highest per capita infection rates in the world. However, this second wave has differed in many aspects from the first. Whereas the first wave showed particularly high infection rates in the regions of Switzerland bordering with Italy and France, the second wave hit the whole country more homogenously, with many rural regions being intensely affected. Furthermore, owing to more widespread testing, many more cases in mildly symptomatic, younger patients have been identified, resulting in an overall decrease in case fatality rates.
Although COVID-19 predominately affects the respiratory tract, the cardiovascular system may also be involved in a number of ways. Since SARS-Cov-2 enters the cell via the angiotensin converting-enzyme-2 (ACE2), there initially was controversy about the role of the renin-angiotensin-aldosterone system (RAAS) for infection susceptibility and infection course. There was uncertainty about the danger of pre-existing cardiovascular disease, the possible deleterious effect of direct myocardial and vascular infection and the trigger of acute coronary syndromes by COVID-19. More recently, concern about lasting damage to the heart came into the forefront. A plethora of publications, some of them rather anecdotal and some of them contradictory, was published within a few months. In this review, we will try to summarise and put into perspective the current understanding of what is known, and also what is not yet known, regarding cardiovascular aspects in COVID-19.
Clinical manifestations of COVID-19
SARS-CoV-2 is the seventh coronavirus known to be pathogenic to humans. Four Coronaviridae (229E, OC43, NL63 and HKU1) manifest as a “common cold”, whereas the other three are more pathogenic and may lead to severe pneumonia and death. The SARS-CoV virus was responsible for the severe acute respiratory syndrome (SARS) epidemic of 2002 and the MERS-CoV virus for the Middle East respiratory syndrome (MERS) of 2012. Just like SARS-CoV and MERS-CoV, SARS-CoV-2 is thought to have originated in bats and to have been transferred to humans via an intermediate host [2, 3]. SARS-CoV-2 interacts with two surface proteins on human cells in order to gain entry into the cell, one being the transmembrane proteinase serine 2 (TMPRSS2), the second being ACE2 [4]. TMPRSS2 activates the spike protein of SARS-CoV-2, which subsequently binds to ACE2 and allows fusion of the viral and cellular membranes [4]. The SARS-CoV-2 spike protein / ACE2 complex then enters the cell through endocytosis. ACE2 is expressed in high levels in the upper airway (ciliated epithelial cells, goblet cells), the lungs (alveolar type II epithelial cells), but also in the heart (cardiofibroblasts, cardiomyocytes, endothelial cells, pericytes, epicardial adipose tissue), the vasculature (endothelial cells, migratory angiogenic cells, vascular smooth muscle cells), the gastrointestinal epithelium, the kidneys (glomerular endothelial cells, podocytes, proximal tubular endothelial cells), the liver (cholangiocytes, hepatocytes), the nervous system, the testis and the eye, explaining in part the multi-organ dysfunction present in severe cases of COVID-19 [5, 6].
Infection occurs via the respiratory tract as droplet or aerosolised transmission, similarly to influenza [7]. The incubation time ranges from 2–13 days (median 4–7 days). The time period of infectivity is uncertain, as is the rate of transmission by asymptomatic patients and the time period during which an individual remains contagious [2, 8, 9]. The viral RNA levels in specimens of the respiratory tract appear to be the highest at the time of symptom onset [2, 8]. Although infected individuals may remain contagious for up to 42 days [9], reassuringly the virus can no longer be detected in >90% of patients after 10 days [10]. There is a huge variability in the clinical manifestations of COVID-19, and there is an age-dependent association with disease severity [2].
The frequency of clinical symptoms of COVID-19 are summarised in table 1. Fever (80–90%), dry cough (60–70%) and shortness of breath (53–80%) are the most common early symptoms [11]. Fatigue and myalgia are also frequent. In contrast, rhinorrhoea occurs in only about 5% of patients. Loss of taste and smell is also prevalent among COVID-19 patients (88% and 86%, respectively) [12], and may be the sole presenting symptom [11]. Common laboratory abnormalities among patients requiring hospitalisation include lymphopenia, elevated C-reactive protein and abnormal coagulation parameters (elevated D-dimer, thrombocytopenia, low fibrinogen) [13].
Table 1 Frequency of clinical symptoms of COVID-19.
| Fever 80–90% |
| Dry cough 60–70%, productive cough 30–40% |
| Fatigue 40–60% |
| Loss of appetite 30–40% |
| Myalgia 20–30% |
| Dyspnoea 20–30% |
| Headache 10–20% |
| Sore throat 10–20% |
| Chills 10–20% |
| Diarrhea 5–15% |
| Palpitations, thoracic oppression 5–15% |
| Nausea/emesis 5–10% |
| Nasal congestion 5% |
To understand the disease course three clinical stages of COVID-19 have been proposed [14]. The clinical stage I is the stage of early infection of the patient with mild respiratory and systemic symptoms. Stage II is the pulmonary phase, characterised by viral multiplication and localised inflammation of the lungs. Cough, fever and hypoxia are the leading symptoms. Laboratory findings in this stage may include lymphopenia, increased liver enzymes and low procalcitonin. Clinical stage III is termed the hyperinflammation phase, during which markers of systemic inflammation are elevated, particularly inflammatory cytokines, C-reactive protein and D-dimer. Systemic organ involvement such as kidney failure and myocardial injury manifest during this stage [14]. Many clinical trials have been performed or are ongoing to investigate potentially beneficial antiviral medications or anti-inflammatory interventions, a selection of which is summarised in table 2. Of course, disease control will ultimately only be possible with a potent vaccine.
Table 2 Drugs under evaluation for COVID-19 treatment.
|
Drug
|
Mechanism
|
|
Disease modulating
|
| Camostat mesylate |
Blocks TMPRSS2
Inhibits entry of SARS-CoV-2 into the cell |
| Hydroxychloroquine |
Changes endosomal pH
Reduces fusion of the virus with the cell |
| Recombinant human ACE2 (APN01) |
Enzymatic cleavage of angiotensin-II. Production of angiotensin-(1–7)
Reduces pulmonary injury |
| Losartan |
AT1 receptor blocker
Reduces pulmonary and myocardial injury |
| Interferon |
Immune activator |
| Methylprednisolone |
Anti-inflammatory |
Tocilizumab
Sarilumab |
Anti-interleukin-6 receptor antibody
Anti-inflammatory. Improves ARDS |
|
Antiviral action
|
| Lopinavir//ritonavir |
Lopinavir = protease inhibitor
Ritonavir blocks CYP3A
Anti-HIV drugs |
| Remdesivir |
RNA-polymerase inhibitor
Anti-Ebola drug |
| Ribavirin |
Nucleotide analogue, virostatic
Anti-hepatitis C drug |
| Favipiravir |
RNA-polymerase inhibitor
Anti-influenza drug |
| ACE = angiotensin converting-enzyme; ARDS = acute respiratory distress syndrome; AT1 = angiotensin-II type 1 receptor; HIV = human immunodeficiency virus; TMPRSS2 = transmembrane proteinase serine 2 |
COVID-19 and the renin-angiotensin-aldosterone system
Early reports demonstrated an increased prevalence of COVID-19 among patients with hypertension and cardiovascular disease (17% and 5% of infected patients, respectively), and that hypertensive patients were at increased risk for severe disease [15]. In the large registry report including 72,314 cases from the “Chinese Centre for Disease Control and Prevention”, the case fatality rate was 6% among hypertensive patients and 10.5% in patients with cardiovascular disease, compared to 2.3% in the whole cohort [16]. These observations and the frequent use of angiotensin converting-enzyme inhibitors (ACE-Is) and angiotensin-receptor blockers (ARBs) [16] in these high-risk subgroups, coupled with the fact that SARS-CoV-2 uses the ACE2 protein to enter the cell, led to concerns that these drugs might increase susceptibility and disease severity through upregulation of ACE2 [17]. Some scientists even suggested stopping the use of ACE-Is and ARBs to slow pandemic spreading, an idea that was quickly taken up by the lay press and received wide publicity [18, 19]. However, other experts have taken a contrary position, suggesting that these drugs might be protective [20, 21]. Both are reasonable hypotheses considering the complexity of the RAAS, with regulative and endogenous counter-regulative measures present at multiple levels. An increased expression of membrane bound ACE2 might increase infection susceptibility and on the other hand an increased or preserved ACE2 function might be protective against RAAS-mediated damage in the course of the disease [22].
What is the evidence that chronic ACE-I or ARB treatment increases infection susceptibility toward SARS-CoV-2 by increasing ACE2 expression? In cell cultures and animal models ACE-Is and ARBs have shown mixed effects and thus there is no consistent evidence on the upregulation of ACE2 expression by RAAS inhibitors [20, 23]. In humans there is insufficient evidence for an upregulation of ACE2 by RAAS inhibitors [20, 23]. In fact, recent studies found that RAAS inhibitors were associated with lower, not higher, soluble ACE2 [24, 25] and lower ACE2 expression in the lung [26]. Furthermore, four large population-based case-control and/or retrospective studies in humans have found no increased risk of SARS-CoV-2 infection in patients on RAAS inhibitors [27–30]. Similarly, there is no evidence from retrospective studies that RAAS inhibitor use is associated with increased disease severity or mortality in COVID-19 [23]. Continuing ACE-I/ARB treatment in COVID-19 patients similarly did not cause harm in two studies [31, 32] and one study showed a tendency for a benefit [33]. So far, only one randomised controlled trial has tested whether continuation of RAAS blockers or their suspension is beneficial in COVID-19 patients [34]. The primary endpoint was days alive and out of the hospital at 30 days and was not different among the 659 randomised patients [34].
In order to better understand the role of RAAS and the potential beneficial effect of angiotensin-(1–7) in COVID-19, we need to keep in mind its complex nature (fig. 1). Inflammation can stimulate the RAAS and increase angiotensin-II. Angiotensin-II binds to the angiotensin-II type 1 (AT1) receptor resulting in vasoconstriction, increased vascular permeability, fibrosis, cell proliferation, and subsequently acute pulmonary injury and negative myocardial remodelling. These effects of angiotensin-II on the AT1 receptor are balanced by two counteractive circuits. First, angiotensin-II also activates the AT2 receptor, which – although less abundant – triggers opposing effects such as vasodilation and anti-proliferation [36]. Second, ACE2 is a potent counter-regulatory enzyme that degrades angiotensin-II to angiotensin-(1–7), which opposes the effects of angiotensin-II by binding to the MAS1 receptor. Activation of MAS1 by angiotensin-(1–7) triggers antifibrotic, antihypertrophic, vasodilatory, anti-inflammatory and antioxidant effects [22]. During SARS-CoV-2 infection, angiotensin-(1–7) may exert beneficial effects by reducing alveolar cell apoptosis, endothelial activation and pulmonary oedema, and limiting the production of pro-inflammatory and profibrotic cytokines [35]. However, SARS-CoV-2 infection might reduce ACE2 expression and activity by endocytosis and by shedding of membrane-bound ACE2 [6, 35]. The reduction of ACE2 results in an imbalance between ACE2/angiotensin-1–7/MAS receptor and angiotensin-II/AT1 receptor that aggravates the viral pneumonia or lung injury [23].
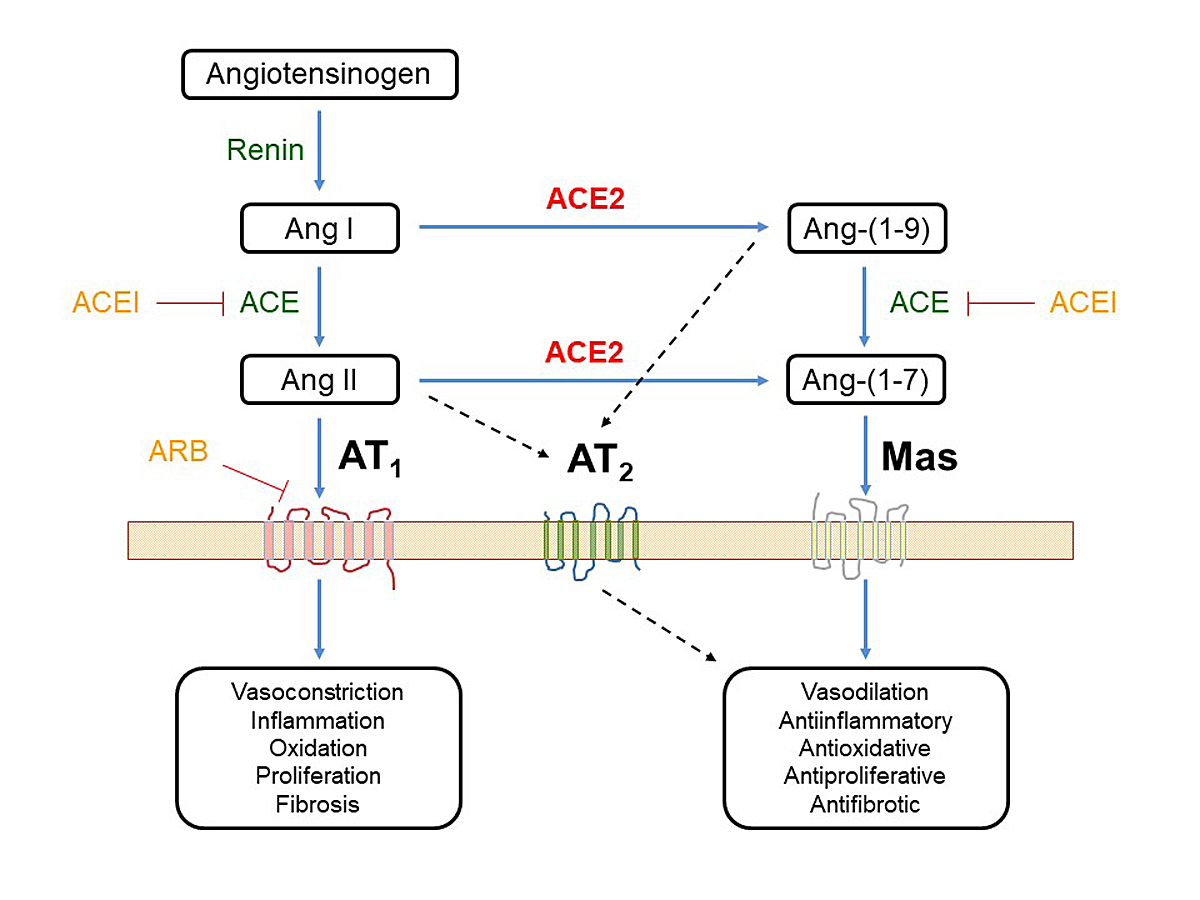
Figure 1
Role of angiotensin converting-enzyme-2 (ACE2) in the renin-angiotensin-aldosterone system (RAAS). The main metabolic pathway of the RAAS is driven by the angiotensin-converting enzyme ACE and the main effects are exerted by angiotensin II (AngII) via the angiotensin II type 1 receptor (AT1). The effects can be inhibited by ACE inhibitors (ACEI) or by AT1 receptor blockers (ARB). The effects of Ang II are balanced (1) by a simultaneous stimulation of Ang II type 2 receptors (AT2), which exert regulatory effects and (2) by ACE2 that cleaves Ang II into angiotensin-(1–7) (Ang-(1–7)) which exerts its effects via the MAS receptor (MAS). The biological effects of this second axis through ACE2/Ang-(1–7)/MAS counteract or modulate the effects of the classical axis [35].
In the SARS epidemic of 2002, the protective effect of ACE2 and angiotensin-(1–7) was recognised as crucial to the disease course [37]. Therefore, it would not be surprising to find beneficial effects of ACE2 upregulation during COVID-19 [20, 22]. Indeed, in animal models virus infection was shown to induce loss of ACE2 in the cell membrane, and the subsequently increased angiotensin-II activity contributed to local tissue damage [37]. This negative effect could be experimentally attenuated by treatment with ARBs [37]. Furthermore, in preliminary studies recombinant ACE2 reduced lung injury after influenza H5N1 infections and in acute respiratory distress syndrome [38]. Currently, trials investigating the effect of the ARB losartan and of recombinant human ACE2 in COVID-19 patients are ongoing (table 2) [39].
In summary, there is currently insufficient evidence to support the withdrawal of ACE-Is or ARBs, and as yet – despite promising preclinical data – no controlled trials indicating a therapeutic benefit from their use.
Pre-existing cardiovascular disease and COVID-19
Early reports from China indicated that pre-existing cardiovascular disease is a common comorbidity in COVID-19 [9, 10, 15–17, 40–42]. When patients with mild or moderate disease were compared with patients with severe or critical illness, cardiovascular disease, specifically hypertension, diabetes, coronary artery disease and heart failure, were associated with worse prognosis [9, 10, 16, 41]. This prompted health authorities to declare any person with pre-existing cardiovascular disease high risk for COVID-19 with respect to susceptibility as well as to a worse course of the disease. With respect to susceptibility to a SARS-Cov-2 infection, the data remain debated. In a meta-analysis of seven early studies from China comprising 1576 infected patients, the prevalence of hypertension was 21.1%, diabetes 9.7%, and cardiovascular disease 8.4% [15]. In the adult Chinese population the prevalence of hypertension is 23.2% and that of diabetes 10.9% [43, 44]. Therefore, these data do not support the notion of an increased susceptibility of patients with hypertension and/or diabetes for COVID-19. In an Italian study comparing a large number of COVID-19 patients with an unaffected population group with similar risk profile, pre-existing cardiovascular disease was not associated with increased risk for COVID-19 (hazard ratio 1.01, 95% confidence interval 0.91–1.10). In the same study, patients were scored according to the presence or absence of 31 diseases. Not surprisingly, the higher the number of pre-existing comorbidities, the higher was the odds ratio for contracting COVID-19.
The studies reporting a worse hospital course with hypertension, diabetes and pre-existing cardiovascular disease were unadjusted for other clinical characteristics, most notably age. In a study in severely ill patients, the age difference between survivors and non-survivors was 17 years [40]. In a small study of 201 patients examining risk factors for the development of acute respiratory distress syndrome (ARDS) and progression to death, the age difference between the groups was 10.5 years and unadjusted comparisons showed hypertension and diabetes to be associated with development of ARDS [41]. In a bivariate Cox model, hypertension and diabetes were still associated with development of ARDS, but were not independent risk factors for death [41]. Similarly, in a study examining the role of clinical characteristics on mortality, coronary disease, hypertension and diabetes were all associated with increased risk of death in a univariate comparison, but not after multivariate analysis [42]. Age, sequential organ failure assessment (SOFA) score (a mortality prediction score used in the intensive care unit based on the degree of dysfunction of six organ systems), lymphopenia and increased D-dimer levels were associated with higher risk of in-hospital mortality [42]. In summary, evidence suggests that hypertension, diabetes or coronary disease alone probably do not increase susceptibility for COVID-19, albeit multiple pre-existing comorbidities might play a certain role. With regard to disease course, age, grade of inflammation and pro-thrombotic state are the best predictors of multi-organ failure, whereas pre-existing cardiovascular morbidities can be regarded as modulating factors.
Myocardial injury during COVID-19
Myocardial injury, defined as an increase in myocardial enzymes such as troponin and/or decrease in pump function of the heart, can be caused by several putative mechanisms (fig. 2). The myocardial injury may be categorised into two groups: direct myocardial infection or myocardial damage secondary to effects of the pulmonary and systemic infection. In addition, there have been reports of COVID-19 triggering tako-tsubo myopathy [45, 46].
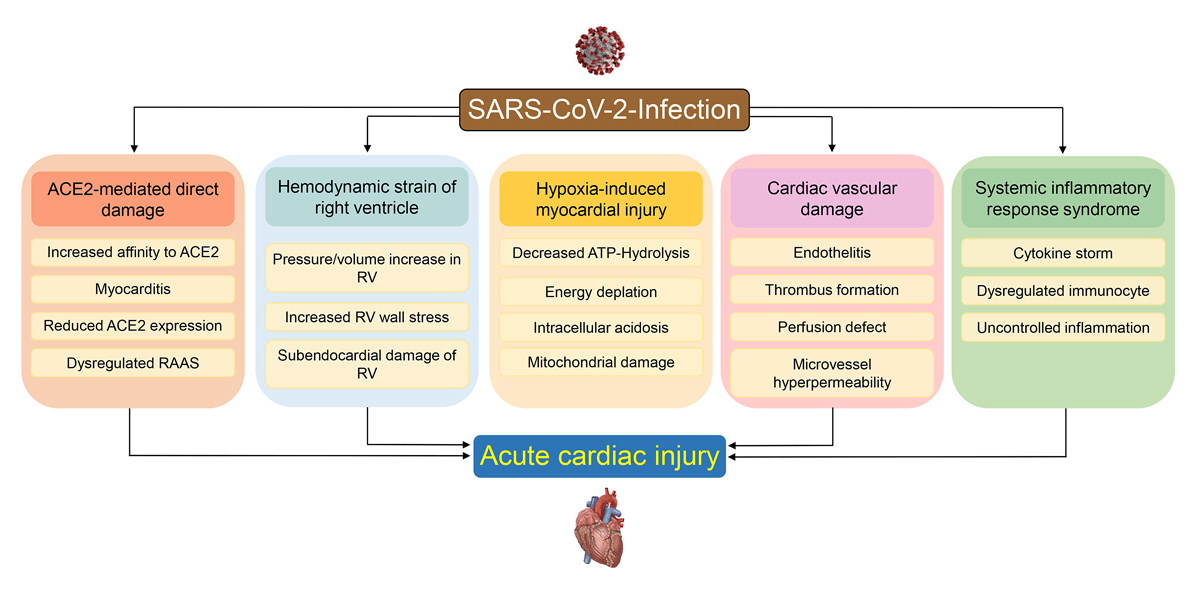
Figure 2
Potential mechanisms of SARS-CoV-2-mediated myocardial injury. Myocardial injury might occur secondary to the infection of the myocardium or secondary to mechanisms related to the pulmonary and systemic infection. These mechanisms include hypoxia induced decrease of oxygen supply to the heart, right ventricular pressure and volume overload, infectious damage of the macro-and microvasculature, and cytokine-induced injury. See text for details.
Myocarditis-like acute cardiac injury
ACE2 is expressed in the heart [5] and therefore a direct infection resulting in myocarditis is possible (fig. 2). Currently we do not know the invasiveness of SARS-Cov-2 in human hearts, neither do we know which cells in the human heart are preferentially infected. Cell types that carry ACE2 are cardiomyocytes, pericytes, cardiofibroblasts, endothelial cells and epicardial adipose cells [6, 47–49]. In addition, in the vasculature, endothelial cells, smooth muscle cells and migratory angiogenic cells express ACE2 [6].
A direct infection of cardiomyocytes would result in acute myocarditis. An early report form Wuhan, China reported clinical myocarditis being present in 7% of COVID-19 deaths [50]. Of 10 patients with “myocarditis-like acute cardiac syndrome” undergoing cardiac magnetic resonance imaging (MRI), eight were diagnosed as having myocarditis according to the 2018 Lake Louise criteria [51]. Interestingly, myocarditis in COVID-19 is characterised by unusual features, most notably the lack of widespread myocyte damage [49, 51]. A few cases with a predominant clinical picture of myocarditis, suggesting a direct myocardial infection, have been reported [52–57].
In pathology studies, some level of viral RNA could be detected in 5/12 (42%) and in 24/39 (61%) autopsies of COVID-19 victims [58, 59]. Despite the high prevalence of viral genome, no patient had acute myocarditis and the virus was assumed to be present in the interstitial space and in macrophages [58]. Studies examining which cells of the myocardium carry the virus are sparse [60]. In an 11-year-old boy who presented with multi-system inflammatory syndrome (MIS-C) and died of cardiac failure, electron microscopy detected viral particles in myocytes, endothelial cells and fibroblasts of the heart [52]. In one patient with rapid onset of myocardial damage caused by SARS-CoV-2 infection, the myocardial biopsy showed viral particles in the interstitial tissue, but not in the myocytes [57]. In a study of an acute lymphocytic myocarditis, molecular analysis showed absence of the SARS-CoV-2 genome within the myocardium [46], whereas in another study of two patients with myocarditis 4 weeks after COVID-19 infection, SARS-CoV-2 genome was found in the heart [61]. In three COVID-19 patients widespread endotheliitis was found, but no lymphocytic myocarditis [49]. In an international multicentre study examining heart tissue from autopsies of 21 consecutive patients only 3 (14%) showed lymphocytic myocarditis, whereas 18 (86%) showed increased macrophage infiltration [62]. The summed findings of 277 cardiac autopsies from 22 published studies revealed that myocarditis is rare in COVID-19, occurring in 7.2% of cases when myocarditis was defined as any nonspecific inflammatory infiltrates and in 1.4% when defined as lymphocytic infiltrates [63]. This low incidence of pathology-confirmed myocarditis contrasts with the dramatically high incidence of 60% myocarditis and 78% of cardiac abnormalities in MRI studies of COVID-19 patients 2 months after the infection [64] or of 26% myocarditis, 11% perimyocarditis and 3% pericarditis in another as yet unreviewed MRI study in 139 healthcare workers late after COVID-19 infection [65]. It is difficult to reconcile the findings of pathology studies with the MRI studies, with the first being reassuring and the latter worrisome with respect to lasting damage to the heart in recovered COVID-19 patients.
Indirect myocardial Injury
Cardiac injury as evidenced by an increase in cardiac biomarkers, such as high sensitivity (hs) troponin, is present in up to 27% of COVID-19 patients and portends a poorer prognosis. Except in the rare cases of acute myocarditis, a biphasic pattern of troponin elevation was usually observed. An early and mild elevation of hs-troponin during the early and pulmonary phases (stage I and II) is followed by a marked increase in troponin in the case of further deterioration and a hyperinflammatory course (stage III) of the disease [9, 14, 42, 66]. An elevation of cardiac troponin has been observed in other infections with myotropic viruses such as influenza or coxsackie [67, 68] and in pneumonia [69]. The modest increase in troponin levels frequently found in COVID-19 is probably not due to viral infection of the heart, as outlined above. Furthermore, increased troponin levels do not correlate with deterioration of systolic left ventricular function, even in the event of massive troponin increase or clinical deterioration [70, 71]. In a systematic echocardiographic study of COVID-19 patients at different levels of disease severity, elevated troponin levels were associated with right ventricular dysfunction, suggesting that the most common mechanism of increased troponin is acute right ventricular overload secondary to parenchymal or vascular lung disease resulting in subendocardial damage of the right ventricular myocardium (fig. 2) [71]. In a pathology study, high levels of troponin were associated with right ventricular myocardial injury most probably resulting from right ventricular strain [62]. In that study, 19% of deceased COVID-19 patients had histological signs of right ventricular strain [62]. Interestingly, clinical deterioration, including haemodynamic instability and further increase in cardiac troponins, seems to only rarely lead to left ventricular pump failure, whereas in most cases it is accompanied by a further deterioration of right ventricular function and increased right ventricle dilatation [70, 71].
Similar to troponin, increased levels of N-terminal pro-b-type natriuretic peptide (NT-proBNP) have been associated with disease severity and poor prognosis [9]. NT-proBNP and b-type natriuretic peptide (BNP) levels are markers of myocardial wall stress. Although there is a weak correlation with troponin levels, a substantial number of patients will manifest with increased BNP/NT-proBNP levels at normal or minimally increased troponin levels [9, 40, 42, 66]. This may be explained by pre-existing heart failure or predominant increase in right ventricular wall stress increasing BNP levels, but inducing only minimal troponin increase. On the other hand, high levels of BNP/NT-proBNP are known to occur in the setting of sepsis and other syndromes of systemic inflammation in non-cardiac patients, strongly predicting both short- and long-term mortality [72]. Several reports have documented a sharp increase in troponin levels with worsening disease and mortality [9, 40, 42, 66]. It is important to note that about one third of severely ill COVID-19 patients develop acute kidney failure, often necessitating haemodialysis. In these patients, troponin levels are unpredictably elevated owing to the lack of proper renal clearance of troponin, weakening their diagnostic use in the assessment of cardiac injury.
Nevertheless, cardiac injury in addition to right ventricular failure can occur. Several mechanisms may contribute to this injury and are summarised in figure 2. First, as outlined above, in rare cases viral myocarditis may directly damage the heart. Second, COVID-19 is primarily a pulmonary disease and subsequent ARDS results in profound hypoxia. Pulmonary function might be further impaired through occurrence of pulmonary emboli or microthrombi and may contribute to right ventricular overload. A pro-thombotic state, as evidenced by increased levels of D-dimer, is common and portends a poor prognosis [41, 42, 73, 74]. Pulmonary emboli are a feared complication of COVID-19. Currently several clinical trials are ongoing to test anticoagulation strategies to prevent embolic events in COVID-19. In addition, widespread vascular thrombosis, microangiopathy, and occlusion of alveolar capillaries are distinctive features of pulmonic pathology in COVID-19 patients [75, 76] and might further decrease oxygen saturation. Oxygen demand of the myocardium is increased in severely ill COVID-19 patients secondary to the tachycardia, increased cardiac output and increased afterload of the right ventricle. Decreased oxygen supply will result in decreased adenosine-triphosphate (ATP) hydrolysis and in the case of severe energy depletion lead to cell membrane damage, intracellular acidosis and mitochondrial damage, which further decrease ATP synthesis. (fig. 2). The resulting imbalance of increased oxygen demand and reduced supply might well result in subendocardial ischaemia. Such supply-demand ischaemia will induce diastolic dysfunction followed by systolic dysfunction, a scenario fitting with the findings of systemic echocardiographic studies in COVID-19 patients [71]. In the event of pre-existing coronary disease or heart failure supply-demand ischaemia will occur earlier, i.e., at higher oxygen levels than in hearts without pre-existing conditions. Third, even in cases without pre-existing cardiovascular disease, COVID-19 associated endotheliitis and pro-thrombotic state might cause macro- and microvascular damage, microthrombi and myocardial ischaemia, which in conjunction with hypoxia results in myocardial damage [49, 75]. Viral and bacterial infections can trigger a systemic inflammatory response syndrome or a cytokine storm [77]. Cytokine storm leads to profound hypotension and multiorgan failure. In the heart, extravasation of leucocytes and proteins may lead to tissue oedema and impede mechanical cardiac function [14, 78, 79]. The hyperinflammatory response in COVID-19 has several features that distinguish the observed “cytokine storm” from that of bacterial infections. These distinctive features are: modest increase of cytokine levels, most notably of interleukin-6, coagulation abnormalities, activation of endothelial cells rather than macrophages, exhaustion of lymphocytes and T-cell deficiency [80–82]. The lack of a distinct cytokine increase has advanced the notion that, instead of a hyperinflammatory response, COVID-19 may be accompanied by a viral sepsis [81].
Myocardial infarction and COVID-19
Inflammatory syndromes and active infections are known triggers of acute coronary syndromes. Contrary to expectations, there have been no reports of excess myocardial infarctions in the setting of COVID-19. Intriguingly, many centres performing acute coronary interventions even noticed a sudden drop in the frequency of ST-elevation myocardial infarction (STEMI) during the first weeks of the pandemic. Mafham et al. reported a 40% drop in the weekly rate of myocardial infarction admissions in the United Kingdom during the first wave of the pandemic compared with the weekly rate during 2019 [83]. Similarly, a large cohort analysis of myocardial infarction admissions in the United States also observed an initial decline during the first weeks of the pandemic, which returned to normal levels after several weeks. This was associated with higher than expected myocardial infarction mortality rates, especially for STEMI [84]. In our institution, compared with our year-round average, the weekly STEMI rate decreased by 39% in the first 2 weeks after “lockdown” procedures began. This worldwide phenomenon was interpreted to be due primarily to patient’s fear of leaving their homes, or even coming to the emergency rooms because of the media reports of hospitals being flooded with COVID-19 patients. A study from Hong Kong clearly demonstrated that patient delay in STEMI (time from symptom onset to first medical contact) was increased >300% during the COVID-19 pandemic [85]. Interestingly, the system delay component of STEMI treatment (time from hospital admission to reperfusion / stent implantation) was also significantly increased, suggesting protocol changes made in the emergency room to manage the pandemic were impairing other diagnostic and treatment algorithms. The theory that other factors such as decreased air pollution and lifestyle changes such as stress reduction brought about by the “lockdown” enforced by most governments may have also contributed to a worldwide drop in the risk of myocardial infarction seems less likely, but will need further evaluation. During the second COVID-19 wave of autumn 2020, a similar decrease in STEMI hospitalisation rates has so far not been reported.
Multisystem inflammatory syndrome in children (MIS-C)
In children, SARS-CoV-2 infection in general results in a mild form of COVID-19. However, it may lead to a post-infection multi-system inflammatory state with dermatological, mucocutaneous and gastrointestinal manifestations associated with myocardial injury and acute heart failure [86–93]. Children presented with fever or chills, tachycardia, asthenia, gastrointestinal symptoms, adenopathy, skin rashes and mucosal changes. Many of the affected children developed cardiogenic shock necessitating vasopressor support, and some even required veno-arterial extracorporeal membrane oxygenation. The occurrence and severity of cardiac injury seems to be age dependent. In adolescents (age 12–20 years), the majority showed severe cardiac failure or myocarditis [88]. About one third of the children in the various reports had Kawasaki disease-like clinical features and a substantial percentage also showed dilatation of coronary arteries similar to Kawasaki disease [86, 88, 89]. Kawasaki disease, or atypical or incomplete Kawasaki disease were more common in young children (age 0–5 years) [88, 89]. In contrast to Kawasaki disease, where cardiogenic shock is observed in about 5% [94], the hyperinflammation state after COVID-19 in children was associated with cardiogenic shock in at least 50% [86, 88, 89]. Treatment included intravenous immunoglobulin, intravenous steroids and immunomodulators. In most cases this treatment resulted in recovery of the cardiogenic shock and also left ventricular ejection fraction [86]. Whether inflammatory damage or indirect injury by myocardial stunning and oedema causes the acute heart failure in MIS-C remains unresolved [86].
Conclusions
The worldwide COVID-19 pandemic is currently leading to hundreds of thousands new infections daily, resulting in significant morbidity in 5–10% of these patients. A vast inter-individual variation in response to the infection is obvious, and still many aspects of its transmission, pathogenesis and treatment remain unclear. The initial fear of a detrimental effect of RAAS inhibitors has been ruled out. Similarly, COVID-19 does not seem to trigger type-1 myocardial infarction. Patients with pre-existing cardiovascular disease are at increased risk for a more serious disease course and death. Acute SARS-CoV-2 myocarditis is rare, but myocardial injury is observed in many sicker patients, through a variety of mechanisms. Worries remain relating to late cardiac damage in children (MIS-C) and in adults in view of the widespread, subclinical abnormalities found in cardiac MRI studies.
References
1
https://www.bag.admin.ch/bag/de/home/krankheiten/ausbrueche-epidemien-pandemien/aktuelle-ausbrueche-epidemien/novel-cov/situation-schweiz-und-international.html.
2
Andersen
KG
,
Rambaut
A
,
Lipkin
WI
,
Holmes
EC
,
Garry
RF
. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–2. doi:.https://doi.org/10.1038/s41591-020-0820-9
3
Zhang
T
,
Wu
Q
,
Zhang
Z
. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr Biol. 2020;30(7):1346–1351.e2. doi:.https://doi.org/10.1016/j.cub.2020.03.022
4
Hoffmann
M
,
Kleine-Weber
H
,
Schroeder
S
,
Krüger
N
,
Herrler
T
,
Erichsen
S
, et al.
SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271–280.e8. doi:.https://doi.org/10.1016/j.cell.2020.02.052
5
Hamming
I
,
Timens
W
,
Bulthuis
ML
,
Lely
AT
,
Navis
G
,
van Goor
H
. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7. doi:.https://doi.org/10.1002/path.1570
6
Gheblawi
M
,
Wang
K
,
Viveiros
A
,
Nguyen
Q
,
Zhong
JC
,
Turner
AJ
, et al.
Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res. 2020;126(10):1456–74. doi:.https://doi.org/10.1161/CIRCRESAHA.120.317015
7McIntosh K, Hirsch M, Bloom A. Coronavirus Disease 19 (COVID-19). UpToDate. 2020.
8
Bavishi
C
,
Maddox
TM
,
Messerli
FH
. Coronavirus Disease 2019 (COVID-19) Infection and Renin Angiotensin System Blockers. JAMA Cardiol. 2020;5(7):745–7. doi:.https://doi.org/10.1001/jamacardio.2020.1282
9
Guo
T
,
Fan
Y
,
Chen
M
,
Wu
X
,
Zhang
L
,
He
T
, et al.
Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):811–8. doi:.https://doi.org/10.1001/jamacardio.2020.1017
10
Guan
WJ
,
Ni
ZY
,
Hu
Y
,
Liang
WH
,
Ou
CQ
,
He
JX
, et al.; China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382(18):1708–20. doi:.https://doi.org/10.1056/NEJMoa2002032
11
Wiersinga
WJ
,
Rhodes
A
,
Cheng
AC
,
Peacock
SJ
,
Prescott
HC
. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;324(8):782–93. doi:.https://doi.org/10.1001/jama.2020.12839
12
Lechien
JR
,
Chiesa-Estomba
CM
,
De Siati
DR
,
Horoi
M
,
Le Bon
SD
,
Rodriguez
A
, et al.
Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur Arch Otorhinolaryngol. 2020;277(8):2251–61. doi:.https://doi.org/10.1007/s00405-020-05965-1
13
Levi
M
,
Thachil
J
,
Iba
T
,
Levy
JH
. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438–40. doi:.https://doi.org/10.1016/S2352-3026(20)30145-9
14
Siddiqi
HK
,
Mehra
MR
. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J Heart Lung Transplant. 2020;39(5):405–7. doi:.https://doi.org/10.1016/j.healun.2020.03.012
15
Yang
J
,
Zheng
Y
,
Gou
X
,
Pu
K
,
Chen
Z
,
Guo
Q
, et al.
Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–5. doi:.https://doi.org/10.1016/j.ijid.2020.03.017
16
Wu
Z
,
McGoogan
JM
. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239–42. doi:.https://doi.org/10.1001/jama.2020.2648
17
Zheng
YY
,
Ma
YT
,
Zhang
JY
,
Xie
X
. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60. doi:.https://doi.org/10.1038/s41569-020-0360-5
18
Diaz
JH
. Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med. 2020;27(3):taaa041. doi:.https://doi.org/10.1093/jtm/taaa041
19
Watkins
J
. Preventing a covid-19 pandemic. BMJ. 2020;368:m810. doi:.https://doi.org/10.1136/bmj.m810
20
Vaduganathan
M
,
Vardeny
O
,
Michel
T
,
McMurray
JJV
,
Pfeffer
MA
,
Solomon
SD
. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N Engl J Med. 2020;382(17):1653–9. doi:.https://doi.org/10.1056/NEJMsr2005760
21
Kuster
GM
,
Pfister
O
,
Burkard
T
,
Zhou
Q
,
Twerenbold
R
,
Haaf
P
, et al.
SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19?
Eur Heart J. 2020;41(19):1801–3. doi:.https://doi.org/10.1093/eurheartj/ehaa235
22
Wang
K
,
Gheblawi
M
,
Oudit
GY
. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation. 2020;142(5):426–8. doi:.https://doi.org/10.1161/CIRCULATIONAHA.120.047049
23
Zhang
J
,
Wang
M
,
Ding
W
,
Wan
J
. The interaction of RAAS inhibitors with COVID-19: Current progress, perspective and future. Life Sci. 2020;257:118142. doi:.https://doi.org/10.1016/j.lfs.2020.118142
24
Oudit
GY
,
Pfeffer
MA
. Plasma angiotensin-converting enzyme 2: novel biomarker in heart failure with implications for COVID-19. Eur Heart J. 2020;41(19):1818–20. doi:.https://doi.org/10.1093/eurheartj/ehaa414
25
Sama
IE
,
Ravera
A
,
Santema
BT
,
van Goor
H
,
Ter Maaten
JM
,
Cleland
JGF
, et al.
Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur Heart J. 2020;41(19):1810–7. doi:.https://doi.org/10.1093/eurheartj/ehaa373
26
Milne
S
,
Yang
CX
,
Timens
W
,
Bossé
Y
,
Sin
DD
. SARS-CoV-2 receptor ACE2 gene expression and RAAS inhibitors. Lancet Respir Med. 2020;8(6):e50–1. doi:.https://doi.org/10.1016/S2213-2600(20)30224-1
27
Dauchet
L
,
Lambert
M
,
Gauthier
JP
,
Poissy
J
,
Faure
K
,
Facon
A
, et al.
ACE inhibitors, ATI receptor blockers and COVID-19: clinical epidemiology evidences for a continuation of treatments. The ACER-COVID study. medRxiv. 2020.
28
Mancia
G
,
Rea
F
,
Ludergnani
M
,
Apolone
G
,
Corrao
G
. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N Engl J Med. 2020;382(25):2431–40. doi:.https://doi.org/10.1056/NEJMoa2006923
29
Mehta
N
,
Kalra
A
,
Nowacki
AS
,
Anjewierden
S
,
Han
Z
,
Bhat
P
, et al.
Association of Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5(9):1020–6. doi:.https://doi.org/10.1001/jamacardio.2020.1855
30
Reynolds
HR
,
Adhikari
S
,
Pulgarin
C
,
Troxel
AB
,
Iturrate
E
,
Johnson
SB
, et al.
Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N Engl J Med. 2020;382(25):2441–8. doi:.https://doi.org/10.1056/NEJMoa2008975
31
Huang
Z
,
Cao
J
,
Yao
Y
,
Jin
X
,
Luo
Z
,
Xue
Y
, et al.
The effect of RAS blockers on the clinical characteristics of COVID-19 patients with hypertension. Ann Transl Med. 2020;8(7):430. doi:.https://doi.org/10.21037/atm.2020.03.229
32
Li
J
,
Wang
X
,
Chen
J
,
Zhang
H
,
Deng
A
. Association of Renin-Angiotensin System Inhibitors With Severity or Risk of Death in Patients With Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. 2020;5(7):825–30. doi:.https://doi.org/10.1001/jamacardio.2020.1624
33
Zhang
P
,
Zhu
L
,
Cai
J
,
Lei
F
,
Qin
JJ
,
Xie
J
, et al.
Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ Res. 2020;126(12):1671–81. doi:.https://doi.org/10.1161/CIRCRESAHA.120.317134
34
Lopes
RD
,
Macedo
AVS
,
de Barros E Silva
PGM
,
Moll-Bernardes
RJ
,
Feldman
A
,
D’Andréa Saba Arruda
G
, et al.; BRACE CORONA investigators. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)--The BRACE CORONA Trial. Am Heart J. 2020;226:49–59. doi:.https://doi.org/10.1016/j.ahj.2020.05.002
35
Santos
RAS
,
Sampaio
WO
,
Alzamora
AC
,
Motta-Santos
D
,
Alenina
N
,
Bader
M
, et al.
The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol Rev. 2018;98(1):505–53. doi:.https://doi.org/10.1152/physrev.00023.2016
36
Te Riet
L
,
van Esch
JH
,
Roks
AJ
,
van den Meiracker
AH
,
Danser
AH
. Hypertension: renin-angiotensin-aldosterone system alterations. Circ Res. 2015;116(6):960–75. doi:.https://doi.org/10.1161/CIRCRESAHA.116.303587
37
Kuba
K
,
Imai
Y
,
Rao
S
,
Gao
H
,
Guo
F
,
Guan
B
, et al.
A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–9. doi:.https://doi.org/10.1038/nm1267
38
Zou
Z
,
Yan
Y
,
Shu
Y
,
Gao
R
,
Sun
Y
,
Li
X
, et al.
Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014;5(1):3594. doi:.https://doi.org/10.1038/ncomms4594
39
Clerkin
KJ
,
Fried
JA
,
Raikhelkar
J
,
Sayer
G
,
Griffin
JM
,
Masoumi
A
, et al.
COVID-19 and Cardiovascular Disease. Circulation. 2020;141(20):1648–55. doi:.https://doi.org/10.1161/CIRCULATIONAHA.120.046941
40
Chen
T
,
Wu
D
,
Chen
H
,
Yan
W
,
Yang
D
,
Chen
G
, et al.
Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091. doi:.https://doi.org/10.1136/bmj.m1091
41
Wu
C
,
Chen
X
,
Cai
Y
,
Xia
J
,
Zhou
X
,
Xu
S
, et al.
Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–43. doi:.https://doi.org/10.1001/jamainternmed.2020.0994
42
Zhou
F
,
Yu
T
,
Du
R
,
Fan
G
,
Liu
Y
,
Liu
Z
, et al.
Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. doi:.https://doi.org/10.1016/S0140-6736(20)30566-3
43
Hu
S
,
Gao
R
,
Liu
L
, et al.
Summary of the 2018 report on cardiovascular disease in China. Chin Circ J. 2019;34:209.
44
Liu
M
,
Liu
SW
,
Wang
LJ
,
Bai
YM
,
Zeng
XY
,
Guo
HB
, et al.
Burden of diabetes, hyperglycaemia in China from to 2016: Findings from the 1990 to 2016, global burden of disease study. Diabetes Metab. 2019;45(3):286–93. doi:.https://doi.org/10.1016/j.diabet.2018.08.008
45
Meyer
P
,
Degrauwe
S
,
Van Delden
C
,
Ghadri
JR
,
Templin
C
. Typical takotsubo syndrome triggered by SARS-CoV-2 infection. Eur Heart J. 2020;41(19):1860. doi:.https://doi.org/10.1093/eurheartj/ehaa306
46
Sala
S
,
Peretto
G
,
Gramegna
M
,
Palmisano
A
,
Villatore
A
,
Vignale
D
, et al.
Acute myocarditis presenting as a reverse Tako-Tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur Heart J. 2020;41(19):1861–2. doi:.https://doi.org/10.1093/eurheartj/ehaa286
47
Chen
L
,
Li
X
,
Chen
M
,
Feng
Y
,
Xiong
C
. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116(6):1097–100. doi:.https://doi.org/10.1093/cvr/cvaa078
48
Nicin
L
,
Abplanalp
WT
,
Mellentin
H
,
Kattih
B
,
Tombor
L
,
John
D
, et al.
Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur Heart J. 2020;41(19):1804–6. doi:.https://doi.org/10.1093/eurheartj/ehaa311
49
Varga
Z
,
Flammer
AJ
,
Steiger
P
,
Haberecker
M
,
Andermatt
R
,
Zinkernagel
AS
, et al.
Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8. doi:.https://doi.org/10.1016/S0140-6736(20)30937-5
50
Ruan
Q
,
Yang
K
,
Wang
W
,
Jiang
L
,
Song
J
. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–8. doi:.https://doi.org/10.1007/s00134-020-05991-x
51
Esposito
A
,
Palmisano
A
,
Natale
L
,
Ligabue
G
,
Peretto
G
,
Lovato
L
, et al.
Cardiac Magnetic Resonance Characterization of Myocarditis-Like Acute Cardiac Syndrome in COVID-19. JACC Cardiovasc Imaging. 2020;13(11):2462–5. doi:.https://doi.org/10.1016/j.jcmg.2020.06.003
52
Dolhnikoff
M
,
Ferreira Ferranti
J
,
de Almeida Monteiro
RA
,
Duarte-Neto
AN
,
Soares Gomes-Gouvêa
M
,
Viu Degaspare
N
, et al.
SARS-CoV-2 in cardiac tissue of a child with COVID-19-related multisystem inflammatory syndrome. Lancet Child Adolesc Health. 2020;4(10):790–4. doi:.https://doi.org/10.1016/S2352-4642(20)30257-1
53
Hu
H
,
Ma
F
,
Wei
X
,
Fang
Y
. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur Heart J. 2020;ehaa190. doi:.https://doi.org/10.1093/eurheartj/ehaa190
54
Inciardi
RM
,
Lupi
L
,
Zaccone
G
,
Italia
L
,
Raffo
M
,
Tomasoni
D
, et al.
Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):819–24. doi:.https://doi.org/10.1001/jamacardio.2020.1096
55
Kim
IC
,
Kim
JY
,
Kim
HA
,
Han
S
. COVID-19-related myocarditis in a 21-year-old female patient. Eur Heart J. 2020;41(19):1859. doi:.https://doi.org/10.1093/eurheartj/ehaa288
56
Doyen
D
,
Moceri
P
,
Ducreux
D
,
Dellamonica
J
. Myocarditis in a patient with COVID-19: a cause of raised troponin and ECG changes. Lancet. 2020;395(10235):1516. doi:.https://doi.org/10.1016/S0140-6736(20)30912-0
57
Tavazzi
G
,
Pellegrini
C
,
Maurelli
M
,
Belliato
M
,
Sciutti
F
,
Bottazzi
A
, et al.
Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020;22(5):911–5. doi:.https://doi.org/10.1002/ejhf.1828
58
Lindner
D
,
Fitzek
A
,
Bräuninger
H
,
Aleshcheva
G
,
Edler
C
,
Meissner
K
, et al.
Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020;5(11):1281–5. doi:.https://doi.org/10.1001/jamacardio.2020.3551
59
Wichmann
D
,
Sperhake
JP
,
Lütgehetmann
M
,
Steurer
S
,
Edler
C
,
Heinemann
A
, et al.
Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann Intern Med. 2020;173(4):268–77. doi:.https://doi.org/10.7326/M20-2003
60
Oudit
GY
,
Kassiri
Z
,
Jiang
C
,
Liu
PP
,
Poutanen
SM
,
Penninger
JM
, et al.
SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39(7):618–25. doi:.https://doi.org/10.1111/j.1365-2362.2009.02153.x
61
Wenzel
P
,
Kopp
S
,
Göbel
S
,
Jansen
T
,
Geyer
M
,
Hahn
F
, et al.
Evidence of SARS-CoV-2 mRNA in endomyocardial biopsies of patients with clinically suspected myocarditis tested negative for COVID-19 in nasopharyngeal swab. Cardiovasc Res. 2020;116(10):1661–3. doi:.https://doi.org/10.1093/cvr/cvaa160
62
Basso
C
,
Leone
O
,
Rizzo
S
,
De Gaspari
M
,
van der Wal
AC
,
Aubry
MC
, et al.
Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J. 2020;41(39):3827–35. doi:.https://doi.org/10.1093/eurheartj/ehaa664
63
Halushka
MK
,
Vander Heide
RS
. Myocarditis is rare in COVID-19 autopsies: cardiovascular findings across 277 postmortem examinations. Cardiovasc Pathol. 2020;50:107300. doi:.https://doi.org/10.1016/j.carpath.2020.107300
64
Puntmann
VO
,
Carerj
ML
,
Wieters
I
,
Fahim
M
,
Arendt
C
,
Hoffmann
J
, et al.
Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5(11):1265–73. doi:.https://doi.org/10.1001/jamacardio.2020.3557
65
Eiros
R
,
Barreriro-Perez
M
,
Martin-Garcia
A
,
Almeida
J
,
Villacorta
E
,
Perez-Pons
A
, et al.
Pericarditis and myocarditis long after SARS-CoV-2 infection: a cross-sectional descriptive study in health-care workers. medRxiv. doi:.https://doi.org/10.1101/2020.07.12.20151316
66
Liu
PP
,
Blet
A
,
Smyth
D
,
Li
H
. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation. 2020;142(1):68–78. doi:.https://doi.org/10.1161/CIRCULATIONAHA.120.047549
67
Gao
C
,
Wang
Y
,
Gu
X
,
Shen
X
,
Zhou
D
,
Zhou
S
, et al.; Community-Acquired Pneumonia–China Network. Association Between Cardiac Injury and Mortality in Hospitalized Patients Infected With Avian Influenza A (H7N9) Virus. Crit Care Med. 2020;48(4):451–8. doi:.https://doi.org/10.1097/CCM.0000000000004207
68
Valaperti
A
,
Nishii
M
,
Liu
Y
,
Naito
K
,
Chan
M
,
Zhang
L
, et al.
Innate immune interleukin-1 receptor-associated kinase 4 exacerbates viral myocarditis by reducing CCR5(+) CD11b(+) monocyte migration and impairing interferon production. Circulation. 2013;128(14):1542–54. doi:.https://doi.org/10.1161/CIRCULATIONAHA.113.002275
69
Flores
D
,
Walter
J
,
Wussler
D
, et al.
Direct comparison of high-sensitivity cardiac troponin T and I for prediction of mortality in patients with pneumonia. J Clin Chem Lab Med. 2019;2:2.
70
Bhatraju
PK
,
Ghassemieh
BJ
,
Nichols
M
,
Kim
R
,
Jerome
KR
,
Nalla
AK
, et al.
Covid-19 in Critically Ill Patients in the Seattle Region - Case Series. N Engl J Med. 2020;382(21):2012–22. doi:.https://doi.org/10.1056/NEJMoa2004500
71
Szekely
Y
,
Lichter
Y
,
Taieb
P
,
Banai
A
,
Hochstadt
A
,
Merdler
I
, et al.
Spectrum of Cardiac Manifestations in COVID-19: A Systematic Echocardiographic Study. Circulation. 2020;142(4):342–53. doi:.https://doi.org/10.1161/CIRCULATIONAHA.120.047971
72
Khoury
J
,
Arow
M
,
Elias
A
,
Makhoul
BF
,
Berger
G
,
Kaplan
M
, et al.
The prognostic value of brain natriuretic peptide (BNP) in non-cardiac patients with sepsis, ultra-long follow-up. J Crit Care. 2017;42:117–22. doi:.https://doi.org/10.1016/j.jcrc.2017.07.009
73
Huang
C
,
Wang
Y
,
Li
X
,
Ren
L
,
Zhao
J
,
Hu
Y
, et al.
Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:.https://doi.org/10.1016/S0140-6736(20)30183-5
74
Tang
N
,
Li
D
,
Wang
X
,
Sun
Z
. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7. doi:.https://doi.org/10.1111/jth.14768
75
Ackermann
M
,
Verleden
SE
,
Kuehnel
M
,
Haverich
A
,
Welte
T
,
Laenger
F
, et al.
Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120–8. doi:.https://doi.org/10.1056/NEJMoa2015432
76
Magro
C
,
Mulvey
JJ
,
Berlin
D
,
Nuovo
G
,
Salvatore
S
,
Harp
J
, et al.
Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res. 2020;220:1–13. doi:.https://doi.org/10.1016/j.trsl.2020.04.007
77
Tisoncik
JR
,
Korth
MJ
,
Simmons
CP
,
Farrar
J
,
Martin
TR
,
Katze
MG
. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16–32. doi:.https://doi.org/10.1128/MMBR.05015-11
78
Chau
VQ
,
Oliveros
E
,
Mahmood
K
,
Singhvi
A
,
Lala
A
,
Moss
N
, et al.
The Imperfect Cytokine Storm: Severe COVID-19 With ARDS in a Patient on Durable LVAD Support. JACC Case Rep. 2020;2(9):1315–20. doi:.https://doi.org/10.1016/j.jaccas.2020.04.001
79
Mehta
P
,
McAuley
DF
,
Brown
M
,
Sanchez
E
,
Tattersall
RS
,
Manson
JJ
; HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–4. doi:.https://doi.org/10.1016/S0140-6736(20)30628-0
80
Kang
S
,
Tanaka
T
,
Inoue
H
,
Ono
C
,
Hashimoto
S
,
Kioi
Y
, et al.
IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. 2020;117(36):22351–6. doi:.https://doi.org/10.1073/pnas.2010229117
81
Kox
M
,
Waalders
NJB
,
Kooistra
EJ
,
Gerretsen
J
,
Pickkers
P
. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA. 2020;324(15):1565–7. doi:.https://doi.org/10.1001/jama.2020.17052
82
Tang
Y
,
Liu
J
,
Zhang
D
,
Xu
Z
,
Ji
J
,
Wen
C
. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front Immunol. 2020;11:1708. doi:.https://doi.org/10.3389/fimmu.2020.01708
83
Mafham
MM
,
Spata
E
,
Goldacre
R
,
Gair
D
,
Curnow
P
,
Bray
M
, et al.
COVID-19 pandemic and admission rates for and management of acute coronary syndromes in England. Lancet. 2020;396(10248):381–9. doi:.https://doi.org/10.1016/S0140-6736(20)31356-8
84
Gluckman
TJ
,
Wilson
MA
,
Chiu
ST
,
Penny
BW
,
Chepuri
VB
,
Waggoner
JW
, et al.
Case Rates, Treatment Approaches, and Outcomes in Acute Myocardial Infarction During the Coronavirus Disease 2019 Pandemic. JAMA Cardiol. 2020. doi:.https://doi.org/10.1001/jamacardio.2020.3629
85
Tam
CF
,
Cheung
KS
,
Lam
S
,
Wong
A
,
Yung
A
,
Sze
M
, et al.
Impact of Coronavirus Disease 2019 (COVID-19) Outbreak on ST-Segment-Elevation Myocardial Infarction Care in Hong Kong, China. Circ Cardiovasc Qual Outcomes. 2020;13(4):e006631. doi:.https://doi.org/10.1161/CIRCOUTCOMES.120.006631
86
Belhadjer
Z
,
Méot
M
,
Bajolle
F
,
Khraiche
D
,
Legendre
A
,
Abakka
S
, et al.
Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic. Circulation. 2020;142(5):429–36. doi:.https://doi.org/10.1161/CIRCULATIONAHA.120.048360
87European Centre for Disease Prevention and Control. Rapid risk assessment: paediatric inflammatory multisystem syndrome and SARS-CoV-2 infection in children. 2020 May 15. Available from : https://www.ecdc.europa.eu/en/publications-data/paediatric-inflammatory-multisystem-syndrome-and-sars-cov-2-rapid-risk-assessment.
88
Dufort
EM
,
Koumans
EH
,
Chow
EJ
,
Rosenthal
EM
,
Muse
A
,
Rowlands
J
, et al.; New York State and Centers for Disease Control and Prevention Multisystem Inflammatory Syndrome in Children Investigation Team. Multisystem Inflammatory Syndrome in Children in New York State. N Engl J Med. 2020;383(4):347–58. doi:.https://doi.org/10.1056/NEJMoa2021756
89
Feldstein
LR
,
Rose
EB
,
Horwitz
SM
,
Collins
JP
,
Newhams
MM
,
Son
MBF
, et al.; Overcoming COVID-19 Investigators; CDC COVID-19 Response Team. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N Engl J Med. 2020;383(4):334–46. doi:.https://doi.org/10.1056/NEJMoa2021680
90Royal College of Paediatrics and Child Health. Guidance: paediatric multisystem inflammatory syndrome temporally associated with COVID-19. 2020. Available from : https://www.rcpch.ac.uk/sites/default/files/2020-05/COVID-19-Paediatric-multisystem-%20inflammatory%20syndrome-20200501.pdf.
91
Jones
VG
,
Mills
M
,
Suarez
D
,
Hogan
CA
,
Yeh
D
,
Segal
JB
, et al.
COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp Pediatr. 2020;10(6):537–40. doi:.https://doi.org/10.1542/hpeds.2020-0123
92
Riphagen
S
,
Gomez
X
,
Gonzalez-Martinez
C
,
Wilkinson
N
,
Theocharis
P
. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395(10237):1607–8. doi:.https://doi.org/10.1016/S0140-6736(20)31094-1
93
Verdoni
L
,
Mazza
A
,
Gervasoni
A
,
Martelli
L
,
Ruggeri
M
,
Ciuffreda
M
, et al.
An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020;395(10239):1771–8. doi:.https://doi.org/10.1016/S0140-6736(20)31103-X
94
McCrindle
BW
,
Rowley
AH
,
Newburger
JW
,
Burns
JC
,
Bolger
AF
,
Gewitz
M
, et al.; American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiovascular Surgery and Anesthesia; and Council on Epidemiology and Prevention. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation. 2017;135(17):e927–99. doi:.https://doi.org/10.1161/CIR.0000000000000484