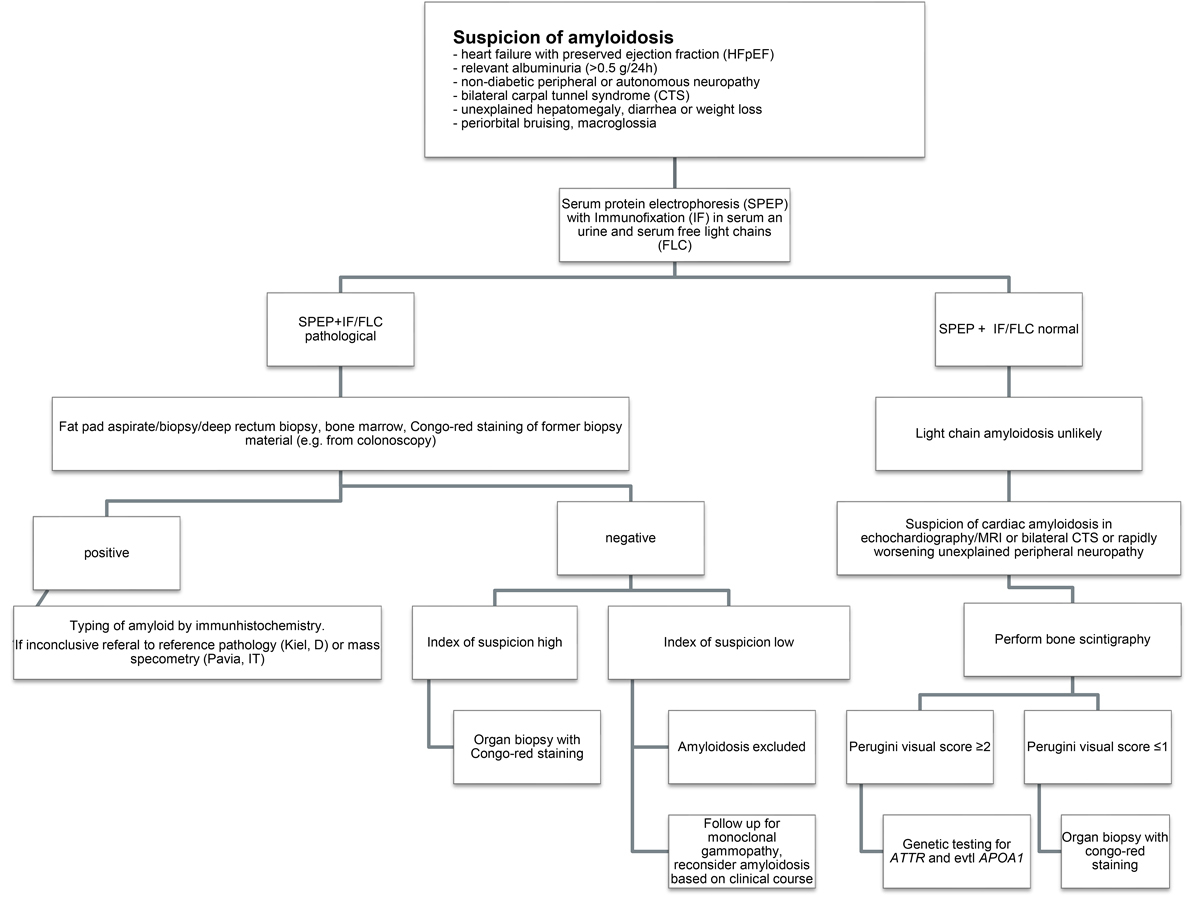
Figure 1 Flow chart of the diagnostic work-up.
DOI: https://doi.org/10.4414/smw.2020.20364
Systemic amyloidosis is a heterogeneous group of diseases associated with protein misfolding into insoluble beta-sheet rich structures that deposit extracellularly in different organs, eventually compromising their function. There are more than 30 different proteins known to be amyloidogenic in vivo with “light chain” (AL)-amyloidosis being the most common type, followed by transthyretin (ATTR)- and amyloid protein A (AA)-amyloidosis [1]. Systemic amyloidosis is a rare disease with an incidence of around 10 patients in 1 million inhabitants [2, 3]. Recently several new therapeutic options have been developed for subgroups of amyloidosis patients, and the introduction of novel therapies for plasma cell myeloma has led to an increase in the therapeutic armamentarium for plasma cell disorders, including AL-amyloidosis. Among them, proteasome inhibitors, immunomodulatory agents (“-imids”) and monoclonal antibodies have been successfully introduced into clinical practice.
Still, high-quality data from randomised controlled trials regarding the benefit of these cost-intensive drugs in AL-amyloidosis are widely lacking, and owing to the rarity of the disease many physicians will not gain routine experience in the management of these frail patients. The diagnosis of AL-amyloidosis relies on a close collaboration between clinicians, pathologists, imaging experts and sometimes geneticists. Diagnosis and treatment options in this complex disorder should be discussed by dedicated multidisciplinary boards. The current Swiss expert recommendations aim at helping decision making for teams caring for patients with AL-amyloidosis.
In January 2020, the first meeting of the Swiss Amyloidosis Network (SAN) took place in Zurich, Switzerland. One aim of this meeting was to establish a consensus guideline regarding the diagnostic work-up and treatment recommendations for systemic amyloidosis, tailored to the Swiss healthcare system. Forty-five participants from haematology, oncology, cardiology, nephrology, neurology, hepatology, nuclear medicine and pathology, as well as geneticists and scientists engaged in basic research, discussed many aspects of amyloidosis. The five University Hospitals of Basel, Bern, Geneva, Lausanne and Zurich, as well as the five large tertiary hospitals Bellinzona, Chur, Luzern, St Gallen and City Hospital Waid and Triemli Zurich were represented.
These Swiss Amyloidosis Network recommendations focus on diagnostic work-up and treatment of AL-amyloidosis.
The precursor proteins of AL-amyloidosis are monoclonal immunoglobulin light chains, deriving from an underlying plasma cell neoplasm, including plasma cell myeloma or, less commonly, B-cell lymphoma. The tumour load, i.e., the degree of bone marrow infiltration by neoplastic cells, does not necessarily correlate with AL load and is often low. Due to the comparatively low plasma cell burden, the free light chains measurable in the serum are often only slightly elevated. Not all monoclonal light chains are amyloidogenic, and there is a predominance of lambda over kappa light chains in AL-amyloidosis [4]. In about 50% of amyloidosis patients, the affected monoclonal plasma cells carry a translocation t(11;14), which is only present in about 15% of plasma cell myeloma patients [5]. Organ dysfunction is a result of amyloid deposition in the tissue, but there is evidence of direct toxicity of the free-light chain, for example on cell metabolism and cardiac function [6].
One of the main limitations for effective treatment is the delay between first symptoms and the diagnosis of AL-amyloidosis. First symptoms of AL-amyloidosis are often nonspecific, and without a high level of awareness most patients will be diagnosed at an advanced stage, where treatment is usually more difficult and less effective [7]. Therefore, it is essential to make the diagnosis of AL-amyloidosis as early as possible. The majority of AL-amyloidosis patients have abnormal free light chain (FLC) studies with elevated dFLC (difference involved minus uninvolved FLC) levels that steadily increase over time for years prior to diagnosis [8, 9]. This is the reason why the SAN recommends regular screening of patients with monoclonal gammopathy of undetermined significance (MGUS) and smouldering plasma cell myeloma according to International Myeloma Working Group (IMWG) 2010 guidelines [10] (initial follow up after 6 months, if stable annually) for the presence of amyloid cardiomyopathy (elevated N-terminal pro-B-type natriuretic peptide [NTproBNP]) [11] and nephropathy (especially albuminuria) or any other clinical signs of amyloidosis. In the event of abnormal findings, work-up for systemic amyloidosis should be initiated. Routine amyloid staining of the bone marrow is not indicated in patients with MGUS or multiple myeloma without suspicion of AL-amyloidosis [12].
The most commonly involved organs are heart, kidneys, nervous system, soft tissue and liver [13, 14]. Suspicion of a diagnosis of AL-amyloidosis should be raised in patients with MGUS or plasma cell myeloma who present with heart failure with preserved ejection fraction (HFpEF), albuminuria, unexplained weight loss, autonomous and peripheral neuropathy (specifically small fibre neuropathy), hepatomegaly, chronic diarrhoea or carpal tunnel syndrome, especially if bilateral. A history of spontaneous periorbital bruising (“raccoon eyes”), macroglossia or swelling of the shoulder (shoulder pad sign) are pathognomonic, but late, signs for the disease and should prompt a work-up for systemic AL-amyloidosis [4] (table 1)
Table 1 Organ involvement.
| Organ | Occurrence | Clinical Symptoms | Biomarkers | Diagnostics |
|---|---|---|---|---|
| Heart | 75% | Heart failure Arrhythmia Syncope Sudden cardiac death |
NTproBNP Troponin T |
ECG: low voltage, pseudoinfarction pattern, atrial fibrillation Echocardiography: biventricular wall thickening, thickening of the interatrial septum, pericardial effusion, preserved ejection fraction with diastolic dysfunction including signs of increased filling pressures or atrial enlargement, “apical sparing” pattern by global longitudinal peak strain or speckle tracking MRI: thickening of the myocardium, atrial enlargement, late gadolinium enhancement, increased native T1, extracellular volume and T2 mapping. Pericardial and pleural effusion |
| Kidney | 50–70% | Oedema Foamy urine |
Proteinuria/albuminuria Estimated glomerular filtration rate |
24-hour urine collection Spot urine |
| Nerve involvement | 35% Presenting symptoms: 7.5% |
Small-fibre neuropathy: paraesthesia, numbness, pain All-fibre neuropathy: at later stages Autonomic: gastroparesis, bladder/bowel dysfunction, orthostatic hypotension, vasomotor symptoms, hyperhidrosis, sicca syndrome, erectile dysfunction Bilateral carpal tunnel syndrome |
– | Nerve conduction study (normal if only small fibres involved) Electrophysiological autonomic tests: R-R interval, sympathetic skin reflex Sudoscan®, laser evoked potentials if available Schellong test Skin biopsy (small fibre neuropathy) Nerve biopsy if large fibres involved and no other organ accessible to biopsy Tissue biopsy if surgery is performed |
| Gastrointestinal involvement | 10% | Diarrhoea Constipation Nausea Vomiting Weight loss |
– | Colonoscopy Gastroscopy |
| Liver | 20% | – | Alkaline phosphatase | Hepatomegaly (ultrasound) If liver biopsy is performed it should be done as a transjugular biopsy and not percoutaneously owing to the increased bleeding risk |
| Skin / soft tissue | 10–15% | Periorbital purpura (racoon eyes) Macroglossia Swelling of the shoulder pad (“Shoulder pad sign”) |
– | Clinical examination |
| Bleeding diathesis | – | Easy bruising / spontaneous bleeding due to acquired factor X deficiency / acquired Von Willebrand disease, amyloid infiltration in blood vessels | Coagulation factor X von Willebrand factor (functional) Quick, activated partial thromboplastin time |
Coagulation testing |
MRI = magnetic resonance imaging; NTproBNP = N-terminal pro-B-type natriuretic peptide
AL-amyloidosis may affect one single site [15, 16]. In localised amyloidosis, a monoclonal component in plasma is usually absent. It is hypothesised that a B-cell / plasma cell clone develops at a site of chronic inflammation, producing amyloidogenic light chains, with additional local factors favouring their deposition as insoluble amyloid. Typical sites for localised amyloidosis are the skin, the upper airways in particular and the respiratory tract in general, the genital and urinary system, the eye, the lymph nodes and the gastrointestinal tract [17]. Localised amyloidosis almost never progresses to a systemic form, and its prognosis is excellent. The treatment of this “amyloidoma” is surgical excision when possible.
The SAN recommends a thorough work-up for patients with suspected localised amyloidosis to exclude systemic involvement. This includes a search for monoclonal free light chains / monoclonal protein by electrophoresis with immunofixation of serum and urine, and nephelometric measurement of serum free light chain levels. In the event of abnormalities, a bone marrow biopsy examination with Congo red staining, Congo red staining of an abdominal fat biopsy, as well as work up for cardiac and renal involvement (NTproBNP, echocardiography including analysis of longitudinal left ventricular deformation [“strain”] and/or cardiac magnetic resonance imaging [MRI]), as well as determination of albumin/creatinine and protein/creatinine ratios) should be performed. The SAN recommends follow-up of these patients annually, at least initially, in order to detect local reoccurrence [18].
When a patient presents with clinical symptoms compatible with AL-amyloidosis, a thorough work-up for monoclonal gammopathy must be performed including serum protein electrophoresis, serum FLCs and immunofixation of the serum and urine. In the presence of a monoclonal gammopathy, especially if a pathological FLC ratio is present, a tissue sample for amyloid detection with Congo red staining and immunohistochemistry or mass spectrometry is required to establish the diagnosis and to allow for subtyping of the amyloid. In the published literature, an abdominal fat tissue aspirate together with a bone marrow biopsy of at least 1.5 cm length is recommended, and should secure the diagnosis in 80% of patients with AL-amyloidosis [19]. Minor salivary gland biopsy is also sensitive in the detection of amyloid (68.4%) [20].
At the SAN meeting, experts agreed that this high yield of positive fat pad aspirates is true for large amyloidosis referral centres, where fat pad aspiration is the recommended first option to search for amyloid deposits, but cannot be achieved by smaller centres and might reflect differences in sampling and processing of the material. Furthermore, fat pad aspirates are helpful for detecting amyloid by Congo red staining, but the material is not suitable for immunohistochemistry, which is needed to type the amyloid and is performed almost exclusively on tissue biopsies. Immunoelectron microscopy or mass spectrometry can be successfully performed on fat pad aspirates, but are currently not available in Switzerland. The SAN recommends fat pad biopsies, minor salivary gland or deep rectum biopsies for diagnosis. If a patient has had a tissue biopsy for other reasons (e.g., polypectomy during a colonoscopy), these samples can be retrospectively screened for amyloid by Congo red staining. If a bone marrow biopsy is performed in a work-up for MGUS in a patient with suspicion of AL-amyloidosis, it must be thoroughly evaluated for amyloid deposition. If all samples stain negative for amyloid and suspicion remains high, an organ biopsy should be performed [1]. We should keep in mind that, owing to vascular amyloid deposits and possible factor X deficiency due to adsorption to amyloid fibrils [21, 22], bleeding risk can be increased. Given the high bleeding risk, liver biopsy should be performed only through a venous trans-jugular access.
The presence of a plasma cell neoplasm in combination with histological documentation of amyloid by Congo red staining alone does not allow the diagnosis of AL-amyloidosis. Amyloid typing is crucial, as other forms of amyloidosis are increasingly recognised to be quite common in the elderly population, especially ATTR-amyloidosis. In these elderly patients, plasma cell dyscrasia might be an innocent bystander unrelated to amyloidosis [23]. As a matter of fact, patients with ATTR-amyloidosis have a very high incidence of concomitant MGUS (23%) [24]. Notably, studies have demonstrated the high diagnostic accuracy of bone scintigraphy in the non-invasive assessment of cardiac ATTR-amyloidosis [25]. However, in the presence of MGUS, bone scintigraphy alone cannot be used to rule out AL-amyloidosis and a tissue biopsy is mandatory [26].
The SAN concludes that amyloid typing is crucial to establish the diagnosis. Mass spectrometry is considered the gold standard [1]. However, in Switzerland mass spectrometry is not available in this context and typing is mainly done by means of immunohistochemistry. Immunohistochemistry is challenging, and one Swiss centre, the Institute of Pathology, University Hospital Basel, has established an immunohistochemistry panel with non-commercially available antibodies for amyloid typing [27], similar to the ones used by the German reference pathology laboratory in Kiel. Equivocal cases can be sent for a second opinion to a reference centre outside of Switzerland (for example, to the German reference pathology laboratory in Kiel). Mass spectrometry can be performed in Pavia, Italy, although the logistical obstacles (e.g., transport, cost coverage by the Swiss health insurance) can delay diagnosis.
The diagnostic work-up is summarised in figure 1.
Figure 1 Flow chart of the diagnostic work-up.
As soon as the diagnosis of AL-amyloidosis is established, the general health status of the patient, the biology of the disease and the extent of organ involvement have to be determined. The SAN recommends a structured baseline assessment for all patients as presented in table 2 .
Table 2 Baseline evaluation.
| General | Haematology | Cardiology | Nephrology | Neurology | Gastroenterology | |
|---|---|---|---|---|---|---|
| Laboratory work-up | CRP TSH, fT4 Protein Albumin Vitamin B12 Hepatitis A+B HIV |
Bloodcount with microscopic differentiation Reticulocytes INR, aPTT Coagulation factor X protein electrophoresis immunfixation* serum free light chain levels Immunoglobulins Type and screen† |
NTproBNP Troponin T | Creatinine Electrolytes Urine sediment Protein/creatinine‡ Albumin/creatinine‡ optl 24-hour urine - protein - albumin - immunofixation |
(Anti-MAG antibodies) | Liver parameters Cholestatic parameters |
| Clinical examination | Macroglossia? Racoon eyes? Shoulder pad sign? Nail changes? |
Bruises | Blood pressure Heart rate Schellong test Peripheral oedema Signs of heart failure ECG ECG Holter Spiroergometry |
Screening for polyneuropathy (sensitivity to light touch, pin-prick and temperature, vibration and position sense, deep tendon reflexes) | Hepatomegaly Splenomegaly |
|
| Imaging | – | Low-dose CT if osteolysis is suspected optl whole body MRI |
Echocardiography optl MRI |
optl kidney ultrasound | NCS and neurophysiological tests for small nerve fibres if screening is positive | Abdominal ultrasound optl fibroscan optl endoscopy |
| Intervention | Bone marrow tap - cytology - histology including Congo red stain - flowcytometry (minimal residual disease if possible) - FISH |
For specific questions (e.g., differentiation ATTR vs AL) Endomyocardial biopsy |
aPTT = activated partial thromboplastin time; CRP = C-reactive protein; CT = computed tomography; FISH = fluorescence in situ hybridisation; fT4 = free thyroxine; HIV = human immunodeficiency virus; INR = international normalised ratio; MAG = myelin associated glycoprotein; MRI = magnetic resonance imaging; NCS = nerve conduction studies; NTproBNP = N-terminal pro-B-type natriuretic peptide; optl = optional; TSH = thyroid stimulating hormone * Serum and urine; † especially prior to daratumumab; ‡ spot urine Scintigraphy for ATTR-amyloidosis only (not necessary in work-up for AL-amyloidosis)
The global prognosis of patients suffering from AL-amyloidosis has improved over time as a result of better therapeutic options targeting plasma cells [28]. Nevertheless, early mortality remains a challenge [29, 30]. The clinical course of patients with AL-amyloidosis depends mainly on the type and extent of the organ involvement. Cardiac involvement is the main determinant for adverse outcome and prognostic models have implemented cardiac biomarkers such as troponin T or troponin I, NTproBNP and BNP, as powerful predictors at diagnosis for survival duration and relapse [31]. In Switzerland, centres mainly use the revised Mayo risk prediction model of 2012 to predict outcome and adapt therapy, and a NTproBNP cut-off of >8500 ng/l to identify a subset of patient with a particularly poor outcome [32] (table 3). In patients with predominantly renal involvement, a distinct staging system can help predict the renal outcome [33] (table 4).
Table 3 Staging of AL-amyloidosis (revised Mayo Model 2012, Kumar et al., JCO 2012 [31]).
| Risk factors | Difference between involved and uninvolved serum free light chains >180 mg/l | |
| Cardiac troponin T ≥25 ng/l | ||
| NTproBNP ≥1800 ng/l | ||
| Score (1 point per risk factor) | Stage | Median survival (months) |
| 0 | I | 94.1 |
| 1 | II | 40.3 |
| 2 | III | 14.0 |
| 3 | IV | 5.8 |
NTproBNP = N-terminal pro-B-type natriuretic peptide
Table 4 Renal staging system (Palladini et al., Blood 2014 [33]).
| Risk factors | Estimated glomerular filtration rate <50 ml/min | |
| Proteinuria >5 g/24h | ||
| Score (1 point per risk factor) | Stage | 2-year risk for dialysis |
| 0 | I | 0–3% |
| 1 | II | 11–25% |
| 2 | III | 60–75% |
Additional patient and disease characteristics associated with poor outcome and survival include systolic blood pressure <100 mm Hg, intraventricular septum thickness >15 mm, myocardial tissue characterisation by cardiac MRI (transmural extent of late gadolinium enhancement [34], extracellular volume >45% by T1 mapping [35] or evidence of myocardial oedema by T2 mapping [36]), classical myeloma end-organ damage (hypercalcaemia, renal insufficiency, anaemia, bone lesions) and/or bone marrow plasma cells ≥10%, >50% del17p in interphase fluorescence in situ hybridisation (iFISH), immunoparesis, presence of circulating plasma cells and elevated lactate dehydrogenase [37–41].
Patient and disease characteristics associated with better outcome and survival are eligibility for high-dose melphalan and autologous stem cell transplantation (ASCT) [42, 43], low and therefore non-evaluable difference of involved and uninvolved free light chains (dFLC) at diagnosis (<50 mg/l) [44], depth of haematological response after induction therapy (≥very good partial remission, VGPR) and depth of organ response [45].
Predictive markers: Baseline iFISH can predict the efficacy of the first-line therapy in AL-amyloidosis. Translocation t(11;14) is detected in roughly half of the patients and is associated with lower haematological event-free survival and overall survival in patients treated with bortezomib [46, 47]. However, treatment with high-dose melphalan + ASCT is able to reverse the negative impact of t(11;14) on overall survival [48]. Conversely, gain of 1q is an independent adverse prognostic factor in AL-amyloidosis patients treated with melphalan [49]. In the bone marrow work-up, the SAN recommends cytogenetic analysis including iFISH +/− aCGH (array-based comparative genomic hybridisation) as well as immunophenotyping at baseline, iFISH should be performed in a CD138+ plasma cell enriched sample.
In AL-amyloidosis, one has to differentiate between haematological response and organ response. The haematological response measures the effect of the plasma cell-directed therapy, and is subdivided into four response categories based on the changes in dFLC and serum and urine electrophoresis with immunofixation (table 5). The deeper the response the better the outcome, but obtaining at least a VGPR should be the goal of every treatment regimen (overall survival 80–90% after 3 years) [50]. The effectiveness of therapy should be assessed regularly. As FLC have a short half-life (hours), response is measurable shortly after treatment initiation and the treatment regimen should be changed rapidly if inefficient (decrease in dFLC <50% after 2–3 cycles). Organ response is the ultimate goal of therapy but is usually delayed and can take as long as 9.4 months (heart), 6 months (kidney) and 6.1 months (liver) after obtaining a complete haematological response (table 5) [51]. The SAN recommends a structured assessment and documentation of the haematological response and of the organ response at least every 3 months. During immuno-/chemotherapy haematologialc response (dFLC) should be determined at least on day one of every treatment cycle and at the end of treatment.
Table 5 Response criteria (Palladini et al., JCO 2012 [50]).
| Haematological | |
| Complete response (CR) | Negative serum and urine immunofixation, normal serum FLC ratio |
| Very good partial response (VGPR) | dFLC <40 mg/l |
| Partial response (PR) | Reduction of dFLC ≥50% |
| No response | Reduction of dFLC <50% |
| Organ response parameters | |
| Non-responder | ≤30% reduction in organ response parameters: - Cardiac: NTproBNP - Renal: proteinuria - Hepatic: alkaline phosphatase |
| Partial responders | 31–60% reduction |
| Very good partial responders | >60% |
| Complete responders | Nadir NTproBNP ≤400 ng/l Nadir proteinuria ≤200 mg/24h Nadir alkaline phosphatase ≤2 × upper limit of reference range |
| dFLC = difference involved minus uninvolved FLC; FLC = free light chain; NTproBNP = N-terminal pro-B-type natriuretic peptide | |
Therapy is indicated in all patients with confirmed systemic AL-amyloidosis. There are few exceptions in patients with a coincidental finding of amyloid deposits in tissue samples with absence of systemic organ involvement. These subjects with asymptomatic amyloid deposits do not require treatment, but should be carefully followed up to detect potential organ dysfunction in a timely manner [9]. The goal of treatment is to suppress the malignant plasma cell clone in order to prevent further FLC production and deposition of amyloid in the already altered organs. Although the pathological plasma cell clone is generally sensitive to current treatment approaches, the therapeutic agents are often poorly tolerated and morbidity and mortality, especially at the beginning of treatment, can be significant. Frequent adjustment of the therapeutic regimen is common and symptom management is an integral part of therapy. Therefore, the SAN recommends a multidisciplinary approach involving amyloidosis specialists from different fields in internal medicine to make treatment decisions, even in experienced centres [1].
The available clinical data to drive treatment decisions in AL-amyloidosis consist mainly of phase II studies, retrospective comparisons and case series. The treatment protocols (see figs 2 and 3 ) are similar to those used for plasma cell myeloma, although dose adaptations are common to allow better tolerability. The choice of the initial therapy is also driven by the labelling and reimbursement of cost by the Swiss healthcare insurances, as there are no drugs specifically approved for AL-amyloidosis in the absence of an underlying multiple myeloma.
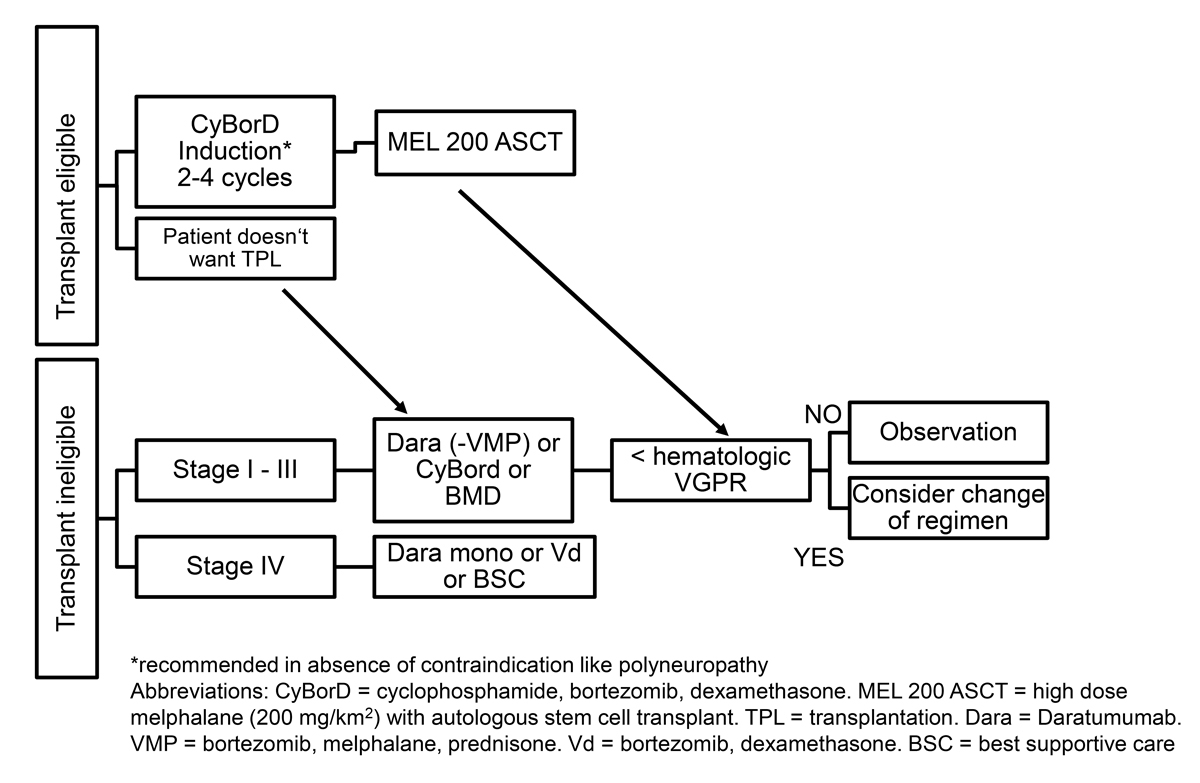
Figure 2 First-line treatment.
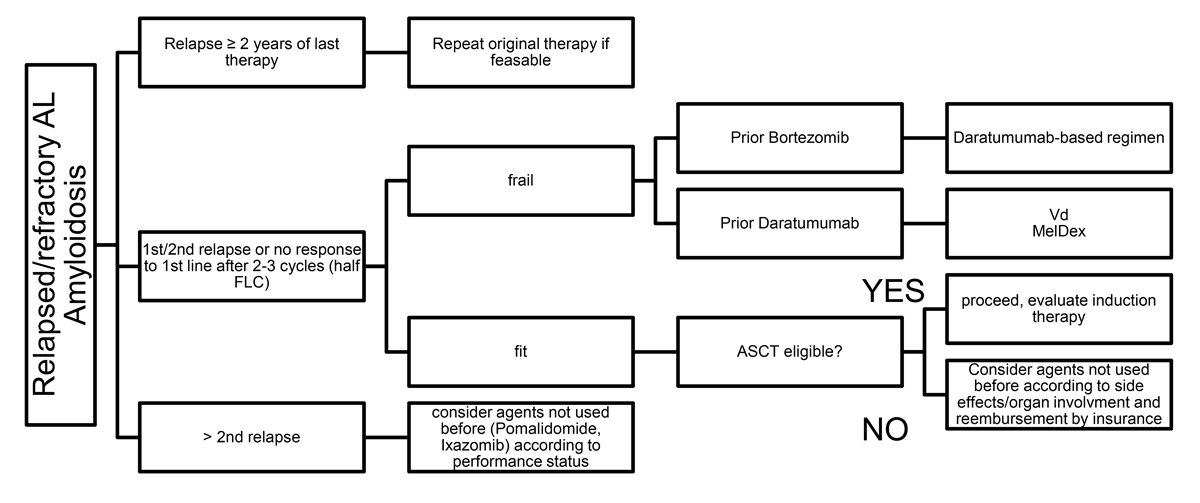
Figure 3 Treatment in the relapse/refractory setting.
One of the most important treatment decisions is whether a given patient is “fit” enough to tolerate high-dose melphalan + ASCT. Historically, transplant-related mortality in less stringently selected patients with AL-amyloidosis was as high as 24%, mainly because of the inclusion of patients with advanced cardiac involvement [52]. As a consequence, many centres have adapted their selection criteria for high-dose melphalan + ASCT with special attention to sufficient cardiac reserve [53]. Selection criteria for ASCT in AL amyloidosis have undergone significant modification over time [54]. The SAN experts agreed on the Onkopedia Guidelines established by the University Hospital Heidelberg as selection criteria for patients eligible for ASCT (table 6). Further, troponin T levels >60 ng/l are associated with adverse outcome as expected day-100 mortality rate is 25% or higher. Therefore, these patients should not be considered viable candidates for ASCT [55]. With this more restrictive patient selection, only 25% of patients are eligible for high-dose melphalan + ASCT. Provided the procedure is performed in an experienced centre, transplant-related mortality is significantly reduced to <5%, with a favourable outcome especially in patients who achieve a complete response (3 year overall survival 84% vs 59% in the non-transplant group). If high-dose melphalan + ASCT is used, the SAN recommends a bortezomib-based induction therapy prior to stem cell collection, although the data to support this approach are scarce [53, 56]. Further, high-dose melphalan + ASCT should only be used in patients fit enough for full dose melphalan, as dose reduction is associated with adverse outcome (lower complete response rates, higher transplant-related mortality, lower survival rate) [57]. Induction therapy allows an immediate start of therapy, which is important since the prompt clearance of the toxic circulating light chains will potentially improve patient outcome and will reveal valuable information with regard to physical fitness prior to the more strenuous ASCT. In patients with polyneuropathy, the SAN considers skipping induction to avoid additional bortezomib-induced neurotoxicity. The SAN agrees that high-dose melphalan + ASCT should only be performed at centres with experience in the treatment of AL-amyloidosis as it has been shown that outcome is better in centres performing more than four AL transplants a year [57].
Table 6 Selection criteria for autologous stem cell transplantation (Onkopedia Leitlinien September 2017).
| Age <65–70 years |
| Eastern Cooperative Oncology Group performance status score <2 |
| Revised Mayo cardiac stage <3, New York Heart Association class <III |
| Systolic blood pressure >90 mm Hg |
| No cardiac related effusions |
| Creatinine clearance >30 ml/min |
The introduction of proteasome inhibitors, especially bortezomib, has played a decisive role in the treatment of AL-amyloidosis and has significantly contributed to improvement in outcome of AL-amyloidosis patients. It is used as an induction regimen prior to high-dose melphalan + ASCT in combination with dexamethasone and cyclophosphamide (CyBorD) with haematological response rates between 50 and 70% [53, 58]. Preliminary data of a prospective randomised controlled phase III trial comparing melphalan and dexamethasone with bortezomib, melphalan and dexamethasone (BMDex) in non-transplant-eligible patients showed an improved haematological response in the BMDex group (84% vs 57%, p = 0.005) [59].
The dose and frequency of bortezomib administration varied in the different studies, the most commonly used dosing being 1.3 mg/ m2 once or twice weekly. In patients with cardiac involvement, therapy with bortezomib might worsen orthostatic dysregulation, especially in high risk patients (revised Mayo stage III with NTproBNP levels >8500 ng/l and/or New York Hear Association [NYHA] class III) [32] To improve tolerability of therapy the SAN recommends starting with 1 mg/m2 once weekly in patients with cardiac involvement (revised Mayo stage III), with a dose increase if well tolerated. Bortezomib should be administered subcutaneously and not be used in patients suffering from neuropathy (peripheral and autonomic). In transplant-eligible patients, the SAN recommends the non-melphalan containing regimen CyBorD as an induction regimen prior to high-dose melphalan + ASCT to reduce stem cell toxicity. In non-transplant eligible patients the SAN prefers the combination of bortezomib and melphalan plus dexamethasone (BMDex) to overcome the previously discussed lower response rates associated with doublet therapy in patients with t(11;14) or gain of 1q [60, 61].
Carfilzomib, a second-generation proteasome inhibitor, is a standard agent for patients with plasma cell myeloma. In a multicentre phase I/II trial in patients with AL-amyloidosis, the overall haematological response rate was 63% in these relapsed/refractory patients, but all-cause Common Terminology Criteria for Adverse Events (CTCAE) grade 3 (severe) and 4 (life-threatening) where as high as 71% with several cardiac or pulmonary events [62]. Therefore, the SAN recommends using carfilzomib only in the absence of other options and only in patients with AL-amyloidosis without cardiac involvement.
Ixazomib, an oral proteasome inhibitor, is currently approved in Switzerland in combination with lenalidomide and dexamethasone as a second and third line treatment in patients with plasma cell myeloma. In 2014, a phase I/II study demonstrating a haematological response rate of 52% with less neurotoxicity when compared with bortezomib led to the US Food and Drug Administration (FDA) approval of ixazomib and dexamethasone in AL-amyloidosis patients [63]. In a phase III trial (TOURMALINE-AL1) [64], ixazomib was studied in previously treated AL-amyloidosis patients in combination with dexamethasone and did not demonstrate significant improvement of haematological response compared with standard therapy, which led to the discontinuation of the study. Ixazomib + dexamethasone led to an overall response rate of 53% vs 51% in the physician’s choice group. The majority of patients (71 of 83) in the control arm where treated with either melphalan + dexamethasone or lenalidomide + dexamethasone. Both combinations are effective, with haematological response rates between 51% and76% (melphalan + dexamethasone), and 41% and 67% (lenalidomide + dexamethasone), so this result is not surprising [65, 66]. Ixazomib was well tolerated in the trial population and remains an option in patients with neuropathy if a proteasome inhibitor is the agent of choice.
Daratumumab, a monoclonal antibody targeting CD38, appears to be highly effective in the treatment of AL-amyloidosis patients [67–69]. The antibody shows rapid and deep response rates with an excellent tolerability profile, even in stage IV patients. So far, there were only reports from retrospective case series, all reporting haematological response rates between 63% and 83% in a generally pretreated patient population [70, 71]. A phase III trial comparing daratumumab -CyBorD with CyBorD enrolled patients with daratumumab given subcutaneously (NCT03201965). Results have been presented at EHA 2020, and show higher rates of overall haematological response (92% vs 77%) in the experimental arm and very good partial response or better (≥VGPR 79% vs 49%). The 6-month cardiac response rate was 42% for daratumumab -CyBorD and 22% for CyBorD (p = 0.0029); 6-month renal response rates were 54% and 27%, respectively (p <0.0001) [72].
Daratumumab commonly causes profound hypogammaglobulinaemia and increases susceptibility for infections, including pneumonia. Intravenous or subcutaneous immunoglobulin substitution should be established in patients with infectious complications, and vaccination against invasive pneumococcal disease and influenza must be given prior to treatment initiation [67]. With respect to these precautionary measures, the SAN recommends implementation of daratumumab treatment early in the course of the disease, as monotherapy or in combination with a bortezomib-based regimen. In Switzerland, daratumumab in combination with bortezomib, melphalan and prednisone is approved for first-line treatment of transplant-ineligible patients with plasma cell myeloma. This is valuable in negotiations with the health insurance company to obtain cost coverage.
For elotuzumab, an anti-SLAM-F7 antibody, there are only sporadic data in patients with AL-amyloidosis. Elotuzumab cannot be recommended as a standard treatment for AL-amyloidosis as yet.
Despite the abundance of new effective treatment options, alkylating agents such as melphalan, cyclophosphamide or bendamustine are still therapeutic options for AL-amyloidosis. Haematological response rates with melphalan can be as high as 76% (combination with dexamethasone 40 mg daily for 4 days) with good tolerability [66]. Time to response is usually longer than with newer agents. It is most suitable for elderly patients with revised Mayo stage I/II, where the need to start immediate treatment is lower. Cyclophosphamide is mainly used in combination with bortezomib and dexamethasone (CyBorD) as an induction regimen prior to high-dose melphalan + ASCT and upfront in transplant-ineligible patients as discussed above. Oral administration once weekly (300 mg/m2) is a typical mode of application. Bendamustine with or without rituximab is mainly used in IgM-AL-amyloidosis, where the pathological cell clone is usually at a B-cell maturation step (e.g., IgM MGUS, Waldenström's macroglobulinaemia, lymphoplasmocytic lymphoma, marginal zone lymphoma, chronic lymphatic lymphoma). It has proven to be reasonably well tolerated and effective with haematological response rates of 57% [73].
Immunomodulators such as lenalidomide and pomalidomide are given orally, usually as combination therapies with corticosteroids (“doublets”) and have shown efficacy mainly in case series of pretreated AL-amyloidosis patients. Lenalidomide combined with dexamethasone shows haematological response rates of 41–67% [63, 64], without further improvement when used as a “triplets” for example together with melphalan (overall response rate 50–58%) or cyclophosphamide (overall response rate 46–60%). Pomalidomide has been combined with dexamethasone, showing haematological response rates of 48–60% [74]. Unlike in plasma cell myeloma, the tolerability of these drugs in AL-amyloidosis is limited, and the discontinuation rate is high (>40%). The SAN experts agree on mandatory initial dose reductions. Typical initial dose levels for lenalidomide are 10 mg/d (5–15 mg) on days 1 to day 21, and for pomalidomide 2 mg (1–4 mg) on days 1 to 21. Lenalidomide and pomalidomide have several haematological and non-haematological toxicities, including neutropenia, infections, worsening of kidney function and a rise of NT-pro BNP levels [75]. Both agents are legitimate second-line agents when used with caution in patients with cardiac and/or renal involvement.
Venetoclax is a B cell lymphoma 2 (BCL-2) inhibitor active in plasma cell myeloma, particularly those patients harbouring t(11;14) associated with high BCL-2 expression. Approximately 50% of patients with AL-amyloidosis have t (11;14), making venetoclax a suitable agent to consider in this rare disease. So far, limited case series of mostly heavily pretreated patients confirmed efficacy as single agent and in combination with an acceptable safety profile. Further studies are warranted [76–78].
Symptom management is an integral part of therapy. It includes anti-infective strategies, diuretics, antiarrhythmic drugs, pain medication and agents that control bowel dysfunction.
Anti-infectious strategies: Prior to treatment initiation vaccination against invasive pneumococcal disease should be offered to all patients, as well as a yearly vaccination for seasonal influenza. Immunoglobulin substitution should be considered in patients with hypogammaglobulinaemia (IgG <6 g/l) and related infections. Prophylaxis against varicella zoster virus and pneumocystis pneumonia are part of most immuno/chemotherapy schemes.
If the heart is affected, heart failure with preserved ejection fraction (HFpEF), with diastolic dysfunction and its consequences, is the main clinical issue. Fluid retention leads to dyspnoea, leg oedema, ascites, weight gain and loss of appetite. Furthermore, high left ventricular filling pressure, together with structural alterations in the atrium, increases the likelihood for atrial fibrillation and thus thromboembolic stroke. Besides atrial fibrillation, amyloid deposits may lead to heart blocks and ventricular tachycardia, the latter being an important contributor to mortality. Traditional heart failure therapy such as beta-blockers or angiotensin converting-enzyme inhibitors are not well tolerated and may even worsen clinical symptoms and outcome. Diuretics are the cornerstone of heart failure management in amyloidosis patients; however, doses must be titrated carefully. As a result of the altered pressure/volume relationship in the “stiff” heart with severe diastolic dysfunction, “over diuresis” might lead to profound hypotension, whereas “under diuresis” worsens dyspnoea and increases the risk for cardiac decompensations. Antiarrhythmic therapy should be restricted to amiodarone, rate control with a beta-blocker is an option, but heart rate should not be lowered too much (cardiac output in patients with cardiac amyloidosis depends on heart rate, because stroke volume is fixed owing to amyloid deposits). Regular screening for atrial fibrillation is mandatory in patients with cardiac amyloidosis, and all patients with atrial fibrillation should receive oral anticoagulation, irrespective of the CHA2DS2-VASc score, given the very high risk for thromboembolic events. The SAN experts recommend that selected patients with an atrial mechanical dysfunction and a restrictive filling pattern (severe diastolic dysfunction), after careful evaluation of bleeding risk, receive anticoagulation, irrespective of whether atrial fibrillation is present or not [79]. Particular attention should be given to ventricular arrhythmias with their risk for sudden cardiac death. Although an implantable cardioverter defibrillator may provide life-saving treatments, its long-term benefit on mortality has not been proven in cardiac amyloidosis and should be made on an individual basis in selected cases [80, 81].
In patients with renal AL-amyloidosis and proteinuria, the SAN does not recommend anti-proteinuric treatment with renin-angiotensin-aldosterone system blocking agents. It is unknown whether they provide a benefit, and they might be harmful in patients with co-existing autonomic dysfunction and cardiac amyloidosis. Nephrotoxic drugs should be avoided whenever possible.
Doxycycline, an antibiotic that has been used as a prophylaxis in the ASCT [82] setting, has been shown to have a fibril-stabilising effect in a transgenic mouse model [83]. Currently, a phase III study is comparing doxycycline with standard supportive therapy in newly diagnosed patients undergoing bortezomib-based therapy (NCT03474485). The addition of doxycycline to induction therapy can be considered. Patients should be instructed about phototoxicity, sunscreen SPF 50 should be applied. The results of a randomised trial comparing epigallocatechin-3-gallate (EGCG, “green tea”) to placebo conducted in Heidelberg and presented at the ISA 2018 conference in Kumamoto did not show benefit in the EGCG group.
In recent years, research also focused on the clearance of already deposited amyloid fibrils. There have been three antibodies under development, but so far none of them has shown clinical efficacy leading to commercialisation. The antibody NEOD001 showed promising results in a phase I/II trial [84], but further development was halted when a phase IIb trial did not meet its primary and secondary endpoints [85]. The entire phase III programme was then discontinued, based on futility analysis [86]. Carboxy-pyrrolidinyl-hexanoyl-pyrrolidine-carboxylate (CPHPC), a small molecule that binds to serum amyloid P in the plasma in combination with an anti-SAP antibody [87] to remove the deposited amyloid in the tissue, was able to show clearance of amyloid in 15 patients. The phase II trial had some major safety problems and had to be halted [88]. The third antibody under development is called 11-1F4. The results from the phase I trial are promising, showing early organ response [89]. A phase II trial is planned.
It is worth noting that therapies that target protein aggregation are not trivial. For instance, dissolution of fibrils may lead to formation of soluble oligomers, which may have a negative effect if these species are responsible for toxicity. It is therefore crucial to develop compounds that do not generally target protein aggregates but specifically decrease the concentration of the most toxic species. This specific targeting requires a fundamental understanding of the molecular mechanisms responsible for amyloid formation and dissolution, as well as of the relationship between aggregation and toxicity. This is an area where close interactions between academia and medical doctors are crucial to advance the field [90].
The goal of treatment is to reduce/eliminate toxic FLC production in order to achieve organ response and eventually prolong survival [45]. Even among patients achieving haematological response, there is a significant proportion in whom organ damage is irreversible at the time of diagnosis or will continue to deteriorate owing to residual production of toxic immunoglobulins [91], sometimes even undetected by standard methods. For years, the role of solid organ transplant as an option in end-stage organ failure for patients with AL-amyloidosis was questioned, mainly because of the lack of effective plasma cell-directed therapy, complications with the allograft and organ donor shortage [92]. With the introduction of highly effective therapies and improvement in the management of transplant-related complications and supportive care, the perception changed. Recently published data show that outcome of organ transplantation in AL-amyloidosis in patients with predominantly single organ involvement is good (5-year survival with kidney transplants 67–86% [82, 93], heart transplants 60–77% [94–98]). The main limitation is the retrospective nature of the data. Efforts should be made to establish specific selection criteria for solid organ transplantation, which should be guided by a multidisciplinary team in specialised centres.
There have been recent major improvements in the management of AL-amyloidosis with improved outcome. Despite some recent disappointments and setbacks in the field of amyloid-targeting therapies, mortality has decreased over time and especially new plasma cell-directed therapies allow long-term control of the disease with good quality of life. We still face the challenge of early diagnosis and allowing access to the more effective treatment options. Expert groups such as the SAN might help in increasing awareness of the disease and guidance in tailored treatment for healthcare providers not as familiar with the disease. Furthermore, the SAN should take the role of providing an interface between academia, pharmaceutical companies, regulatory agencies and health insurance to optimise patient treatment with respect to financial resources of the Swiss healthcare system.
The first meeting of the Swiss Amyloidosis Network was supported by Alnylam, Janssen and Pfizer. The funders did not participate at the meeting, and had no role in the decision to publish or preparation of the manuscript.
RS: fees from Alnylam and Pfizer and an unrestricted research grant from Pfizer relevant for this article as well as Janssen, Mundipharma and Takeda unrelated to this article. AJF: fees from Alnylam and Pfizer relevant for this article, as well as fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Fresenius, Imedos Systems, Mundipharma, Novartis, Orion Pharma, Roche, Schwabe Pharma, Vifor, and Zoll, unrelated to this article. HHJ fees from Alnylam related to this article as well as Biogen, Sanofi-Aventis, Ipsen, Alexion, Mitsubishi, Allergan and CSL Behring unrelated to this article. NL: fees from Alnylam and Pfizer, relevant for this article. PM: fees from Pfizer not relevant for this article. MT: fees from Alnylam relevant for this article, research grant from Pfizer not related to this article, fees from Biogenldec, Sanofi, Merck, Roche not related to this article. AS: fees from Takeda, Janssen, Celgene, Sanofi, Amgen and Novartis unrelated to this article. SFS: fees from Alnylam and Pfizer relevant for this article, as well as fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Fumedica, and Novartis unrelated to this article. BG. fees from Alnylam, Pfizer, Janssen and an unrestricted research grant from Pfizer relevant for this article, funding for accredited continuing medical education from Alnylam, Axonlab, Bayer, Bristol Myers Squibb, Celgene, Daiichi-Sankyo, Janssen, Mitsubishi Tanabe Pharma, NovoNordisk, Octapharma, Takeda, Sanofi, SOBI; and non-financial support from Axonlab and ThermoFisher not related to this article. APP: fees from GE Healthcare and Bayer unrelated to this article. GS: fees from Alnylam related to this article. OP: fees from Pfizer relevant for this article, as well as fees from AstraZeneca, Bayer, Novartis, Vifor, Boehringer Ingelheim, MSD unrelated to this article.
1 Vaxman I , Dispenzieri A , Muchtar E , Gertz M . New developments in diagnosis, risk assessment and management in systemic amyloidosis. Blood Rev. 2020;40:100636. doi:.https://doi.org/10.1016/j.blre.2019.100636
2 Quock TP , Yan T , Chang E , Guthrie S , Broder MS . Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046–53. doi:.https://doi.org/10.1182/bloodadvances.2018016402
3 Kyle RA , Larson DR , Kurtin PJ , Kumar S , Cerhan JR , Therneau TM , et al. Incidence of AL Amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin Proc. 2019;94(3):465–71. doi:.https://doi.org/10.1016/j.mayocp.2018.08.041
4 Palladini G , Perfetti V , Merlini G . Therapy and management of systemic AL (primary) amyloidosis. Swiss Med Wkly. 2006;136(45-46):715–20.
5 Bochtler T , Hegenbart U , Heiss C , Benner A , Moos M , Seckinger A , et al. Hyperdiploidy is less frequent in AL amyloidosis compared with monoclonal gammopathy of undetermined significance and inversely associated with translocation t(11;14). Blood. 2011;117(14):3809–15. doi:.https://doi.org/10.1182/blood-2010-02-268987
6 Diomede L , Rognoni P , Lavatelli F , Romeo M , di Fonzo A , Foray C , et al. Investigating heart-specific toxicity of amyloidogenic immunoglobulin light chains: A lesson from C. elegans. Worm. 2014;3(3):e965590. doi:.https://doi.org/10.4161/21624046.2014.965590
7 Chee CE , Lacy MQ , Dogan A , Zeldenrust SR , Gertz MA . Pitfalls in the diagnosis of primary amyloidosis. Clin Lymphoma Myeloma Leuk. 2010;10(3):177–80. doi:.https://doi.org/10.3816/CLML.2010.n.027
8 Weiss BM , Hebreo J , Cordaro DV , Roschewski MJ , Baker TP , Abbott KC , et al. Increased serum free light chains precede the presentation of immunoglobulin light chain amyloidosis. J Clin Oncol. 2014;32(25):2699–704. doi:.https://doi.org/10.1200/JCO.2013.50.0892
9 Merlini G , Palladini G . Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology (Am Soc Hematol Educ Program). 2012;2012(1):595–603. doi:.https://doi.org/10.1182/asheducation.V2012.1.595.3798563
10 Kyle RA , Durie BGM , Rajkumar SV , Landgren O , Blade J , Merlini G , et al.; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–7. doi:.https://doi.org/10.1038/leu.2010.60
11 Palladini G , Campana C , Klersy C , Balduini A , Vadacca G , Perfetti V , et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107(19):2440–5. doi:.https://doi.org/10.1161/01.CIR.0000068314.02595.B2
12 Siragusa S , Morice W , Gertz MA , Kyle R , Greipp PR , Lust JA , et al. Asymptomatic Amyloidosis at the Time of Diagnostic Bone Marrow Biopsy in Newly Diagnosed Patients with Multiple Myeloma and Smoldering Multiple Myeloma. Blood. 2009;114(22):2803. doi:.https://doi.org/10.1182/blood.V114.22.2803.2803
13 Kyle RA , Gertz MA . Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59.
14 Dispenzieri A , Gertz MA , Buadi F . What do I need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev. 2012;26(4):137–54. doi:.https://doi.org/10.1016/j.blre.2012.03.001
15 Gertz MA , Comenzo R , Falk RH , Fermand JP , Hazenberg BP , Hawkins PN , et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79(4):319–28. doi:.https://doi.org/10.1002/ajh.20381
16 Westermark P . Localized AL amyloidosis: a suicidal neoplasm? Ups J Med Sci. 2012;117(2):244–50. doi:.https://doi.org/10.3109/03009734.2012.654861
17 Hamidi Asl K , Liepnieks JJ , Nakamura M , Benson MD . Organ-specific (localized) synthesis of Ig light chain amyloid. J Immunol. 1999;162(9):5556–60.
18 Benson MD , Buxbaum JN , Eisenberg DS , Merlini G , Saraiva MJM , Sekijima Y , et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–9. doi:.https://doi.org/10.1080/13506129.2018.1549825
19 Garcia Y , Collins AB , Stone JR . Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Hum Pathol. 2018;72:71–9. doi:.https://doi.org/10.1016/j.humpath.2017.11.001
20 Lecadet A , Bachmeyer C , Buob D , Cez A , Georgin-Lavialle S . Minor salivary gland biopsy is more effective than normal appearing skin biopsy for amyloid detection in systemic amyloidosis: A prospective monocentric study. Eur J Intern Med. 2018;57:e20–1. doi:.https://doi.org/10.1016/j.ejim.2018.07.026
21 Choufani EB , Sanchorawala V , Ernst T , Quillen K , Skinner M , Wright DG , et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: incidence, bleeding manifestations, and response to high-dose chemotherapy. Blood. 2001;97(6):1885–7. doi:.https://doi.org/10.1182/blood.V97.6.1885
22 Thompson CA , Kyle R , Gertz M , Heit J , Pruthi R , Pardanani A . Systemic AL amyloidosis with acquired factor X deficiency: A study of perioperative bleeding risk and treatment outcomes in 60 patients. Am J Hematol. 2010;85(3):171–3. doi:.https://doi.org/10.1002/ajh.21603
23 Kyle RA , Therneau TM , Rajkumar SV , Larson DR , Plevak MF , Offord JR , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–9. doi:.https://doi.org/10.1056/NEJMoa054494
24 Phull P , Sanchorawala V , Connors LH , Doros G , Ruberg FL , Berk JL , et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018;25(1):62–7. doi:.https://doi.org/10.1080/13506129.2018.1436048
25 Treglia G , Glaudemans AWJM , Bertagna F , Hazenberg BPC , Erba PA , Giubbini R , et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging. 2018;45(11):1945–55. doi:.https://doi.org/10.1007/s00259-018-4013-4
26 Gillmore JD , Maurer MS , Falk RH , Merlini G , Damy T , Dispenzieri A , et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12. doi:.https://doi.org/10.1161/CIRCULATIONAHA.116.021612
27 Menter T , Bachmann M , Grieshaber S , Tzankov A . A more accurate approach to amyloid detection and subtyping: Combining in situ congo red staining and immunohistochemistry. Pathobiology. 2017;84(1):49–55. doi:.https://doi.org/10.1159/000447304
28 Barrett CD , Dobos K , Liedtke M , Tuzovic M , Haddad F , Kobayashi Y , et al. A Changing Landscape of Mortality for Systemic Light Chain Amyloidosis. JACC Heart Fail. 2019;7(11):958–66. doi:.https://doi.org/10.1016/j.jchf.2019.07.007
29 Wechalekar AD , Gillmore JD , Hawkins PN . Systemic amyloidosis. Lancet. 2016;387(10038):2641–54. doi:.https://doi.org/10.1016/S0140-6736(15)01274-X
30 Muchtar E , Gertz MA , Kumar SK , Lacy MQ , Dingli D , Buadi FK , et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017;129(15):2111–9. doi:.https://doi.org/10.1182/blood-2016-11-751628
31 Kumar S , Dispenzieri A , Lacy MQ , Hayman SR , Buadi FK , Colby C , et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–95. doi:.https://doi.org/10.1200/JCO.2011.38.5724
32 Wechalekar AD , Schonland SO , Kastritis E , Gillmore JD , Dimopoulos MA , Lane T , et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121(17):3420–7. doi:.https://doi.org/10.1182/blood-2012-12-473066
33 Palladini G , Hegenbart U , Milani P , Kimmich C , Foli A , Ho AD , et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. 2014;124(15):2325–32. doi:.https://doi.org/10.1182/blood-2014-04-570010
34 Fontana M , Pica S , Reant P , Abdel-Gadir A , Treibel TA , Banypersad SM , et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132(16):1570–9. doi:.https://doi.org/10.1161/CIRCULATIONAHA.115.016567
35 Banypersad SM , Fontana M , Maestrini V , Sado DM , Captur G , Petrie A , et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J. 2015;36(4):244–51. doi:.https://doi.org/10.1093/eurheartj/ehu444
36 Kotecha T , Martinez-Naharro A , Treibel TA , Francis R , Nordin S , Abdel-Gadir A , et al. Myocardial Edema and Prognosis in Amyloidosis. J Am Coll Cardiol. 2018;71(25):2919–31. doi:.https://doi.org/10.1016/j.jacc.2018.03.536
37 Feng J , Zhang C , Shen K , Sun J , Fang Q , Zhang L , et al. Outcome of cardiac light-chain amyloidosis in the era of novel therapy: A single-center cohort study of 227 patients. Circ J. 2019;83(4):775–82. doi:.https://doi.org/10.1253/circj.CJ-18-1048
38 Kourelis TV , Kumar SK , Gertz MA , Lacy MQ , Buadi FK , Hayman SR , et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013;31(34):4319–24. doi:.https://doi.org/10.1200/JCO.2013.50.8499
39 Wong SW , Palladini G , Hegenbart U , Landau H , Warner M , Jaccard A , et al. The Finding of Del 17p in Marrow Plasma Cells from Patients with Light-Chain Amyloidosis (AL) May Confer a Worse Prognosis. Blood. 2015;126(23):3049. doi:.https://doi.org/10.1182/blood.V126.23.3049.3049
40 Sidana S , Tandon N , Dispenzieri A , Gertz MA , Dingli D , Jevremovic D , et al. Prognostic significance of circulating plasma cells by multi-parametric flow cytometry in light chain amyloidosis. Leukemia. 2018;32(6):1421–6. doi:.https://doi.org/10.1038/s41375-018-0063-7
41 He H , Liu J , Jiang H , Du J , Li L , Lu J , et al. High serum lactate dehydrogenase adds prognostic value to cardiac biomarker staging system for light chain amyloidosis. J Cancer. 2019;10(23):5622–7. doi:.https://doi.org/10.7150/jca.30345
42 Sanchorawala V , Sun F , Quillen K , Sloan JM , Berk JL , Seldin DC . Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood. 2015;126(20):2345–7. doi:.https://doi.org/10.1182/blood-2015-08-662726
43 Rosengren S , Mellqvist UH , Nahi H , Forsberg K , Lenhoff S , Strömberg O , et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation in Sweden, long-term results from all patients treated in 1994-2009. Bone Marrow Transplant. 2016;51(12):1569–72. doi:.https://doi.org/10.1038/bmt.2016.249
44 Dittrich T , Bochtler T , Kimmich C , Becker N , Jauch A , Goldschmidt H , et al. AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis. Blood. 2017;130(5):632–42. doi:.https://doi.org/10.1182/blood-2017-02-767475
45 Muchtar E , Dispenzieri A , Leung N , Lacy MQ , Buadi FK , Dingli D , et al. Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia. 2018;32(10):2240–9. doi:.https://doi.org/10.1038/s41375-018-0060-x
46 Hayman SR , Bailey RJ , Jalal SM , Ahmann GJ , Dispenzieri A , Gertz MA , et al. Translocations involving the immunoglobulin heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood. 2001;98(7):2266–8. doi:.https://doi.org/10.1182/blood.V98.7.2266
47 Muchtar E , Dispenzieri A , Kumar SK , Ketterling RP , Dingli D , Lacy MQ , et al. Interphase fluorescence in situ hybridization in untreated AL amyloidosis has an independent prognostic impact by abnormality type and treatment category. Leukemia. 2017;31(7):1562–9. doi:.https://doi.org/10.1038/leu.2016.369
48 Bochtler T , Hegenbart U , Kunz C , Benner A , Kimmich C , Seckinger A , et al. Prognostic impact of cytogenetic aberrations in AL amyloidosis patients after high-dose melphalan: a long-term follow-up study. Blood. 2016;128(4):594–602. doi:.https://doi.org/10.1182/blood-2015-10-676361
49 Bochtler T , Hegenbart U , Kunz C , Benner A , Seckinger A , Dietrich S , et al. Gain of chromosome 1q21 is an independent adverse prognostic factor in light chain amyloidosis patients treated with melphalan/dexamethasone. Amyloid. 2014;21(1):9–17. doi:.https://doi.org/10.3109/13506129.2013.854766
50 Palladini G , Dispenzieri A , Gertz MA , Kumar S , Wechalekar A , Hawkins PN , et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541–9. doi:.https://doi.org/10.1200/JCO.2011.37.7614
51 Muchtar E , Dispenzieri A , Leung N , Lacy MQ , Buadi FK , Dingli D , et al. Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia. 2018;32(10):2240–9. doi:.https://doi.org/10.1038/s41375-018-0060-x
52 Jaccard A , Moreau P , Leblond V , Leleu X , Benboubker L , Hermine O , et al.; Myélome Autogreffe (MAG) and Intergroupe Francophone du Myélome (IFM) Intergroup. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083–93. doi:.https://doi.org/10.1056/NEJMoa070484
53 Sidiqi MH , Aljama MA , Buadi FK , Warsame RM , Lacy MQ , Dispenzieri A , et al. Stem Cell Transplantation for Light Chain Amyloidosis: Decreased Early Mortality Over Time. J Clin Oncol. 2018;36(13):1323–9. doi:.https://doi.org/10.1200/JCO.2017.76.9554
54 Sidiqi MH , Aljama MA , Buadi FK , Warsame RM , Lacy MQ , Dispenzieri A , et al. Stem Cell Transplantation for Light Chain Amyloidosis: Decreased Early Mortality Over Time. J Clin Oncol. 2018;36(13):1323–9. doi:.https://doi.org/10.1200/JCO.2017.76.9554
55 Gertz M , Lacy M , Dispenzieri A , Hayman S , Kumar S , Buadi F , et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk Lymphoma. 2008;49(1):36–41. doi:.https://doi.org/10.1080/10428190701684518
56 Afrough A , Saliba RM , Hamdi A , Honhar M , Varma A , Cornelison AM , et al. Impact of Induction Therapy on the Outcome of Immunoglobulin Light Chain Amyloidosis after Autologous Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2018;24(11):2197–203. doi:.https://doi.org/10.1016/j.bbmt.2018.07.010
57 Sanchorawala V , Skinner M , Quillen K , Finn KT , Doros G , Seldin DC . Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood. 2007;110(10):3561–3. doi:.https://doi.org/10.1182/blood-2007-07-099481
58 Kastritis E , Anagnostopoulos A , Roussou M , Toumanidis S , Pamboukas C , Migkou M , et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92(10):1351–8. doi:.https://doi.org/10.3324/haematol.11325
59 Kastritis E , Leleu X , Arnulf B , Zamagni E , Cibeira MT , Kwok F , et al. A Randomized Phase III Trial of Melphalan and Dexamethasone (MDex) Versus Bortezomib, Melphalan and Dexamethasone (BMDex) for Untreated Patients with AL Amyloidosis. Blood. 2016;128(22):646. doi:.https://doi.org/10.1182/blood.V128.22.646.646
60 Bochtler T , Hegenbart U , Kunz C , Granzow M , Benner A , Seckinger A , et al. Translocation t(11;14) is associated with adverse outcome in patients with newly diagnosed AL amyloidosis when treated with bortezomib-based regimens. J Clin Oncol. 2015;33(12):1371–8. doi:.https://doi.org/10.1200/JCO.2014.57.4947
61 Bochtler T , Hegenbart U , Kunz C , Benner A , Seckinger A , Dietrich S , et al. Gain of chromosome 1q21 is an independent adverse prognostic factor in light chain amyloidosis patients treated with melphalan/dexamethasone. Amyloid. 2014;21(1):9–17. doi:.https://doi.org/10.3109/13506129.2013.854766
62 Cohen AD , Landau H , Scott EC , Liedtke M , Kaufman JL , Rosenzweig M , et al. Safety and Efficacy of Carfilzomib (CFZ) in Previously-Treated Systemic Light-Chain (AL) Amyloidosis. Blood. 2016;128(22):645. doi:.https://doi.org/10.1182/blood.V128.22.645.645
63 Kastritis E , Terpos E , Roussou M , Gavriatopoulou M , Pamboukas C , Boletis I , et al. A phase 1/2 study of lenalidomide with low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL amyloidosis. Blood. 2012;119(23):5384–90. doi:.https://doi.org/10.1182/blood-2011-12-396903
64 Dispenzieri A , Kastritis E , Wechalekar AD , Schönland SO , Kim K , Sanchorawala V , et al. Primary Results from the Phase 3 Tourmaline-AL1 Trial of Ixazomib-Dexamethasone Versus Physician’s Choice of Therapy in Patients (Pts) with Relapsed/Refractory Primary Systemic AL Amyloidosis (RRAL). Blood. 2019;134(Supplement_1):139. doi:.https://doi.org/10.1182/blood-2019-124409
65 Dispenzieri A , Lacy MQ , Zeldenrust SR , Hayman SR , Kumar SK , Geyer SM , et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109(2):465–70. doi:.https://doi.org/10.1182/blood-2006-07-032987
66 Palladini G , Milani P , Foli A , Obici L , Lavatelli F , Nuvolone M , et al. Oral melphalan and dexamethasone grants extended survival with minimal toxicity in AL amyloidosis: long-term results of a risk-adapted approach. Haematologica. 2014;99(4):743–50. doi:.https://doi.org/10.3324/haematol.2013.095463
67 Kimmich CR , Terzer T , Benner A , Dittrich T , Veelken K , Carpinteiro A , et al. Daratumumab for systemic AL amyloidosis: prognostic factors and adverse outcome with nephrotic-range albuminuria. Blood. 2020;135(18):1517–30. doi:.https://doi.org/10.1182/blood.2019003633
68 Roussel M , Merlini G , Chevret S , Arnulf B , Stoppa AM , Perrot A , et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood. 2020;135(18):1531–40. doi:.https://doi.org/10.1182/blood.2019004369
69 Sanchorawala V , Sarosiek S , Schulman A , Mistark M , Migre ME , Cruz R , et al. Safety, tolerability, and response rates of daratumumab in relapsed AL amyloidosis: results of a phase 2 study. Blood. 2020;135(18):1541–7. doi:.https://doi.org/10.1182/blood.2019004436
70 Schwotzer R , Manz MG , Pederiva S , Waibel C , Caspar C , Lerch E , et al. Daratumumab for relapsed or refractory AL amyloidosis with high plasma cell burden. Hematol Oncol. 2019;37(5):595–600. doi:.https://doi.org/10.1002/hon.2677
71 Roussel M , Merlini G , Chevret S , Arnulf B , Stoppa AM , Perrot A , et al. A prospective phase 2 study of daratumumab in previously-treated systemic light-chain (AL) amyloidosis. Blood. 2020;135(18):1531–40. doi:.https://doi.org/10.1182/blood.2019004369
72 Palladini G , Kastritis E , Maurer MS , Zonder J , Minnema MC , Wechalekar AD , et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71–80. doi:.https://doi.org/10.1182/blood.2019004460
73 Milani P , Schönland S , Merlini G , Kimmich C , Foli A , Dittrich T , et al. Treatment of AL amyloidosis with bendamustine: a study of 122 patients. Blood. 2018;132(18):1988–91. doi:.https://doi.org/10.1182/blood-2018-04-845396
74 Sanchorawala V , Shelton AC , Lo S , Varga C , Sloan JM , Seldin DC . Pomalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 1 and 2 trial. Blood. 2016;128(8):1059–62. doi:.https://doi.org/10.1182/blood-2016-04-710822
75 Palladini G , Merlini G . Current treatment of AL amyloidosis. Haematologica. 2009;94(8):1044–8. doi:.https://doi.org/10.3324/haematol.2009.008912
76 Leung N , Thomé SD , Dispenzieri A . Venetoclax induced a complete response in a patient with immunoglobulin light chain amyloidosis plateaued on cyclophosphamide, bortezomib and dexamethasone. Haematologica. 2018;103(3):e135–7. doi:.https://doi.org/10.3324/haematol.2017.183749
77 Ghilardi G , Stussi G , Mazzucchelli L , Röcken C , Rossi D , Gerber B . Venetoclax plus daratumumab induce hematological CR and organ response in an AL amyloidosis patient with t(11;14). Amyloid. 2019;26(3):173–4. doi:.https://doi.org/10.1080/13506129.2019.1615428
78 Le Bras F , Dupuis J , Lemonnier F , Oghina S , Bodez D , Ladaique A , et al. Venetoclax induces sustained complete responses in refractory/relapsed patients with cardiac AL amyloidosis. J Clin Oncol. 2019;37(15_suppl):e19538. doi:.https://doi.org/10.1200/JCO.2019.37.15_suppl.e19538
79 Feng D , Syed IS , Martinez M , Oh JK , Jaffe AS , Grogan M , et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119(18):2490–7. doi:.https://doi.org/10.1161/CIRCULATIONAHA.108.785014
80 Rezk T , Whelan CJ , Lachmann HJ , Fontana M , Sachchithanantham S , Mahmood S , et al. Role of implantable intracardiac defibrillators in patients with cardiac immunoglobulin light chain amyloidosis. Br J Haematol. 2018;182(1):145–8. doi:.https://doi.org/10.1111/bjh.14747
81 Grogan M , Dispenzieri A . Natural history and therapy of AL cardiac amyloidosis. Heart Fail Rev. 2015;20(2):155–62. doi:.https://doi.org/10.1007/s10741-014-9464-5
82 Kumar SK , Dispenzieri A , Lacy MQ , Hayman SR , Buadi FK , Dingli D , et al. Doxycycline Used As Post Transplant Antibacterial Prophylaxis Improves Survival in Patients with Light Chain Amyloidosis Undergoing Autologous Stem Cell Transplantation. Blood. 2012;120(21):3138. doi:.https://doi.org/10.1182/blood.V120.21.3138.3138
83 Hawkins PN , Ando Y , Dispenzeri A , Gonzalez-Duarte A , Adams D , Suhr OB . Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–38. doi:.https://doi.org/10.3109/07853890.2015.1068949
84 Gertz MA , Landau H , Comenzo RL , Seldin D , Weiss B , Zonder J , et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol. 2016;34(10):1097–103. doi:.https://doi.org/10.1200/JCO.2015.63.6530
85Prothena. Prothena Discontinues Development of NEOD001 for AL Amyloidosis. Press release 23 April 2018.
86 Varga C , Lentzsch S , Comenzo RL . Beyond NEOD001 for systemic light-chain amyloidosis. Blood. 2018;132(18):1992–3. doi:.https://doi.org/10.1182/blood-2018-07-865857
87 Richards DB , Cookson LM , Berges AC , Barton SV , Lane T , Ritter JM , et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med. 2015;373(12):1106–14. doi:.https://doi.org/10.1056/NEJMoa1504942
88Walmsley E. Press release Third quarter 2018. 2020;(October 2018):1–62.
89Caelum Biosciences. Alexion and Caelum Biosciences Announce Collaboration to Develop Targeted Therapy for Light Chain (AL) amyloidosis. Press release 31 January 2019.
90 Arosio P , Vendruscolo M , Dobson CM , Knowles TPJ . Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol Sci. 2014;35(3):127–35. doi:.https://doi.org/10.1016/j.tips.2013.12.005
91 Palladini G , Massa M , Basset M , Russo F , Milani P , Foli A , et al. Persistence of Minimal Residual Disease By Multiparameter Flow Cytometry Can Hinder Recovery of Organ Damage in Patients with AL Amyloidosis Otherwise in Complete Response. Blood. 2016;128(22):3261. doi:.https://doi.org/10.1182/blood.V128.22.3261.3261
92 Theodorakakou F , Fotiou D , Dimopoulos MA , Kastritis E . Solid organ transplantation in amyloidosis. Acta Haematol. 2020;143(4):352–64. doi:.https://doi.org/10.1159/000508262
93 Angel-Korman A , Stern L , Sarosiek S , Sloan JM , Doros G , Sanchorawala V , et al. Long-term outcome of kidney transplantation in AL amyloidosis. Kidney Int. 2019;95(2):405–11. doi:.https://doi.org/10.1016/j.kint.2018.09.021
94 Kristen AV , Kreusser MM , Blum P , Schönland SO , Frankenstein L , Dösch AO , et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. J Heart Lung Transplant. 2018;37(5):611–8. doi:.https://doi.org/10.1016/j.healun.2017.11.015
95 Grogan M , Gertz M , McCurdy A , Roeker L , Kyle R , Kushwaha S , et al. Long term outcomes of cardiac transplant for immunoglobulin light chain amyloidosis: The Mayo Clinic experience. World J Transplant. 2016;6(2):380–8. doi:.https://doi.org/10.5500/wjt.v6.i2.380
96 Dubrey SW , Burke MM , Hawkins PN , Banner NR . Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23(10):1142–53. doi:.https://doi.org/10.1016/j.healun.2003.08.027
97 Lacy MQ , Dispenzieri A , Hayman SR , Kumar S , Kyle RA , Rajkumar SV , et al. Autologous stem cell transplant after heart transplant for light chain (Al) amyloid cardiomyopathy. J Heart Lung Transplant. 2008;27(8):823–9. doi:.https://doi.org/10.1016/j.healun.2008.05.016
98 Gillmore JD , Goodman HJ , Lachmann HJ , Offer M , Wechalekar AD , Joshi J , et al. Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood. 2006;107(3):1227–9. doi:.https://doi.org/10.1182/blood-2005-08-3253
The first meeting of the Swiss Amyloidosis Network was supported by Alnylam, Janssen and Pfizer. The funders did not participate at the meeting, and had no role in the decision to publish or preparation of the manuscript.
RS: fees from Alnylam and Pfizer and an unrestricted research grant from Pfizer relevant for this article as well as Janssen, Mundipharma and Takeda unrelated to this article. AJF: fees from Alnylam and Pfizer relevant for this article, as well as fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Fresenius, Imedos Systems, Mundipharma, Novartis, Orion Pharma, Roche, Schwabe Pharma, Vifor, and Zoll, unrelated to this article. HHJ fees from Alnylam related to this article as well as Biogen, Sanofi-Aventis, Ipsen, Alexion, Mitsubishi, Allergan and CSL Behring unrelated to this article. NL: fees from Alnylam and Pfizer, relevant for this article. PM: fees from Pfizer not relevant for this article. MT: fees from Alnylam relevant for this article, research grant from Pfizer not related to this article, fees from Biogenldec, Sanofi, Merck, Roche not related to this article. AS: fees from Takeda, Janssen, Celgene, Sanofi, Amgen and Novartis unrelated to this article. SFS: fees from Alnylam and Pfizer relevant for this article, as well as fees from Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Fumedica, and Novartis unrelated to this article. BG. fees from Alnylam, Pfizer, Janssen and an unrestricted research grant from Pfizer relevant for this article, funding for accredited continuing medical education from Alnylam, Axonlab, Bayer, Bristol Myers Squibb, Celgene, Daiichi-Sankyo, Janssen, Mitsubishi Tanabe Pharma, NovoNordisk, Octapharma, Takeda, Sanofi, SOBI; and non-financial support from Axonlab and ThermoFisher not related to this article. APP: fees from GE Healthcare and Bayer unrelated to this article. GS: fees from Alnylam related to this article. OP: fees from Pfizer relevant for this article, as well as fees from AstraZeneca, Bayer, Novartis, Vifor, Boehringer Ingelheim, MSD unrelated to this article.