The renin-angiotensin-aldosterone system inhibitors in COVID-19: from acidosis to ventilation and immunity
DOI: https://doi.org/10.4414/smw.2020.20444
Wan-Hui
Liaoab, Gee-Gwo
Yangbc, Maciej
Hennebergde
a Department of Medical Education, Taipei Veterans General Hospital, Taipei, Taiwan, R. O. C.
b Department of Medical Education, Buddhist Tzu Chi General Hospital and Tzu Chi University, Hualien, Taiwan, R. O. C.
c Division of Chest Medicine, Buddhist Tzu Chi General Hospital, Hualien, Taiwan, R. O. C.
d Institute of Evolutionary Medicine, University of Zurich, Switzerland
e Biological Anthropology and Comparative Anatomy Unit, University of Adelaide, Australia
SARS-CoV-2 has caused a pandemic coronavirus disease 2019 (COVID-19), claiming over a million worldwide lives of people among whom those with cardiovascular disease or hypertension are over-represented. Understanding the mechanism by which coronavirus threatens human health is vital for reducing the current disease burden and even for preventing future attacks. Inspired by the temporal relationship between three coronavirus outbreaks in humans since 2000 and hypertension medications gaining popularity since 2000, we discuss here how hypertension medications might contribute to the COVID-19 pandemic and mortality with a focus on inhibitors of the renin-angiotensin-aldosterone system (RAAS). The fact that RAAS inhibitors increase angiotensin converting enzyme-2 (ACE2) expression in respiratory epithelial cells, the kidneys and heart has stirred a surge of interest in the discussion of the role of RAAS inhibitors in COVID-19 disease, all related to ACE2-mediated effects [1–5]. Here, we abstain from contributing further to this extensive discussion, but expand this discussion beyond ACE2 and centre on how RAAS blockade influences human interaction with SARS-CoV-2 by altering acid-base balance. This expansion is based on our previous experimental work conducted on aldosterone deficient mice [6] and on the fact, supported by accumulated groundwork in physiology, that acid-base homeostasis is maintained by the coordinated work of kidneys and lungs, and is vital for normal physiology and health. RAAS evolved to facilitate humans’ adaptation to land life by conserving salt and water in the intestine and kidney, and facilitating renal acid and other water-soluble waste elimination. As renal acid-excretion effects are largely dependent on the RAAS, inhibiting renal salt absorption by RAAS inhibitors in attempt to reduce blood pressure also slows renal acid elimination (see fig. 1 below).
Hypertension and COVID-19 mortality
The Chinese Centre for Disease Control and Prevention reported that the highest mortality rate (10.5%) of COVID-19 was in people with cardiovascular disease, followed by diabetes, chronic respiratory disease, hypertension and cancer, whereas the overall mortality rate was 2.3% in a sample of nearly 50,000 patients [7–9]. Among conditions classified as “cardiovascular disease” the most common is hypertension. Subsequently, several independent studies from China, Italy and Spain found that up to 80% of COVID-19 mortality cases have arterial hypertension and 30% have type 2 diabetes [10, 11]. According to the American College of Cardiology and the American Heart association some 46% of American adults have high blood pressure, as defined by the College standards (>130/80 mm Hg) [12].
Hypertension, unless it is a hypertensive crisis, does not exert a negative influence on lung tissues or on lung function. Therefore, patients with hypertension should not have been exceptionally susceptible to SARS-CoV-2 infection and its consequences, unless they have accompanying lung disease or generally lower immunity [13]. On the other hand, the commonality in patients with cardiovascular disease and/or diabetes and/or hypertension is treatment with angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin-II receptor blockers (ARBs) as they have been shown to be cardio- and renal protective in clinical trials [14, 15]. Wu and McGooan’s findings suggested a link between ACEI and ARB use and increased case fatality [7].
Mechanism linking hypertension medication to COVID-19 infectivity and pandemic
SARS-CoV-2 is transmitted mainly through the respiratory route and specifically uses its surface spike protein to gain entry into respiratory epithelial cells via binding to ACE2 [16], in the same way as SARS-CoV [17]. The crucial role of ACE2 in mediating SARS-CoV infection was demonstrated previously in a study where knockout of ACE2 granted the mice resistance to SARS-CoV infection, virus replication and the subsequent inflammatory cell infiltrations in their lungs [18]. Similarly, in human lung tissue, SARS-CoV infection depends on ACE2 protein expression levels. The well-differentiated ciliated cells with the most abundant ACE2 protein expression on the apical surface are the main entry and infection site for SARS-CoV [19]. SARS-CoV-2 virus has binding affinity to ACE2 ~10- to 20-fold higher than SARS-CoV [17]. This suggests the greater ACE2 protein expression levels in the respiratory epithelium and the high level of ventilation should define one’s propensity to and the severity of SARS-CoV-2 infection. Common anti-hypertension medications targeting the RAAS – ACEIs and ARBs – upregulate ACE2 expression in lung, heart and kidney [20, 21]. They also reduce glomerular filtration rate, aldosterone synthesis and renal bicarbonate reabsorption in several animal models [22]. Mineralocorticoid receptor blockers used in congestive heart failure also have ACE2-upregulating effects [23] and directly block aldosterone’s acid-excreting action in the renal tubules. By compromising renal acid excretion, RAAS inhibitors expand the baseline body acid pool and consume pH-buffering components, of which the prime one in the blood is the bicarbonate of the bicarbonate / carbonic acid buffer system. This makes the body prone to pH fluctuations and to the sudden rises in pCO2, which demand reflexive increases in ventilation to maintain acid-base balance [6, 24]. RAAS inhibitors therefore may further facilitate interaction of SARS-CoV-2 with our respiratory system by increasing ventilation if the bicarbonate level is low enough to cause pH drops during activities. Moreover, the burdening effect of insufficient renal acid elimination on the cardiorespiratory system is magnified when the body is in a hypermetabolic state, where body CO2 production is massively increased, such as in chronic heart and respiratory disease and cancer [6]. Chronic heart and respiratory disease and cancer themselves contribute to COVID-19 infection mortality [7].
Mechanism linking hypertension medication to COVID-19 mortality
Developing acid-base imbalance
ACEIs and ARBs use should also prime patients to develop metabolic acidosis during SARS-CoV-2 infection because of a further increase in ventilation to overcome ventilation/perfusion mismatch and a sudden increase in endogenous acid production from acute inflammatory processes. Peripheral tissue inflammation is characterised by tissue hypoxia due to small vessel damage, and activation and infiltration of innate immune cells, together with clonal expansion of adaptive immune cells. All these processes contribute to local pH reduction, which can be as low as 5.5 [25]. Activated neutrophils and T lymphocytes rely mainly on glycolytic metabolism for proliferation, differentiation and function, which results in accumulation of lactic acid [26]. Analogous to this scenario, insufficient renal acid elimination due to the use of ACEIs was reported to induce metabolic acidosis during surgical stress [27].
Metabolic acidosis compromising cell-mediated immunity
Once metabolic acidosis has developed, extracellular acidosis impairs cellular immunity, which plays a vital role in the clearance of SARS-CoV-2 and other respiratory viruses [28].
Pulmonary cytotoxic CD8 T cells recognise and induce apoptosis in virus-infected cells through direct (cell-cell contact) and indirect mechanisms with the involvement of secreted cytolytic enzymes perforin and granzymes, as well as the cytokines interferon-γ and tumour necrosis factor [28]. A subpopulation of CD4 T cells is also recruited in parallel, and differentiate into cytotoxic T cells which further expands CD8 T cell pool. Some CD8 cells are even stimulated to gain the memory of CD8 T cell phenotype. Transient increases in both effector and memory CD8 T cells constitutes an effective and efficient response during early viral infection [29]. Low pH has been shown to induce anergy in CD8 T cells, including reduced cell proliferation, cytokine production, and impaired cytotoxic activity [30, 31]. It also suppresses natural killer (NK) and NKT cells to produce cytotoxic responses and interferon-γ [32, 33] and inhibits CD4+ T cell function [34]. Acidosis also increases circulating glucocorticoid levels. Its anti-inflammatory and immunosuppressive properties further compromise immunity against viruses [35, 36]. Lymphopenia, which can be induced by acute metabolic acidosis [37] was reported to predict disease severity and secondary bacterial infection in COVID-19 [8, 38]. Without robust cellular immune responses to clear virus efficiently, evolving host tissue destruction during uncontrolled viral infection elicits persistent innately induced inflammation likely to predispose to the development of cytokine storm, which accelerates the progression to lung failure [39, 40]. Prolonged cytotoxic T cell activity in the lung and other tissues in this scenario exacerbates organ damage by exaggerating immunopathology.
SARS-CoV-2 infection also can cause a coagulopathy, already seen in the early phase, and it has been suggested that this is a result of accumulated proinflammatory responses [41]. Extracellular acidosis selectively dampens homeostatic functions while increasing the pro-inflammatory effects of platelets [42]. It is plausible that dysfunctional platelets caused by acidosis contribute to COVID-19–associated coagulopathy.
Added to RAAS blockade that compromises cellular immunity via acidosis, ACEIs may further spread viral replication and invasion by inhibiting the antigen processing and presentation that are essential to facilitate and enhance adaptive immunity. Intracellular ACE enzymatically prepares self and non-self major histocompatibility complex (MHC) class 1 peptides to be presented onto the surface of virus-infected cells [43]. The catalytic activity of ACE in monocytes and macrophages is also required during their antigen processing and presentation processes [43].
Figures 1 and 2
graphically illustrate the above mechanistic links.
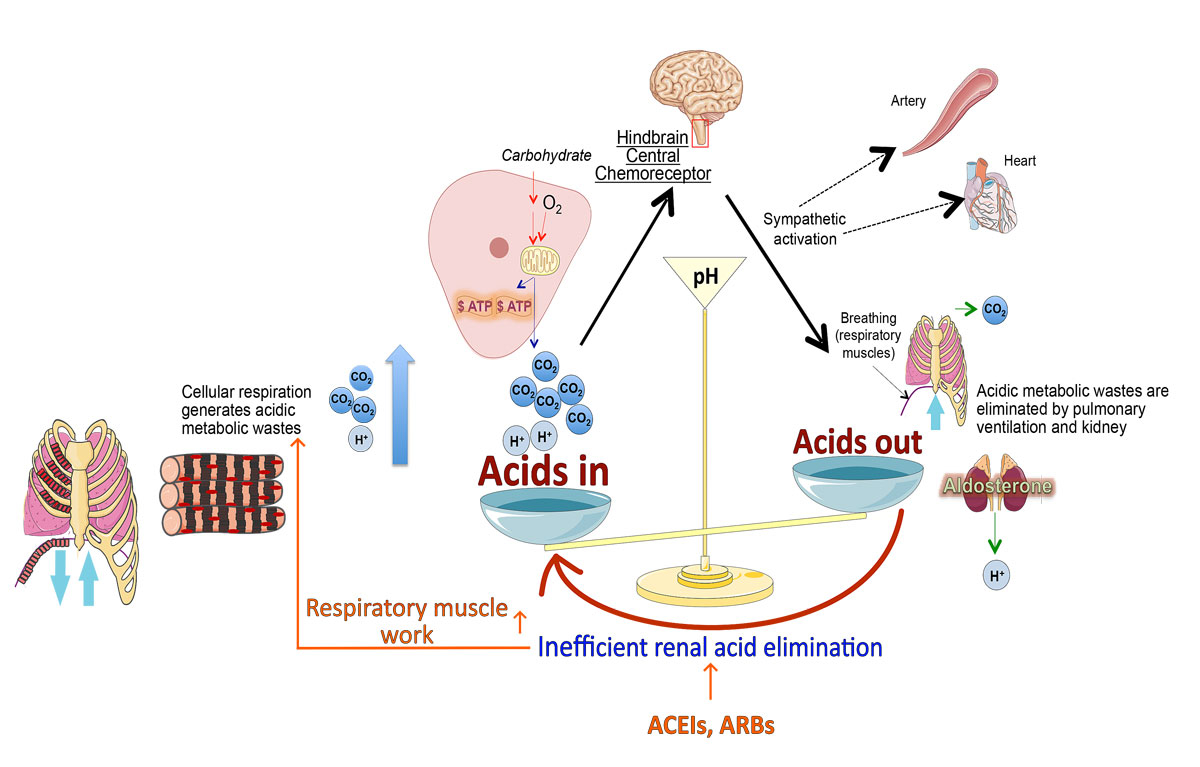
Figure 1
Influence of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin-II receptor blockers (ARBs) used as hypertension medication on acid-base balance regulation. Blocking the renin-angiotensin-aldosterone system with ACEIs or ARBs reduces renal acid elimination, leading to constant activation of brainstem-mediated chemoreceptor reflex to increase acid elimination by ventilation and burdens cardiopulmonary system. (Figure modified from Liao et al. [6])
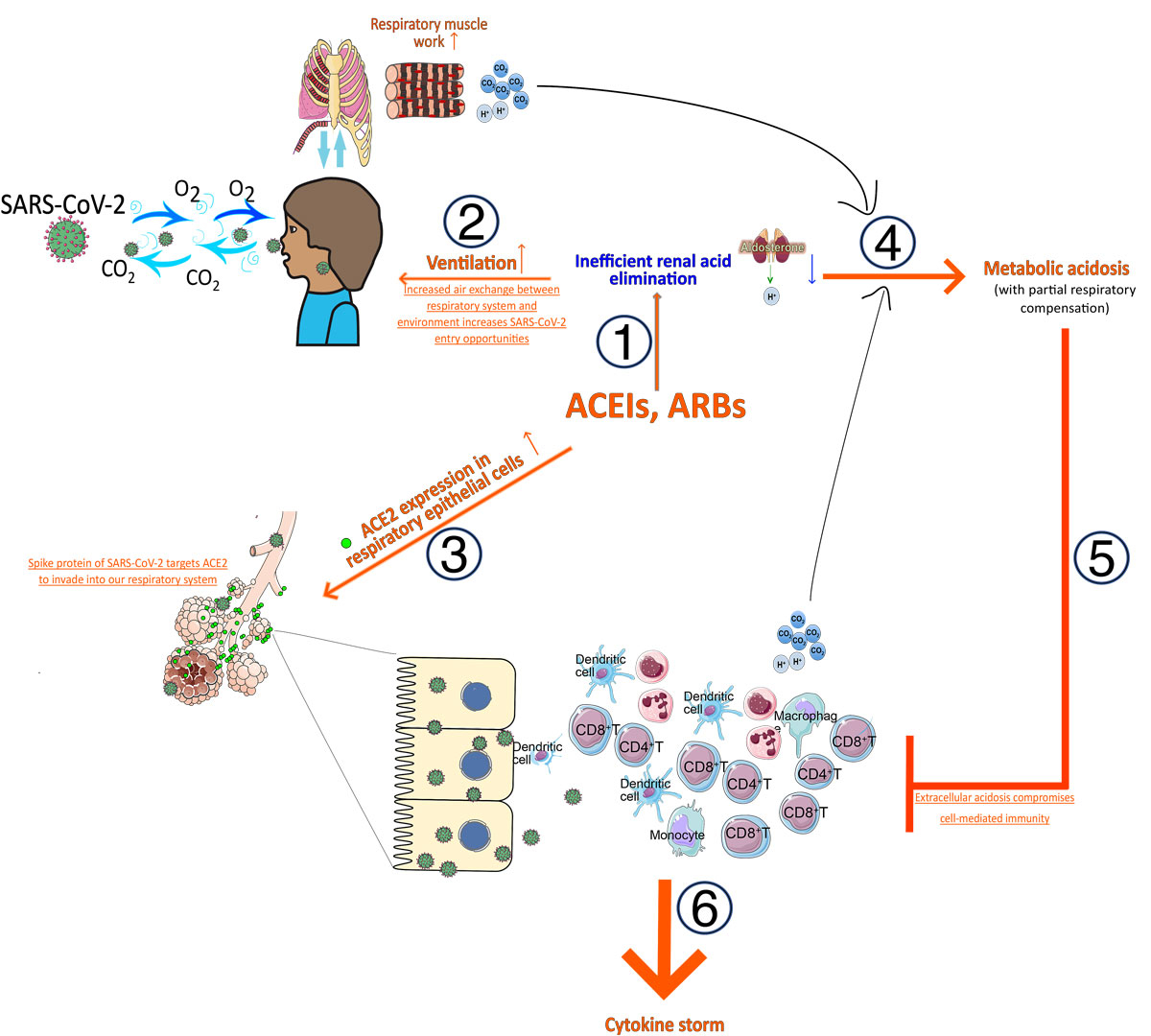
Figure 2
Influence of angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin-II receptor blockers (ARBs) used as hypertension medication on increased susceptibility to and mortality in SARS-CoV-2 infection. (1) Blocking renin-angiotensin-aldosterone system with ACEIs or ARBs reduces renal acid elimination, leading to increased ventilation to maintain acid-base balance. This process also (2) increases opportunity for SARS-CoV-2 to interact with respiratory system and (3) increases angiotensin-converting enzyme 2 (ACE2) expression in respiratory epithelial cells, the target of SARS-CoV-2 to gain access to our body. (4) Inflammatory responses in the respiratory system aiming to eliminate SARS-CoV-2 result in generation of more acidic metabolic wastes, which together with increased respiratory muscular work leads to the development of metabolic acidosis. (5) Metabolic acidosis compromises adaptive cellular immunity and (6) inefficient eradication of SARS-CoV-2 may predispose to the generation of cytokine storm.
Discussion
Coronaviruses have insidiously become human threats since 2000 [44]. Three outbreaks in sequence – severe acute respiratory syndrome (SARS) in 2002, Middle East respiratory syndrome in 2012, and 2020 COVID-19, temporally coincide with hypertension medications gaining popularity [45, 46]. ACEIs and ARBs have stood out among all types of antihypertensive medications as guideline-recommended first line therapy in diabetic hypertension [47, 48] and in secondary prevention for acute coronary syndromes. ACEIs and ARBs potentially attenuate cardiovascular and renal function decline, particularly in diabetic patients [14, 15], but they may cause humans to gradually lose anti-coronavirus immunity by increasing ACE2 expression and precipitating the development of metabolic acidosis, which compromises cell-mediated immunity. The RAAS evolved to facilitate adaptation of terrestrial vertebrates including humans, to land life by conserving salt and water in the intestine and kidney, and facilitating renal acid and other water-soluble waste elimination. Blocking the RAAS’s ability to retain sodium in the kidney to reduce blood pressure also reduces renal ability to excrete acidic waste [49]. This primes the development of full-blown metabolic acidosis during acute stress, such as infection as is the case in COVID-19 disease. Long-term ACEI/ARB use was recently reported to correlate with severe renal dysfunction and acute kidney injury in patients with severe COVID-19 [50]. Low serum bicarbonate levels in both hospitalised patients (<22 mEq/l) and in non-hospitalised patients (<24 mEq/l) carried high risks of acute kidney injury [51]. Metabolic acidosis contributes to acute kidney injury via intrarenal RAAS activation, renal tubular inflammation and ischaemic injury [52, 53]. All these findings together support our hypothesis that ACEIs/ARBs contribute to COVID-19 disease by predisposing their users to the development of metabolic acidosis during infection. Our hypothesis is based on our own experimental results in animals with complete lack of aldosterone. Therapeutic dysregulation of aldostereone levels by ACEIs/ARBs in human patients may have only partial effects on the development of metabolic acidosis.
There has been growing interest in the mechanistic link between up-regulation of ACE2 by ACEIs and ARBs, and acute lung failure in COVID-19 [1–3, 5, 54]. Several groups specifically propose that, by increasing ACE2 levels, ACEIs and ARBs should protect against the acute progression to lung failure in SARS-CoV-2 infection [2, 4, 54]. Their proposition is rooted in the finding by Lami et al. that ACE2 knockout mice were more susceptible to acute lung failure induced by acid aspiration or sepsis secondary to caecal ligation and perforation [55]. Also, elevated ACE2 levels could attenuate the pulmonary vasoconstrictive and inflammatory effects of angiotensin II, the level of which is positively related to SARS-CoV-2 viral load and lung injury [54]. Indeed, perturbing or downregulating ACE2 induces uncontrolled inflammation in hosts. This is the common mechanism employed by respiratory syncytial virus, influenza virus and SARS-CoV [55, 56]. But it also has to be emphasised here that downregulation of ACE2 protein expression in SARS-CoV infection is initiated by the binding of the surface spike protein of SARS-CoV virus to ACE2 [18]. That also means that ACE2 reduction is the direct consequence of the ACE2-mediaed entry by SARS-CoV virus. This sequential mechanistic link explains why ACE2 knockout mice have protection rather than susceptibility to SARS-CoV-induced lung failure, as mentioned in the previous paragraph [55]. It therefore has to be made clear that in the case of SARS-CoV infectious diseases, the high ACE2 expression in respiratory epithelium does not confer protection and enhances the virus entry. The idea of keeping ACE2 levels high after SARS-CoV-2 infection, in order to moderate virus-induced lung injury and slow the disease progression to lung failure, is not new. Sodhi et al. have demonstrated that recombinant ACE2 benefits bacterially infected lungs after an initial inflammatory response [57]. Though ACEIs and ARBs could also increase pulmonary ACE2 protein level expression, the cost of accompanying increases in cardiopulmonary burden, the respiratory tract interaction with SARS-CoV-2, and the predisposition to the development of metabolic acidosis during stress should outweigh the potential benefits. On the other hand, Teijaro et al. revealed that pulmonary endothelial cells are a key regulator of cytokine storm development during influenza virus infection. They even demonstrated that pharmacologically manipulating the sphingosine-1-phosphate receptor signalling successfully inhibits cytokine storm, but it does not affect virus clearance [39]. Before the emergence of effective therapy against SARS-CoV-2 virus, pharmacological alteration of sphingosine-1-phosphate receptor signalling may ameliorate the lung injury caused by cytokine storm.
In the meantime, several retrospective clinical studies have shown that the use of ACEIs and ARBs does not affect SARS-CoV-2 infection [11, 58, 59]. We would like to draw attention to the limitations of their levels of evidence and study design. Case-control studies have an evidence level of hypothesis generation only, not of theory. Post-hoc analysis is even more problematic. The chosen statistical methods and surrogate outcome measures all can produce biases. The method of conditional logistic regression used in two [11, 59] and “propensity score matching” used in one retrospective clinical study [58] are intended to circumvent confounding bias. No one can balance un-recognised causal factors. Propensity score matching has been criticised [60] because it has the bias of treating all recognised confounders as categorical irrespective of their nature. Confounders are not always categorical like sex or ethnicity. Those that also have continuous components, such as quantities of alcohol, betel nut, cigarette use, diet, medication, exposure length and the dose greatly influence their effects. Propensity score matching, however, is unable to take the dose and exposure length into account because it reduces all confounders to presence or absence. This is also true for certain diseases. Ten years on an ACEI, for example lisinopril 10 mg once daily, should be different from another kind of ACEI, for example enalapril 10 mg twice daily only for 3 months, in terms of aldosterone-inhibitory and acid-retaining effects, and effects on ACE2 expression in the respiratory epithelial cells. In the efficiency of lung eliminating CO2 waste, total respiratory tract surface area also varies between a patient with chronic lung disease for 1 year and another for 10 years. Propensity score matching also does not take into account disease-disease interactions and drug-drug interactions. Many medications have been reported to alter acid-base balance [61]. One obvious example is bicarbonate supplementation used to retard chronic kidney disease progression. The bicarbonate supplementation should neutralise the acidosis triggered by ACEIs and ARBs. Moreover, drugs and diseases may also interact synergistically or additively and modify the disease outcomes, which neither propensity score matching nor logistic regression can reveal. Furthermore, the outcomes used in these studies are not objective, such as “admission” or “not admission” to the hospital [11] or to the intensive care unit (ICU), and non-invasive ventilator use or not, as indicators of disease severity [58]. All these interventions are not double-blinded and are subjected to the doctors’ choices and the available resources. Hospitals in different regions and at different time-points of the COVID-19 pandemic differ in the availability and abundance of medical supplies, ICU beds and ICU specialists. Lastly, the study design and statistical analysis should be considered. A non-inferiority design should be used to test an alternative hypothesis (H1) to prove the safety of an intervention, instead of a superiority design and null hypothesis (H0) testing. The main concern brought up about RAAS inhibitors in COVID-19 was that these inhibitors increase COVID-19 risk by increasing ACE2 expression. Therefore, for a clinical study challenging this hypothesis, its statistical analysis should be a non-inferiority test. But all studies discussed here used the superiority test with the null hypothesis being that RAAS inhibitors do not increase COVID-19 risks. To falsify the hypothesis of RAAS inhibitors increasing COVID-19 risk, the clinical study should have a null hypothesis “RAAS inhibitors increase COVID-19 risk”. Even with these pitfalls the study by de Abajo et al. [11] showed that patients without use of any antihypertensive drugs are better off (odds ration [OR] ~0.5) for COVID-19 requiring hospital admission than RAAS inhibitor users (OR ~0.9–1.1, tables 2 and 3 in the original article), irrespective of comparisons with patients on other medications. This finding may be due to the fact that nearly all currently available antihypertensive medications inhibit renal acid excretion to some extent [61] with ACEIs, ARBs and mineralocorticoid antagonists directly targeting aldosterone, the final downstream effector of the RAAS, which has powerful stimulating effects on renal acid excretion in the form of ammonium and through urinary acidification. With the inherent pitfalls in retrospective clinical studies, their conclusion of no association between the use of ACEIs and ARBs and SARS-CoV-2 infection probability and outcomes can only be viewed as hypothesis-generating and not theory-forming. Randomised clinical trials with all confounders equalised prior to the study are warranted to elucidate whether RAAS inhibitors contribute to COVID-19 disease.
Conclusion
To find the true role of RAAS inhibitors in COVID-19, explanatory mechanisms and evidence of outcome modifications from clinical studies should both be provided. Our work explains mechanisms by which RAAS inhibitors may increase susceptibility to and the development of cytokine storm during SARS-CoV-2 infection. These mechanistic links are supported by our previous work in aldosterone deficient mice and accumulated groundwork in physiology in the field of acid-base balance. Our work appears to support the suggestion that patients on ACEIs and ARBs for hypertension control during COVID infection should be switched to other types of antihypertensive medication [3]. This would potentially reduce the interaction of our respiratory tract with SARS-CoV-2 and lower the viral loads. Moreover, by resuming renal elimination of acidic wastes, it would relieve the cardiopulmonary burden of SARS-CoV-2-infected patients. A L-type calcium channel blocker, amlodipine for example, may be a better choice because it has minimal effects on RAAS inhibition [62]. Calcium channel blockers have varying/inconsistent effects on the RAAS [63], as medications in ACEI and ARB groups have on increasing ACE2 levels in different disease backgrounds. However, because our work is not yet backed up by clinical data to demonstrate that RAAS inhibitors alter COVID-19 infectivity and disease outcome by predisposing the development of acidosis during infection, it should only be treated as hypothesis-generating but not theory-forming. Therefore, it is not advised to be used as a sole guide to the treatments currently. Given the complexity of human physiology and pathophysiology, the precise understanding of how ACEIs and ARBs influence the relationship with and immune response to SARS-CoV-2 virus in humans warrants well-planned randomised clinical trials. Several professional societies have recommended continuing ACEIs and ARBs as prescribed, following the mainstream guidelines [64]. With this work, we suggest that in future clinical studies planned to address the role of RAAS inhibitors in COVID-19, data including arterial blood gases, potassium levels, the amount of bicarbonate use and even renal replacement therapy use in the patients should also be collected, as these data analysed all together help the patient outcome assessment and also improve understanding of the effects of RAAS inhibition on COVID-19 disease.
Acknowledgments
We greatly thank Professor Wolfgang Langhans of the Swiss Institute of Technology, Zurich for valuable comments.
Author contributions
Wan-Hui Liao and Maciej Henneberg drafted the manuscript. All authors had intellectual contributions and also edited and approved manuscript for publication.
References
1
Esler
M
,
Esler
D
. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic?
J Hypertens. 2020;38(5):781–2. doi:.https://doi.org/10.1097/HJH.0000000000002450
2
Kuster
GM
,
Pfister
O
,
Burkard
T
,
Zhou
Q
,
Twerenbold
R
,
Haaf
P
, et al.
SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19?
Eur Heart J. 2020;41(19):1801–3. doi:.https://doi.org/10.1093/eurheartj/ehaa235
3
Watkins
J
. Preventing a covid-19 pandemic. BMJ. 2020;368:m810. doi:.https://doi.org/10.1136/bmj.m810
4
Vaduganathan
M
,
Vardeny
O
,
Michel
T
,
McMurray
JJV
,
Pfeffer
MA
,
Solomon
SD
. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N Engl J Med. 2020;382(17):1653–9. doi:.https://doi.org/10.1056/NEJMsr2005760
5
Fang
L
,
Karakiulakis
G
,
Roth
M
. Antihypertensive drugs and risk of COVID-19? - Authors’ reply. Lancet Respir Med. 2020;8(5):e32–3. doi:.https://doi.org/10.1016/S2213-2600(20)30159-4
6
Liao
WH
,
Suendermann
C
,
Steuer
AE
,
Pacheco Lopez
G
,
Odermatt
A
,
Faresse
N
, et al.
Aldosterone deficiency in mice burdens respiration and accentuates diet-induced hyperinsulinemia and obesity. JCI Insight. 2018;3(14):e99015. doi:.https://doi.org/10.1172/jci.insight.99015
7
Wu
Z
,
McGoogan
JM
. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239–42. doi:.https://doi.org/10.1001/jama.2020.2648
8
Zhou
F
,
Yu
T
,
Du
R
,
Fan
G
,
Liu
Y
,
Liu
Z
, et al.
Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. doi:.https://doi.org/10.1016/S0140-6736(20)30566-3
9
Chen
T
,
Wu
D
,
Chen
H
,
Yan
W
,
Yang
D
,
Chen
G
, et al.
Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091. doi:.https://doi.org/10.1136/bmj.m1091
10
Abbatecola
AM
,
Antonelli-Incalzi
R
. COVID-19 Spiraling of Frailty in Older Italian Patients. J Nutr Health Aging. 2020;24(5):453–5. doi:.https://doi.org/10.1007/s12603-020-1357-9
11
de Abajo
FJ
,
Rodríguez-Martín
S
,
Lerma
V
,
Mejía-Abril
G
,
Aguilar
M
,
García-Luque
A
, et al.; MED-ACE2-COVID19 study group. Use of renin-angiotensin-aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: a case-population study. Lancet. 2020;395(10238):1705–14. doi:.https://doi.org/10.1016/S0140-6736(20)31030-8
12
Whelton
PK
,
Carey
RM
,
Aronow
WS
,
Casey
DE, Jr
,
Collins
KJ
,
Dennison Himmelfarb
C
, et al.
2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13–115. doi:.https://doi.org/10.1161/HYP.0000000000000065
13
Glezen
WP
,
Greenberg
SB
,
Atmar
RL
,
Piedra
PA
,
Couch
RB
. Impact of respiratory virus infections on persons with chronic underlying conditions. JAMA. 2000;283(4):499–505. doi:.https://doi.org/10.1001/jama.283.4.499
14
Yusuf
S
,
Sleight
P
,
Pogue
J
,
Bosch
J
,
Davies
R
,
Dagenais
G
; Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342(3):145–53. doi:.https://doi.org/10.1056/NEJM200001203420301
15
Brenner
BM
,
Cooper
ME
,
de Zeeuw
D
,
Keane
WF
,
Mitch
WE
,
Parving
HH
, et al.; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–9. doi:.https://doi.org/10.1056/NEJMoa011161
16
Yan
R
,
Zhang
Y
,
Li
Y
,
Xia
L
,
Guo
Y
,
Zhou
Q
. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–8. doi:.https://doi.org/10.1126/science.abb2762
17
Wrapp
D
,
Wang
N
,
Corbett
KS
,
Goldsmith
JA
,
Hsieh
CL
,
Abiona
O
, et al.
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–3. doi:.https://doi.org/10.1126/science.abb2507
18
Kuba
K
,
Imai
Y
,
Rao
S
,
Gao
H
,
Guo
F
,
Guan
B
, et al.
A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–9. doi:.https://doi.org/10.1038/nm1267
19
Jia
HP
,
Look
DC
,
Shi
L
,
Hickey
M
,
Pewe
L
,
Netland
J
, et al.
ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79(23):14614–21. doi:.https://doi.org/10.1128/JVI.79.23.14614-14621.2005
20
Li
XC
,
Zhang
J
,
Zhuo
JL
. The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol Res. 2017;125(Pt A):21–38. doi:.https://doi.org/10.1016/j.phrs.2017.06.005
21
Yuan
YM
,
Luo
L
,
Guo
Z
,
Yang
M
,
Ye
RS
,
Luo
C
. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. J Renin Angiotensin Aldosterone Syst. 2015;16(2):249–53. doi:.https://doi.org/10.1177/1470320315576256
22
Bobulescu
IA
,
Moe
OW
. Na+/H+ exchangers in renal regulation of acid-base balance. Semin Nephrol. 2006;26(5):334–44. doi:.https://doi.org/10.1016/j.semnephrol.2006.07.001
23
Keidar
S
,
Gamliel-Lazarovich
A
,
Kaplan
M
,
Pavlotzky
E
,
Hamoud
S
,
Hayek
T
, et al.
Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res. 2005;97(9):946–53. doi:.https://doi.org/10.1161/01.RES.0000187500.24964.7A
24
Henger
A
,
Tutt
P
,
Riesen
WF
,
Hulter
HN
,
Krapf
R
. Acid-base and endocrine effects of aldosterone and angiotensin II inhibition in metabolic acidosis in human patients. J Lab Clin Med. 2000;136(5):379–89. doi:.https://doi.org/10.1067/mlc.2000.110371
25
Reeh
PW
,
Steen
KH
. Tissue acidosis in nociception and pain. Prog Brain Res. 1996;113:143–51. doi:.https://doi.org/10.1016/S0079-6123(08)61085-7
26
Menk
AV
,
Scharping
NE
,
Moreci
RS
,
Zeng
X
,
Guy
C
,
Salvatore
S
, et al.
Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018;22(6):1509–21. doi:.https://doi.org/10.1016/j.celrep.2018.01.040
27
Abdelaal Ahmed Mahmoud
A
,
Campbell
M
,
Blajeva
M
. Can ACE-I Be a Silent Killer While Normal Renal Functions Falsely Secure Us?
Case Rep Anesthesiol. 2018;2018:1852016. doi:.https://doi.org/10.1155/2018/1852016
28
Schmidt
ME
,
Varga
SM
. The CD8 T Cell Response to Respiratory Virus Infections. Front Immunol. 2018;9:678. doi:. https://doi.org/10.3389/fimmu.2018.00678
29
Wiesel
M
,
Walton
S
,
Richter
K
,
Oxenius
A
. Virus-specific CD8 T cells: activation, differentiation and memory formation. APMIS. 2009;117(5-6):356–81. doi:.https://doi.org/10.1111/j.1600-0463.2009.02459.x
30
Bosticardo
M
,
Ariotti
S
,
Losana
G
,
Bernabei
P
,
Forni
G
,
Novelli
F
. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur J Immunol. 2001;31(9):2829–38. doi:.https://doi.org/10.1002/1521-4141(200109)31:9<2829::AID-IMMU2829>3.0.CO;2-U
31
Calcinotto
A
,
Filipazzi
P
,
Grioni
M
,
Iero
M
,
De Milito
A
,
Ricupito
A
, et al.
Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72(11):2746–56. doi:.https://doi.org/10.1158/0008-5472.CAN-11-1272
32
Fischer
B
,
Müller
B
,
Fischer
KG
,
Baur
N
,
Kreutz
W
. Acidic pH inhibits non-MHC-restricted killer cell functions. Clin Immunol. 2000;96(3):252–63. doi:.https://doi.org/10.1006/clim.2000.4904
33
Xie
D
,
Zhu
S
,
Bai
L
. Lactic acid in tumor microenvironments causes dysfunction of NKT cells by interfering with mTOR signaling. Sci China Life Sci. 2016;59(12):1290–6. doi:.https://doi.org/10.1007/s11427-016-0348-7
34
Walton
ZE
,
Patel
CH
,
Brooks
RC
,
Yu
Y
,
Ibrahim-Hashim
A
,
Riddle
M
, et al.
Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell. 2018;174(1):72–87.e32. doi:.https://doi.org/10.1016/j.cell.2018.05.009
35
Welbourne
TC
. Acidosis activation of the pituitary-adrenal-renal glutaminase I axis. Endocrinology. 1976;99(4):1071–9. doi:.https://doi.org/10.1210/endo-99-4-1071
36
May
RC
,
Kelly
RA
,
Mitch
WE
. Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J Clin Invest. 1986;77(2):614–21. doi:.https://doi.org/10.1172/JCI112344
37
Simon
D
,
Luke
RG
. Polymorphonuclear leucocytosis and lymphopenia in acute renal failure and metabolic acidosis in the rat. Clin Sci Mol Med. 1973;45(3):397–402. doi:.https://doi.org/10.1042/cs0450397
38
Tan
L
,
Wang
Q
,
Zhang
D
,
Ding
J
,
Huang
Q
,
Tang
YQ
, et al.
Correction: Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. 2020;5(1):61. doi:.https://doi.org/10.1038/s41392-020-0159-1
39
Teijaro
JR
,
Walsh
KB
,
Cahalan
S
,
Fremgen
DM
,
Roberts
E
,
Scott
F
, et al.
Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–91. doi:.https://doi.org/10.1016/j.cell.2011.08.015
40
Ahmadpoor
P
,
Rostaing
L
. Why the immune system fails to mount an adaptive immune response to a COVID-19 infection. Transpl Int. 2020;33(7):824–5. doi:.https://doi.org/10.1111/tri.13611
41
Connors
JM
,
Levy
JH
. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033–40. doi:.https://doi.org/10.1182/blood.2020006000
42
Etulain
J
,
Negrotto
S
,
Carestia
A
,
Pozner
RG
,
Romaniuk
MA
,
D’Atri
LP
, et al.
Acidosis downregulates platelet haemostatic functions and promotes neutrophil proinflammatory responses mediated by platelets. Thromb Haemost. 2012;107(1):99–110. doi:.https://doi.org/10.1160/TH11-06-0443
43
Bernstein
KE
,
Khan
Z
,
Giani
JF
,
Cao
DY
,
Bernstein
EA
,
Shen
XZ
. Angiotensin-converting enzyme in innate and adaptive immunity. Nat Rev Nephrol. 2018;14(5):325–36. doi:.https://doi.org/10.1038/nrneph.2018.15
44
Kahn
JS
,
McIntosh
K
. History and recent advances in coronavirus discovery. Pediatr Infect Dis J. 2005;24(11, Suppl):S223–7, discussion S226. doi:.https://doi.org/10.1097/01.inf.0000188166.17324.60
45
Egan
BM
,
Li
J
,
Hutchison
FN
,
Ferdinand
KC
. Hypertension in the United States, 1999 to 2012: progress toward Healthy People 2020 goals. Circulation. 2014;130(19):1692–9. doi:.https://doi.org/10.1161/CIRCULATIONAHA.114.010676
46
Burnier
M
,
Egan
BM
. Adherence in Hypertension. Circ Res. 2019;124(7):1124–40. doi:.https://doi.org/10.1161/CIRCRESAHA.118.313220
47
American Diabetes Association. Standards of medical care for patients with diabetes mellitus. Diabetes Care. 2002;25(1):213–29. doi:.https://doi.org/10.2337/diacare.25.1.213
48
American Diabetes Association. Standards of Medical Care in Diabetes-2020 Abridged for Primary Care Providers. Clin Diabetes. 2020;38(1):10–38. doi:.https://doi.org/10.2337/cd20-as01
49
Karet
FE
. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009;20(2):251–4. doi:.https://doi.org/10.1681/ASN.2008020166
50
Oussalah
A
,
Gleye
S
,
Clerc Urmes
I
,
Laugel
E
,
Callet
J
,
Barbé
F
, et al.
Long-Term ACE Inhibitor/ARB Use Is Associated with Severe Renal Dysfunction and Acute Kidney Injury in Patients with severe COVID-19: Results from a Referral Center Cohort in the North East of France. Clin Infect Dis. 2020;ciaa677. doi:.https://doi.org/10.1093/cid/ciaa677
51
Kendrick
J
,
Chonchol
M
,
You
Z
,
Jovanovich
A
. Lower serum bicarbonate is associated with an increased risk of acute kidney injury. J Nephrol. 2020. doi:.https://doi.org/10.1007/s40620-020-00747-8
52
Wesson
DE
,
Simoni
J
. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010;78(11):1128–35. doi:.https://doi.org/10.1038/ki.2010.348
53
Atkins
JL
. Effect of sodium bicarbonate preloading on ischemic renal failure. Nephron. 1986;44(1):70–4. doi:.https://doi.org/10.1159/000183915
54
Bavishi
C
,
Maddox
TM
,
Messerli
FH
. Coronavirus Disease 2019 (COVID-19) Infection and Renin Angiotensin System Blockers. JAMA Cardiol. 2020;5(7):745–7. doi:.https://doi.org/10.1001/jamacardio.2020.1282
55
Imai
Y
,
Kuba
K
,
Rao
S
,
Huan
Y
,
Guo
F
,
Guan
B
, et al.
Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–6. doi:.https://doi.org/10.1038/nature03712
56
Gu
H
,
Xie
Z
,
Li
T
,
Zhang
S
,
Lai
C
,
Zhu
P
, et al.
Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci Rep. 2016;6(1):19840. doi:.https://doi.org/10.1038/srep19840
57
Sodhi
CP
,
Nguyen
J
,
Yamaguchi
Y
,
Werts
AD
,
Lu
P
,
Ladd
MR
, et al.
A Dynamic Variation of Pulmonary ACE2 Is Required to Modulate Neutrophilic Inflammation in Response to Pseudomonas aeruginosa Lung Infection in Mice. J Immunol. 2019;203(11):3000–12. doi:.https://doi.org/10.4049/jimmunol.1900579
58
Reynolds
HR
,
Adhikari
S
,
Pulgarin
C
,
Troxel
AB
,
Iturrate
E
,
Johnson
SB
, et al.
Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Covid-19. N Engl J Med. 2020;382(25):2441–8. doi:.https://doi.org/10.1056/NEJMoa2008975
59
Mancia
G
,
Rea
F
,
Ludergnani
M
,
Apolone
G
,
Corrao
G
. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N Engl J Med. 2020;382(25):2431–40. doi:.https://doi.org/10.1056/NEJMoa2006923
60
Reiffel
JA
. Propensity Score Matching: The ‘Devil is in the Details’ Where More May Be Hidden than You Know. Am J Med. 2020;133(2):178–81. doi:.https://doi.org/10.1016/j.amjmed.2019.08.055
61
Kitterer
D
,
Schwab
M
,
Alscher
MD
,
Braun
N
,
Latus
J
. Drug-induced acid-base disorders. Pediatr Nephrol. 2015;30(9):1407–23. doi:.https://doi.org/10.1007/s00467-014-2958-5
62
Tanaka
T
,
Tsutamoto
T
,
Sakai
H
,
Fujii
M
,
Yamamoto
T
,
Horie
M
. Comparison of the effects of efonidipine and amlodipine on aldosterone in patients with hypertension. Hypertens Res. 2007;30(8):691–7. doi:.https://doi.org/10.1291/hypres.30.691
63
Bauer
JH
,
Sunderrajan
S
,
Reams
G
. Effects of calcium entry blockers on renin-angiotensin-aldosterone system, renal function and hemodynamics, salt and water excretion and body fluid composition. Am J Cardiol. 1985;56(16):H62–7. doi:.https://doi.org/10.1016/0002-9149(85)90546-6
64
Bozkurt
B
,
Kovacs
R
,
Harrington
B
. Joint HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. J Card Fail. 2020;26(5):370. doi:.https://doi.org/10.1016/j.cardfail.2020.04.013