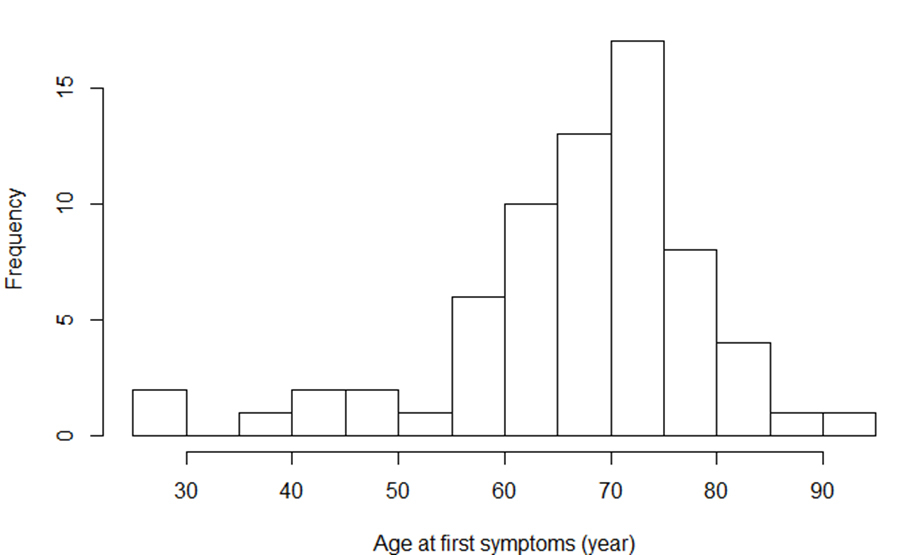
Figure 1 Age distribution at time of first symptoms (years); n = 68 subjects. Median (Q1–Q3): 69.1 (60.8–72.9)
DOI: https://doi.org/10.4414/smw.2020.20258
BACKGROUND: Multidisciplinary management (MDM) of amyotrophic lateral sclerosis (ALS) has been associated with increased survival and improved health-related quality of life (HRQL).
AIMS OF THE STUDY: The aims of this study were (1) to present the specifics and structure of MDM of ALS in our area and the roles of the specialists involved, and (2) to provide the results of MDM concerning aspects such as survival, modalities and timing of ventilator and nutritional support, and HRQL.
METHODS: During a four-year period, systematic data collection on all ALS patients included in our MDM structure was performed at three-month intervals. The data covered neurologic assessment, respiratory function, nutritional status, ENT and phoniatric evaluation, palliative care, HRQL, existing social support, and need for occupational and physical therapy.
RESULTS: 68 patients were included (50% women, 19% with bulbar onset). Mean (SD) age at the time of first symptoms was 66.5 (12.3) years. Median (Q1–Q3) time elapsed from onset of symptoms to diagnosis was 11 (7–18) months. Median survival was 50 (34–95) months from first symptoms and 35 (18–70) months from diagnosis. Riluzole was prescribed in 74% of cases. Noninvasive ventilation was implemented in 28 (41%) and percutaneous gastrostomy in 14 (21%) patients.
CONCLUSIONS: ALS patients and neurologists in private practice adhered to multidisciplinary management of ALS in our area. Implementation of noninvasive ventilation and percutaneous endoscopic gastrostomy could be performed electively in most cases and emergency procedures were seldom required. Decisions on noninvasive ventilation, percutaneous endoscopic gastrostomy, optimal symptom control and advanced care planning were shared among different specialists. The trial was registered at clinicaltrials.gov (trial No NCT03536962).
Keywords: amyotrophic lateral sclerosis; multidisciplinary management; survival; noninvasive ventilation; percutaneous gastrostomy
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease characterised by a loss of motor neurons in the spinal cord, brainstem and motor cortex, leading to progressive muscle weakness, irreversible disability and respiratory failure. The disorder is usually fatal within 2 to 4 years [1–5], although there is a marked heterogeneity in survival among those affected. Incidence is 1–2 cases per 100,000 people/year; prevalence is 3–5 cases per 100,000 people [6–10]. Incidence increases after the age of 40 years, reaching a peak at 70–74 years for men and 65–69 years for women, and declining thereafter. The male:female incidence ratio is 1.25:1 [11].
There is to date no curative treatment for ALS. Riluzole is known to prolong survival by three to six months; a recent review suggests that the impact of riluzole on survival may be more significant [12]. No survival data are available for edaravone, a drug approved by the FDA in 2017 [13].
Observational studies have shown that multidisciplinary management (MDM) of ALS patients is associated with an increased probability of receiving riluzole, improved survival and health-related quality of life (HRQL), and fewer medical complications [14–23]. MDM facilitates access to several specialists within one day and at the same place, providing an integrative approach appreciated by many ALS patients. It also increases the likelihood of noninvasive ventilation (NIV) and reduces the frequency and duration of hospital stays [14, 20, 24]. Weight loss, swallowing problems and speech difficulties may be detected and managed earlier. Occupational therapists and physical therapists help to adapt the patient’s environment to the progression of the disease and provide aids for communication. A palliative team addresses psycho-social and spiritual issues and is important for promoting advanced care planning (ACP) [25–27].
Discrepancies between daily practice and existing recommendations for patients with ALS, particularly regarding artificial nutrition and ventilator support, were noted in Geneva in 2005 [28]. This was one of the reasons that lead to the implementation of an MDM day clinic for ALS at Geneva University Hospitals in 2010.
The goals of this study were (1) to present the specifics and structure of the MDM of ALS in our area and the roles of the specialists involved, and (2) to provide, based on a cohort study, the results of the MDM concerning aspects such as survival, modalities and timing of ventilator and nutritional support, and HRQL.
Between June 2012 and September 2016, an observational cohort study was conducted in our tertiary centre. We systematically and prospectively collected data in the fields of neurology, pulmonology, nutrition, health-related quality of life (HRQL) and ENT (ear, nose and throat)/phoniatric evaluation.
Patients were seen once every three months for a one-day, multidisciplinary clinical evaluation in an outpatient clinic. The specialists involved were a neurologist, a pulmonologist, a medical nutritionist, a dietitian, an ENT specialist, a speech therapist, a palliative care nurse and physician, an occupational therapist, a physical therapist and an ALS nurse. A psychiatrist and/or social worker could intervene on demand. Briefly, after clinical assessment and scoring by the neurologist (ALS-FRS-R), the patients underwent sequential specialised assessments. Table 1 details the tasks and focus of the quarterly monitoring by the specialists involved, derived from EFNS guidelines [15]. Symptomatic treatments for emotional lability, cramps, spasticity and pain were provided. At the end of the MDM day, the neurologist summarised the observations and the therapeutic suggestions.
Table 1 Structure of the multidisciplinary ALS care team and interventions.
| Specialist | Tasks | Tools | Outcome measures and/or endpoint |
|---|---|---|---|
| Neurologist | Diagnosis and treatment Follow-up of clinical and electrophysiological parameters Detection and treatment of spasticity, cramps and pain |
Neurological examination Electromyography and motor evoked potentials at first visit Prescription of riluzole and edaravone |
ALS FRS-R score |
| Pulmonologist | Targeted medical history Detection of sleep-related breathing disorders Follow-up of respiratory and sleep parameters Timing, initiation and monitoring of NIV Implementing of mechanical insufflation/exsufflation and chest therapy when appropriate (see below) |
Pulmonary function assessment (including MIP, SNIP, VC seated and supine, peak cough flow)*
Nocturnal pulsoximetry Daytime arterial (or capillary arterialised) blood gases Epworth Sleepiness Scale (ESS) Transcutaneous capnography (when available) Polygraphy or polysomnography (in specialised centres) |
Correction of daytime ABG and nocturnal hypoxaemia and hypercapnia Improvement of dyspnoea Improvement of sleepiness (ESS score when appropriate) and other symptoms of sleep-related breathing disorders |
| ENT specialist | Evaluation of glottic function and risk of aspiration Treatment of hypersialorrhoea |
Clinical assessment Fiberoptic endoscopic evaluation of swallowing (FEES) Videofluoroscopy Anticholinergic treatment Botulinum toxin injections Radiation therapy of salivary glands |
Control of hypersialorrhoea In coordination with speech therapist, adapting texture and modalities of food and liquid intake to avoid or minimise aspiration |
| Speech therapist | Dysarthria Detection of swallowing problems |
Clinical examination Strategies for avoiding aspiration |
See above Improved intelligibility |
| Palliative care specialist | Symptom control Advance care planning Psycho-social assessment |
Questionnaires (HADS) and symptom scores (ESAS) Adapting treatment for optimal symptom control |
Improved symptom control (ESAS, HADS) Completion of advanced directives and designation of a healthcare surrogate |
| Respiratory and physical therapist | Assessment of mobility Prevention of falls Implementation of devices to assist ambulation Prevention of retractions and spasms Evaluation of cough and airway clearance |
Clinical assessment Passive and active mobilisation Teaching and implementing cough assist devices (mechanical insufflation/exsufflation) and techniques of airway clearing to caregivers |
Improved comfort of breathing and SpO2
Improved airway clearance Decreased muscular and/or joint pain |
| Medical nutritionist and dietitian | Quantification of weight loss and loss of fat-free mass Estimating energy, protein and liquid intake and requirements Follow-up of nutritional state and adaptation of oral or enteral nutritional support Suggestion of PEG when necessary Prescription of oral nutritional supplements or enteral nutrition with or without hydration via gastrostomy |
Measurement of body weight Measurement of body composition (bioelectrical impedance analysis) Estimation of energy, protein and liquid intake: 24 h recall Estimation of energy requirements: resting metabolic rate estimated by the Harris-Benedict formula + 10% for thermogenesis + 10% for hypermetabolism + 10% for physical activity if the patient is mobile Estimation of protein requirements: 1 g/kg/d Estimation of liquid needs: 30 ml/kg/d Assessment of resting metabolic rate by indirect calorimetry at time of PEG placement |
Prevention of weight loss Maintaining optimal balance between intake and requirements Appropriate timing of PEG when required |
| Occupational therapist | In conjunction with physical therapist, implementing devices to assist mobility Communication aids Posture assisting devices |
Adaptation of environment at each stage of disease and anticipation of disease progression Implementing communication aids (i.e. eye-tracking devices, synthetic speech generating devices) |
Preventing institutionalisation Postural comfort in daily life Maintaining ability to communicate with caregivers/family and other social interactions |
| Psychologist and Psychiatrist | Detection of cognitive impairment and mood disturbances Support for caregivers |
Frontal Assessment Battery (FAB) Hospital Anxiety & Depression Scale |
Improvement of mood disturbance (depression, anxiety) |
| Social worker | Organisation of home care services Seeking financial support |
Providing resources for patient and relatives/caregivers through knowledge of local network | Optimising home care and decreasing associated financial burden |
| Specialised nurse (home visits) | Assessment and identification of new problems Support for patients and caregivers |
Clinical evaluation and problem identification at home Active support for caregivers |
Improving patient and caregiver support |
FVC = forced vital capacity; SNIP = sniff nasal inspiratory pressure; MIP = maximal mouth inspiratory pressure; HADS = Hospital Anxiety and Depression Scale; ESAS = Edmonton Symptom Assessment Scale * Details provided in [29]
Time of first symptoms, diagnosis and treatment initiation were recorded. The diagnosis of definite, probable or possible ALS was established by an experienced neurologist according to the revised El Escorial criteria [30]. This neurologist also conducted neurophysiological investigations and, in case of diagnostic confirmation, prescribed riluzole. Patients were classified as “bulbar” or “non-bulbar” according to initial clinical presentation. The revised ALS Functional Rating Scale (ALS-FRS-R) was used to determine the progression of disability [31]. Mood disturbances and HRQL were assessed using the Hospital Anxiety and Depression Scale (HADS) [32, 33] and the Medical Outcomes Study Short Form-36 (SF-36) [34, 35].
Respiratory function: Assessment included measurement of forced expiratory volume in one second (FEV1), forced vital capacity (FVC), seated and supine VC, peak expiratory cough flow, arterialised capillary blood gases measured at the earlobe, inspiratory muscle strength (SNIP: sniff nasal inspiratory pressure; MIP: maximal mouth inspiratory pressure) [29], and nocturnal pulsoximetry [15]. The methodological details and results of these tests have been reported recently [29]. Medical history included a targeted checklist focusing on dyspnoea, orthopnoea, sleepiness (Epworth Sleepiness Scale), glottic dysfunction and recurrent lower-respiratory tract infections.
Initiation of non-invasive ventilation (NIV) was performed electively, either on an outpatient basis or during a short hospital stay, after MDM evaluation. The decision was based on symptoms and functional parameters (VC, SNIP or MIP, ABG, oximetry) [15]. Details are provided in the online supplement.
Mechanical insufflation/exsufflation devices (cough assist), combined with home respiratory therapy, were also implemented for the management of bronchial secretions in the presence of cough dysfunction, low peak cough flow values and poor spontaneous airway clearance [8].
Nutrition: Nutritional assessment included a targeted history, a 24-hour recall of energy, protein and liquid intake encompassing duration of meals, and a measurement of body weight and body composition. Body composition was assessed by bioelectrical impedance analysis (Nutriguard®, Data Input GmbH, Darmstadt, Germany) at 50 kHz [36]. Percutaneous endoscopic gastrostomy (PEG) was systematically suggested when weight loss exceeded 10% of previous body weight, or when glottic dysfunction and/or recurrent aspiration occurred [15, 37]. PEG was performed electively during a short hospital stay.
The ENT specialist [38] and the speech therapist evaluated swallowing by observing the patient during lunch and testing different textures, and provided appropriate recommendations (food texture, thickeners for liquids). The ENT also discussed the management of hypersialorrhoea (anticholinergic drugs, injection of botulinum toxin, radiotherapy of salivary glands) and options such as laryngeal derivation or tracheostomy.
Interventions by the physical therapists and occupational therapists were coordinated with their colleagues intervening at home, and aimed to provide technical support concerning mobility, posture, adapting the patient’s environment and providing communication devices in a timely fashion. The possible options were discussed during the MDM sessions.
Palliative care: A palliative care team (a nurse and a physician) intervened systematically, with a focus on symptom control, psycho-social assessment and advance care planning, including intubation, tracheostomy and palliative sedation. Assisted suicide is legal in Switzerland and was discussed according to the patient’s wishes.
The social worker provided administrative support and financial counselling when needed and optimised the use of the home care network.
The general outlook of our MDM team regarding invasive life support and tracheostomy is detailed in a previous publication [39].
Study data were collected in SecuTrial®, a GCP-compliant electronic data capture system. Statistical analyses were essentially descriptive. Patient characteristics were described as frequencies and percentages for qualitative data and as means (SD) or medians (Q1–Q3) for quantitative data, according to the distribution of the data. Overall survival was estimated according to the Kaplan-Meier method. Cumulative incidences of noninvasive ventilation implementation and percutaneous gastrostomy were estimated using a competing risks analysis. This method allows computation of the cumulative incidence of an event of interest (e.g., implementation of NIV) in the presence of competing risks such as death [40]. All analyses were performed using R software, version 3.5.3 (R Foundation for Statistical Computing, Vienna, Austria; https://www-R-project.org).
Sixty-eight patients were included. All patients provided informed consent. Baseline clinical characteristics are presented in table 2. Age distribution at the onset of symptoms is presented in figure 1. Initial clinical presentation was bulbar in 13 cases (19%), spinal in 27 (41%) and generalised in 28 cases (40%). Four patients (6%) presented with dementia.
Table 2 Baseline characteristics of patients (n = 68) in the ALS cohort at inclusion and major outcomes.
| Age at inclusion, mean ± SD (years) | 68.6 ± 11.9 | |
| Age at first symptoms, mean ± SD (years) | 66.5 ± 12.3 | |
| Gender, female, n (%) | 34 (50) | |
| Time between first symptoms and diagnosis (months), median (Q1–Q3) | 11.1 (6.8–18) | |
| Time between first symptoms and inclusion into study (months), median (Q1–Q3) | 20.4 (12.7–32) | |
| El Escorial Criteria | Clinically definite, n (%) | 31 (46) |
| Clinically probable, n (%) | 15 (22) | |
| Clinically probable – laboratory supported, n (%) | 3 (4) | |
| Clinically possible, n (%) | 12 (18) | |
| Bulbar onset, n (%) | 13 (19) | |
| Dementia, n (%) | 4 (6) | |
| ALS-FRS-R score, median (Q1–Q3) | 39 (32–42) | |
| Treatment | Riluzole, n (%) | 50 (74) |
| Antidepressants, n (%) | 19 (28) | |
| Anticholinergic drugs, n (%) | 3 (4) | |
| Home-dwelling patients at inclusion, n (%) | 64 (98) | |
| Caregivers | Spouse, n (%) | 53 (78) |
| Adult daughter or son, n (%) | 17 (25) | |
| Other family member, n (%) | 9 (13) | |
| None, n (%) | 5 (7) | |
| Patient outcomes | Death during follow-up, n (%) | 30 (44) |
| Non-invasive ventilation, n (%) | 28 (41) | |
| Percutaneous endoscopic gastrostomy, n (%) | 14 (21) | |
| Cause of death | Cardiac and/or respiratory arrest, n (%) | 12 (40) |
| Respiratory failure, n (%) | 7 (23) | |
| Palliative sedation*, n (%) | 6 (20) | |
| Other†, n (%) | 5 (17) | |
* Four of these patients died through assisted suicide; † pneumonia (n = 4), cancer (n = 1)
Figure 1 Age distribution at time of first symptoms (years); n = 68 subjects. Median (Q1–Q3): 69.1 (60.8–72.9)
A mixture of prevalent and incident cases were included in the study. Overall, the median time elapsed between first symptoms and diagnosis was 11 (Q1–Q3: 7–18) months (fig. 2), and the time between diagnosis and inclusion in the cohort was 6 (1–17) months.
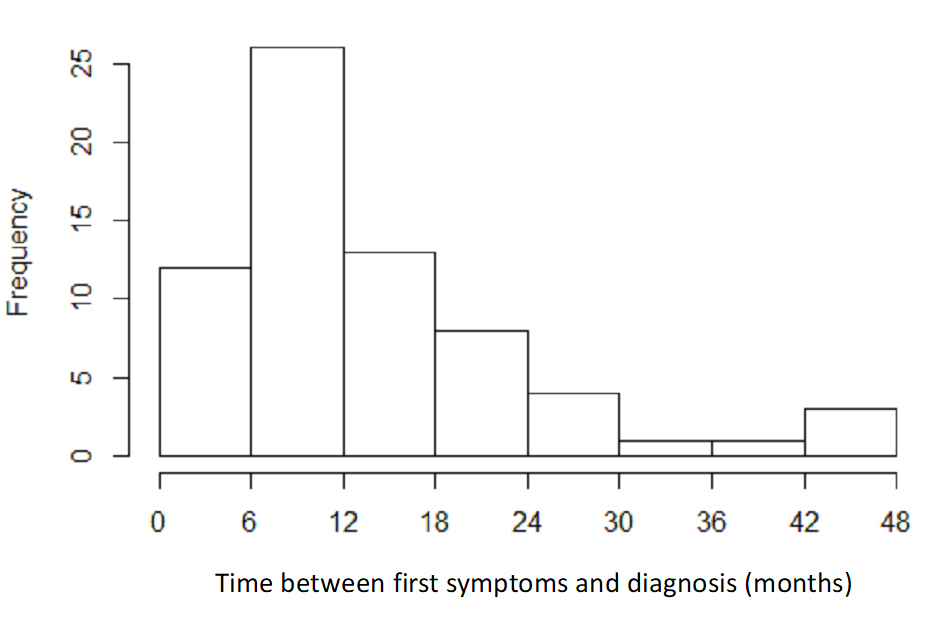
Figure 2 Distribution of delay between first symptoms according to medical history and diagnosis; n = 68 subjects. Median (Q1–Q3) 11.1 (6.8–18) months
Once included in the MDM structure, the median number of visits was 4 (2–6) during a median follow-up of 12 (6–24) months.
The evolution of the ALS-FRS-R scores over time in “bulbar” and “non-bulbar” subjects is detailed in appendix 1.
Survival: Median survival time was 50 (34–95) months from the first symptoms, 35 (18–70) months from diagnosis and 24 (12–31) months from inclusion in the cohort (fig. 3). Detailed survival probabilities are provided in table 3.
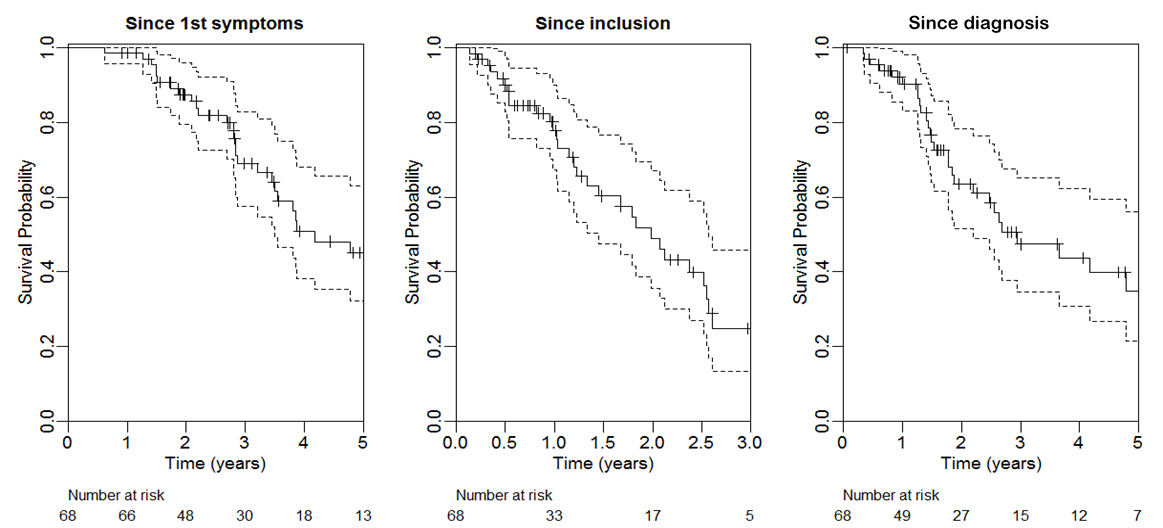
Figure 3 Overall survival from first ALS symptoms, inclusion and diagnosis (Kaplan Meier curves with 95% CI); n = 68 subjects. Median values provided in results section.
Table 3 Overall survival among patients from first symptoms and from diagnosis of ALS; n = 68.
| 1 year | 2 years | 3 years | 4 years | 5 years | |
|---|---|---|---|---|---|
| Overall survival probability (95% CI) since first symptoms | 0.99 (0.96–1.00) |
0.87 (0.80–0.96) |
0.69 (0.57–0.83) |
0.51 (0.38–0.68) |
0.45 (0.32–0.63) |
| Overall survival probability (95% CI) since diagnosis | 0.9 (0.83–0.98) |
0.64 (0.52–0.78) |
0.47 (0.35–0.65) |
0.44 (0.31–0.62) |
0.35 (0.22–0.56) |
| Bulbar (n = 13) | 1 year | 2 years | 3 years | 4 years | 5 years |
| Overall survival probability (95% CI) since first symptoms | 1.00 | 0.91 (0.75–1.00) |
0.45 (0.20–1.00) |
0 | 0 |
| Overall survival probability (95% CI) since diagnosis | 1.00 | 0.39 (0.17–0.94) |
0 | 0 | 0 |
CI = confidence interval
Time to NIV and PEG: Twenty-eight (41%) patients were started on non-invasive ventilation (NIV) during their follow-up, according to previously mentioned criteria. Figure 4 shows cumulative incidence curves for time spent without NIV. From the appearance of first symptoms, cumulative incidence of NIV was 1% (95% CI 0–4%) after one year, 22% (11–33%) after two years and 51% (36–66%) after four years. From diagnosis, cumulative incidence of NIV was 29% (95% CI 17–41%) after one year, 41% (28–54%) after two years and 54% (36–70%) after four years.
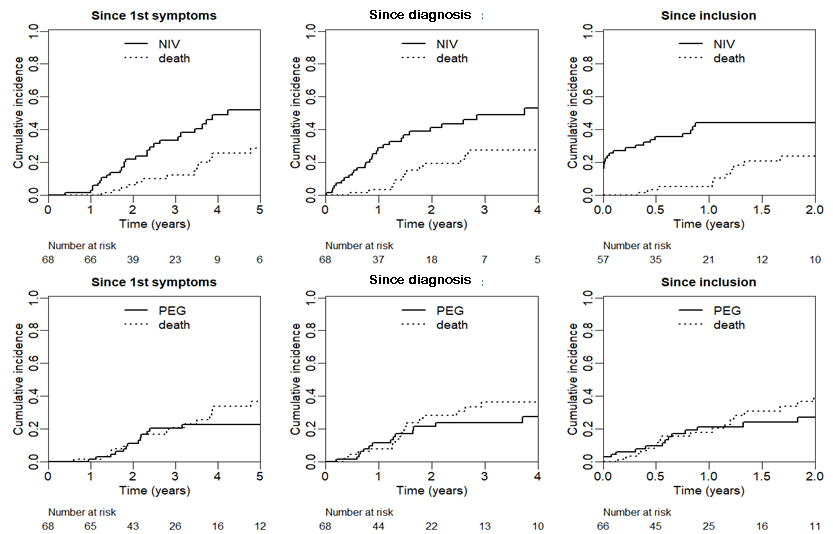
Figure 4 Cumulative incidence curves for time spent without NIV (top) and without PEG (bottom). T0: time of first symptoms (left), time of diagnosis (middle), and time of inclusion in cohort study (right).
Modalities of NIV, settings, and data recorded by the ventilators in 27 patients are provided in the online supplement (data missing for one case). Most implementations of NIV were elective (89%), with only three cases (11%) initiated after admission for acute respiratory failure [28]. No patient was ventilated invasively. One patient refused NIV. One patient had a surgical laryngeal diversion with a tracheotracheal speech fistula because of major secretions not responding to usual care.
Fourteen patients (21%) had a PEG inserted during follow-up, of whom six had a PEG inserted while already under NIV. Seven patients refused the procedure. Cumulative incidence for PEG insertion from appearance of first symptoms was 1% (95% CI 0–4%) after one year and 23% (11–34%) after four years. From diagnosis, cumulative incidence of PEG was 11% (3–20%) after one year and 24% (12–36%) after three years. All procedures were elective and were performed under general anaesthesia. Patients usually stayed under observation for 2–3 days after the procedure. There were no complications of the PEG procedures.
HRQL and mood disorders at inclusion: HADS scores are provided in table 4. Missing values were related to communication impairment. Nineteen percent had abnormal scores for HADS-A (anxiety) and 16% had abnormal scores for HADS-D (depression). Overall, 41 (60%) patients completed the whole SF-36 questionnaire at inclusion. The mean scores for all SF-36 dimensions are provided in figure 5. When compared to reference values for our area [41] scores for “general health”, “vitality”, “physical functioning” and “role-physical” were the most severely impaired.
Table 4 Hospital Anxiety and Depression Scale (HADS) scores at inclusion (n = 43).
| Measure | n = 43 |
|---|---|
| HADS-Depression scores, median (Q1–Q3) | 5 (3–8) (range 0 to 15) |
| Normal (<8), n (%) | 30 (70) |
| Borderline (8–10), n (%) | 6 (14) |
| Abnormal (>10), n (%) | 7 (16) |
| HADS-Anxiety scores, median (Q1–Q3) | 6 (4–8) (range 0 to 16) |
| Normal (<8), n (%) | 28 (65) |
| Borderline (8–10), n (%) | 7 (16) |
| Abnormal (>10), n (%) | 8 (19) |
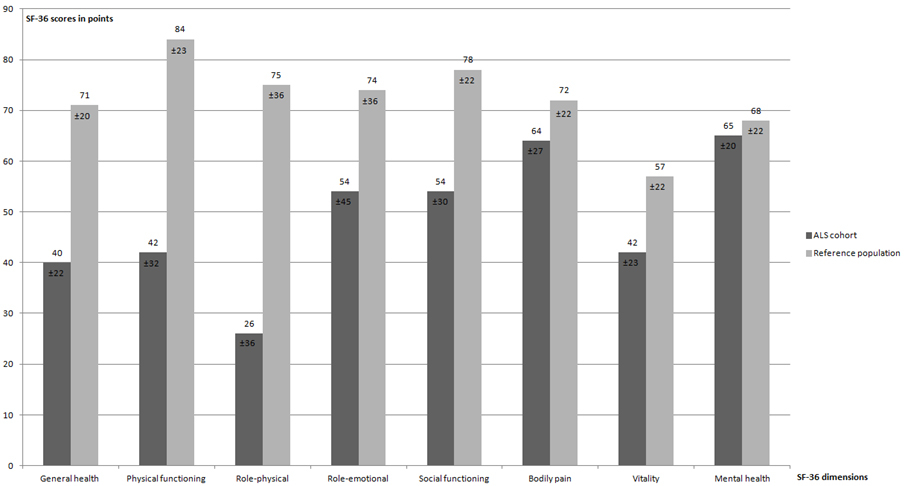
Figure 5 Comparison of SF-36 values (mean ± SD) in our ALS cohort at inclusion with a reference population of healthy individuals in French-speaking Switzerland [41].
Cause of death is provided in table 2. Death occurred at home for 13 patients (40% of the deceased) and in a medical institution (palliative care or hospital ward, long term care institution) for 13 patients (40%) (missing data: n = 4).
Advanced care directives (ACD) were discussed in 49 (72%) cases and written in 23 cases (34%). Cognitive impairment (n = 4; 6% at first visit) and communication were limiting factors for ACD.
The data presented describe the components of a regular, quarterly multidisciplinary follow-up and the management of patients with ALS in an outpatient clinic as of 2012. The MDM structure allowed elective and timely implementation of NIV in most cases: only three (4%) episodes of AHRF leading to the emergency ward occurred during follow-up, which is an improvement compared to prior data reported in our area [28]. Our longitudinal data provides a time course for interventions such as NIV and PEG. Survival was slightly above what is expected based on published data. Health-related quality of life (HRQL) was already affected at the time of inclusion in our MDM structure, with SF-36 scores showing impairments for the sub-scales “general health”, “vitality”, “physical functioning” and “role-physical”, although HADS scores for anxiety and depression were only moderately affected. Most patients were cared for at home (98%), with a heavy burden placed on family members as caregivers. The referral of ALS patients to our MDM structure was well accepted by neurologists in private practice and by MND patients in our area. Finally, although not directly related to the MDM team, we noted a long delay in diagnosis (i.e. time from first symptoms to diagnosis).
This was not a controlled study. However, an earlier study performed in our area provides a historical comparison concerning the care of ALS patients. It suggests that some very significant improvements have occurred, most probably related to the implementation of the MDM structure [28]. Indeed, in this retrospective review of 21 ALS cases who died in Geneva between 1996 and 2002, median survival from diagnosis was only 20.8 months (range: 6–60). Only six patients were treated by NIV, and for two of these NIV was introduced as an emergency procedure. Nutritional assessment and follow-up were seldom performed.
MDM care for ALS patients has been implemented in many Western countries since the turn of the century [14, 16–18, 20–23, 42]. Descriptive studies of tertiary referral centres for ALS suggest improved survival, decreased hospitalisations and increased implementation of NIV and PEG when compared to usual care [16]. A Cochrane review [19] stated that MDM of ALS improved some components of HRQL and reduced hospitalisations, but that the evidence was of “low quality”. A retrospective study performed in the UK and based on a review of 387 cases suggested that MDM clinics had an independent, positive impact on survival [43]. This was also shown in a prospective observational study based on a national registry in Ireland: referral to an ALS clinic was an independent covariate of improved survival, especially in patients with initial bulbar involvement [18]. Randomised studies of MDM care for ALS vs. “usual care” are still lacking, however, and the reason for an independent improvement in survival remains unclear. A recent study by Schellenberg et al. [44], based on semi-structured interviews with ALS patients, showed that integrated care (as in MDM clinics) was considered convenient and was associated with expertise, improved communication between experts, improved information, and possible advocacy for ALS care. Travel, reduced mobility and lengthy appointments were however mentioned as barriers to or disadvantages of MDM attendance.
In our cohort, mean time from first symptoms to diagnosis was 13.7 (SD: 10.2) months (Median 11.1; Q1–Q3: 6.8–18), a delay which is consistent with previous studies (the range reported in the EFNS guidelines is 10–18 months) [15, 45]. As a consequence, treatment with riluzole and optimal management are delayed. More than 50% of ALS patients do not visit a neurologist when the first symptoms of the disease occur, which may be part of the explanation [46]. A further delay may result from postponed referrals by general practitioners and neurologists to the specialised centres and from the lack of available day-care beds.
Median survival time from first symptoms in this cohort was 50 months (Q1–Q3: 34–95). Other studies have reported median survival rates from symptom onset of 27-44 months [1, 3–5, 47–50]. In fact, few studies report median survival rates from symptom onset above 45 months [51–53]. In a study analysing data on 1282 patients over 15 years from an Irish ALS register, median survival was 2.4 years (29 months) from symptom onset and 1.3 years (15 months) from diagnosis [1]. Decreased survival was associated with female gender, bulbar onset, definite or probable El Escorial categories, and PEG insertion. Other reported adverse prognostic markers are increasing age at disease onset and presence of frontotemporal dementia [4, 47, 49–53]. Conversely, protective factors identified in the Irish study were having a familial disease, attendance at an ALS clinic, history of riluzole prescription and, interestingly, greater diagnostic delay. Delayed diagnosis may also be related to slower and thus less typical and less aggressive progression of the disease [50–52].
Appropriate management of nutritional status has a measurable impact on HRQL and survival [54], and early detection and prevention of weight loss (high calorie nutritional supplements and/or PEG) are important components of ALS care [49, 50, 52, 54–56]. Indeed, a higher pre-morbid body mass index (BMI) and maintenance of BMI and nutritional status are significantly related to survival in ALS. The rate of weight loss from onset to diagnosis is a strong and independent prognostic factor in ALS [49, 50, 52]. Nutritional status appears to be related not only to prognosis, but also to physical functioning in ALS. Fat-free mass index, evaluated by bioelectrical impedance, may better predict nutritional requirements, prognosis and respiratory involvement than body mass index or body weight [55]. According to recent European Society for Clinical Nutrition and Metabolism (ESPEN) guidelines, a complete nutritional assessment and body composition analysis are recommended at diagnosis and during follow-up of ALS patients [57]. Percutaneous gastrostomy (PEG) is recommended at an early stage of the disease. Indicators for the placement of gastrostomy include dysphagia, long duration of meals, weight loss, poor respiratory function and risk of choking. The decision is subject to patient acceptance [57]. In this cohort study, PEG was performed in a fifth of our patients, including six patients already on NIV, without complications or major discomfort (details not shown). However, one third of our candidates for PEG (7/21) refused the procedure.
Noninvasive ventilation prolongs survival in ALS and may have a positive impact on HRQL [58, 59]. Appropriate timing of NIV is important for efficacy, acceptance and quality of life. In the UK, the proportion of patients referred for NIV and the success rates for implementing NIV have increased markedly over the past decade [60]. The MDM structure allows: (1) a systematic follow-up of the relevant functional respiratory parameters, (2) anticipating the requirement for NIV; and (3) implementation by a team experienced in NIV. Experience in NIV is mandatory: indeed, prevention of hypoventilation and undesired respiratory events under NIV, and thus selection of the appropriate settings, are predictive of survival [61–63]. Furthermore, NIV in ALS may be associated with specific technical challenges. In our study, NIV could be anticipated and implemented electively in most patients (89%) for whom it was required, either on an outpatient basis (15%) or during short elective hospital stays (85%). Details of ventilator settings are provided in the online supplement. All patients were treated with bi-level, positive pressure ventilators, with daily use increasing progressively over time and reaching 24 hrs/day in a few cases. This was remarkably well tolerated, and none of our cases requested or required invasive ventilation by tracheostomy (TPPV). Because of the tremendous burden placed on caregivers by TPPV, the unavoidable progression towards a locked-in syndrome and the insufficiency of appropriate support for home care and TPPV in Switzerland, our group does not encourage use of TPPV in ALS. Information on the pros and cons of TPPV is systematically provided, however. Younger patients, patients with young children, or with bulbar onset, residual mobility and “failure” of NIV, are possible candidates [39].
This observational study has several limitations: (1) we included a limited number of patients, although our cohort was in agreement with what is epidemiologically expected in our area [7]; (2) longitudinal data for HRQL scores are missing due to difficulties in obtaining HRQL scores in many patients (communication, cognition). We believe, however, that improving knowledge on the organisation and contributions of the multidisciplinary management of patients with ALS is important. As recommended by EFNS guidelines, early referral to specialised clinics must be encouraged to decrease delays in diagnosis and to improve integrated care [15]. Although not emphasised in this manuscript, MDM clinics also offer a platform for clinical research, which can contribute to a better understanding of the disease and to clinical trials of new interventions and treatments.
In summary, this study shows that over a four-year period, ALS patients complied with the modalities of our MDM follow-up without any drop-outs. Furthermore, to our knowledge very few cases were not referred by neurologists in private practice in our area, suggesting that this structure was well accepted by both neurologists and patients. Although cost-effectiveness must be explored further, anticipation of requirements for NIV, PEG and ACP may decrease emergency procedures, an important issue for costs, patient comfort and HRQL. The MDM structure also contributes to increasing the experience and knowledge of the clinicians involved in managing patients suffering from this rare disease. The burden placed on caregivers, mostly family members, remains unacceptably high, and support for home care of ALS must be increased. Also, for patients who are far advanced in their disease, for whom coming to the MDM clinic becomes a logistic problem, the possibility of reaching out to improve home care must be further explored.
All patients were ventilated with bi-level pressure support ventilators (VPAP S9 ST®, VPAP IV ST®, Lumis 150®, Resmed, San Diego, CA, or BiPAP A30® and A40®, Philips Respironics, Eindhoven, NL). Life-support ventilators (with built-in battery and alarms) were used when NIV was required for more than 12 hours/day (Stellar 150®, Resmed), and two ventilators were made available for all patients requiring more than 16 hours/day.
Interfaces used were either nasal prongs, nasal masks or facial masks, and daytime mouthpiece ventilation when required. A combination of masks was suggested when major leaks occurred at night (i.e. nasal mask or prongs during the day, and facial mask at night), or for patient comfort.
For patients under NIV, data recorded by the ventilator were downloaded (i.e. leaks, estimated tidal volume, compliance, residual indices for apnoea and hypopnoea), interfaces and tubing were checked, and ventilator settings and interfaces were adapted, according to the data collected, by the pulmonologist and a specialised nurse at each MDM visit.
Table S1 Ventilator settings, masks and data downloaded from the ventilator.
| n = 27 | |
|---|---|
| Time spent with NIV, months | 12 (5.5–29.5) |
| NIV initiated in an outpatient setting, n (%) | 4 (15) |
| NIV initiated as inpatient, n (%) | 23 (85) |
| Elective initiation of NIV, n (%) | 24 (89) |
| Initiation of NIV during AHRF, n (%) | 3 (11) |
| Ventilator type and settings | |
| - Barometric ventilators, n (%) | 27 (100) |
| o Spontaneous timed mode*, n (%) | 27 (100) |
| o IPAP, cm H2O | 14 (12–16) |
| o EPAP, cm H2O | 5 (4–5.7) |
| o BURR, n | 16 (14–20) |
| Masks and interfaces† | |
| - Facial, n (%) | 18 (66.7) |
| - Nasal, n (%) | 6 (22) |
| - Nasal pillow, n (%) | 12 (44) |
| - Mouthpiece, n (%) | 1 (4) |
| Others equipment | |
| - Oxygen on NIV, n (%) | 4 (15) |
| - Humidifier, n (%) | 27 (100) |
| - Use of 2nd ventilator, n (%) | 11 (41) |
| - Mechanical insufflation/exsufflation device, n (%) | 8 (30) |
| Data downloaded from ventilator software, n | 26 |
| - Compliance, min/24 hrs‡ | 591 (491–1157) |
| - Leaks median, l/min | 8.7 (0.4–12.9) |
| - Leaks 95th percentile, l/min | 22.4 (5.8–30.8) |
| - AHI, n/h | 1.8 (0.6–6.4) |
| - Tidal volume, ml | 375 (300–510) |
| - Minute volume, l/min | 8.2 (5.8–9.8) |
| - Respiratory rate median, n | 18 (16–22) |
| - Δ(RR-BURR), n§ | 0 (0–3) |
| - % spontaneous breathing, % | 30 (12–50) |
AHRF = acute hypercapnic respiratory failure; IPAP = inspiratory airway pressure; EPAP = expiratory airway pressure; AHI = apnoea-hypopnoea index, n/hr; RR = spontaneous respiratory rate; BURR = back-up respiratory rate. Data are presented as median (Q1-Q3) or n (%). * Spontaneous timed mode (S/T) is a pressure support mode in which a back-up respiratory rate is set by the clinician; pressurisation is triggered by the patient unless his/her spontaneous respiratory rate decreases below the BURR. † Patients could use different interfaces at daytime and at night-time. ‡ Six patients used their ventilator more than 20 hours/day. § Difference between RR and BURR: this index shows to what extent RR is controlled by the ventilator. All data provided by the ventilator software shown in the table are the most recent data downloaded. Data not available for one subject (download of data failed).
Figure S1 Evolution of ALS-FRS-R scores during follow-up in patients with bulbar (n = 13, 19%) and non-bulbar (n = 55, 81%) onset of ALS: rate of decline did not differ significantly between the two groups.
The study was supported financially by the Ligue Pulmonaire Genevoise, a non-profit association involved in home care for patients with respiratory disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1 Rooney J , Byrne S , Heverin M , Corr B , Elamin M , Staines A , et al. Survival analysis of irish amyotrophic lateral sclerosis patients diagnosed from 1995-2010. PLoS One. 2013;8(9):e74733. doi:.https://doi.org/10.1371/journal.pone.0074733
2 Kaufmann P , Levy G , Thompson JL , Delbene ML , Battista V , Gordon PH , et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology. 2005;64(1):38–43. doi:.https://doi.org/10.1212/01.WNL.0000148648.38313.64
3 Zoccolella S , Beghi E , Palagano G , Fraddosio A , Guerra V , Samarelli V , et al.; SLAP Registry. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2008;79(1):33–7. doi:.https://doi.org/10.1136/jnnp.2007.118018
4 Pugliatti M , Parish LD , Cossu P , Leoni S , Ticca A , Saddi MV , et al. Amyotrophic lateral sclerosis in Sardinia, insular Italy, 1995-2009. J Neurol. 2013;260(2):572–9. doi:.https://doi.org/10.1007/s00415-012-6681-5
5 Murphy M , Quinn S , Young J , Parkin P , Taylor B . Increasing incidence of ALS in Canterbury, New Zealand: a 22-year study. Neurology. 2008;71(23):1889–95. doi:.https://doi.org/10.1212/01.wnl.0000336653.65605.ac
6 Robberecht W , Philips T . The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–64. doi:.https://doi.org/10.1038/nrn3430
7 Brown RH , Al-Chalabi A . Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377(2):162–72. doi:.https://doi.org/10.1056/NEJMra1603471
8 Niedermeyer S , Murn M , Choi PJ . Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest. 2019;155(2):401–8. doi:.https://doi.org/10.1016/j.chest.2018.06.035
9 Hobson EV , McDermott CJ . Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2016;12(9):526–38. doi:.https://doi.org/10.1038/nrneurol.2016.111
10 Chiò A , Logroscino G , Traynor BJ , Collins J , Simeone JC , Goldstein LA , et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–30. doi:.https://doi.org/10.1159/000351153
11 Logroscino G , Traynor BJ , Hardiman O , Chiò A , Mitchell D , Swingler RJ , et al.; EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385–90. doi:.https://doi.org/10.1136/jnnp.2009.183525
12 Hinchcliffe M , Smith A . Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis. 2017;7:61–70. doi:.https://doi.org/10.2147/DNND.S135748
13 Abe K , Aoki M , Tsuji S , Itoyama Y , Sobue G , Togo M , et al, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505–12. doi:.https://doi.org/10.1016/S1474-4422(17)30115-1
14 Van den Berg JP , Kalmijn S , Lindeman E , Veldink JH , de Visser M , Van der Graaff MM , et al. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology. 2005;65(8):1264–7. doi:.https://doi.org/10.1212/01.wnl.0000180717.29273.12
15 Andersen PM , Abrahams S , Borasio GD , de Carvalho M , Chio A , Van Damme P , et al.; EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360–75. doi:.https://doi.org/10.1111/j.1468-1331.2011.03501.x
16 Chiò A , Bottacchi E , Buffa C , Mutani R , Mora G ; PARALS. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J Neurol Neurosurg Psychiatry. 2006;77(8):948–50. doi:.https://doi.org/10.1136/jnnp.2005.083402
17 Mayadev AS , Weiss MD , Distad BJ , Krivickas LS , Carter GT . The amyotrophic lateral sclerosis center: a model of multidisciplinary management. Phys Med Rehabil Clin N Am. 2008;19(3):619–31, xi. doi:.https://doi.org/10.1016/j.pmr.2008.04.004
18 Traynor BJ , Alexander M , Corr B , Frost E , Hardiman O . Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003;74(9):1258–61. doi:.https://doi.org/10.1136/jnnp.74.9.1258
19 Ng L , Khan F . Multidisciplinary care for adults with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. 2009;(4):CD007425. doi:.https://doi.org/10.1002/14651858.CD007425.pub2
20 Rooney J , Byrne S , Heverin M , Tobin K , Dick A , Donaghy C , et al. A multidisciplinary clinic approach improves survival in ALS: a comparative study of ALS in Ireland and Northern Ireland. J Neurol Neurosurg Psychiatry. 2015;86(5):496–501. doi:.https://doi.org/10.1136/jnnp-2014-309601
21 Marin B , Beghi E , Vial C , Bernard E , Lautrette G , Clavelou P , et al.; EURECALS consortium. Evaluation of the application of the European guidelines for the diagnosis and clinical care of amyotrophic lateral sclerosis (ALS) patients in six French ALS centres. Eur J Neurol. 2016;23(4):787–95. doi:.https://doi.org/10.1111/ene.12941
22 Martin S , Trevor-Jones E , Khan S , Shaw K , Marchment D , Kulka A , et al. The benefit of evolving multidisciplinary care in ALS: a diagnostic cohort survival comparison. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(7-8):569–75. doi:.https://doi.org/10.1080/21678421.2017.1349151
23 Bakker M , Creemers H , Schipper K , Beelen A , Grupstra H , Nollet F , et al. Need and value of case management in multidisciplinary ALS care: A qualitative study on the perspectives of patients, spousal caregivers and professionals. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(3-4):180–6. doi:.https://doi.org/10.3109/21678421.2014.971811
24 O’Brien MR , Whitehead B , Jack BA , Mitchell JD . From symptom onset to a diagnosis of amyotrophic lateral sclerosis/motor neuron disease (ALS/MND): experiences of people with ALS/MND and family carers - a qualitative study. Amyotroph Lateral Scler. 2011;12(2):97–104. doi:.https://doi.org/10.3109/17482968.2010.546414
25 Galvin M , Madden C , Maguire S , Heverin M , Vajda A , Staines A , et al. Patient journey to a specialist amyotrophic lateral sclerosis multidisciplinary clinic: an exploratory study. BMC Health Serv Res. 2015;15(1):571. doi:.https://doi.org/10.1186/s12913-015-1229-x
26 Cheng HWB , Chan OMI , Chan CHR , Chan WH , Fung KS , Wong KY . End-of-life Characteristics and Palliative Care Provision for Patients With Motor Neuron Disease. Am J Hosp Palliat Care. 2018;35(6):847–51. doi:.https://doi.org/10.1177/1049909117735832
27 Bede P , Oliver D , Stodart J , van den Berg L , Simmons Z , O Brannagáin D , et al. Palliative care in amyotrophic lateral sclerosis: a review of current international guidelines and initiatives. J Neurol Neurosurg Psychiatry. 2011;82(4):413–8. doi:.https://doi.org/10.1136/jnnp.2010.232637
28 Pautex S , Janssens JP , Vuagnat H , Conne P , Zulian GB . Management of patients with amyotrophic lateral sclerosis. Swiss Med Wkly. 2005;135(41-42):626–9.
29 Janssens JP , Adler D , Iancu Ferfoglia R , Poncet A , Genton Graf L , Leuchter I , et al. Assessing inspiratory muscle strength for early detection of respiratory failure in motor neuron disease: should we use MIP? SNIP or both? Respiration. 2019;98(2):114–24. doi:.https://doi.org/10.1159/000498972
30 Brooks BR , Miller RG , Swash M , Munsat TL ; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–9. doi:.https://doi.org/10.1080/146608200300079536
31 Cedarbaum JM , Stambler N , Malta E , Fuller C , Hilt D , Thurmond B , et al.; BDNF ALS Study Group (Phase III). The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169(1-2):13–21. doi:.https://doi.org/10.1016/S0022-510X(99)00210-5
32 Lépine JP , Godchau M , Brun P , Lempérière T . [Evaluation of anxiety and depression among patients hospitalized on an internal medicine service]. Ann Med Psychol (Paris). 1985;143(2):175–89.
33 Zigmond AS , Snaith RP . The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361–70. doi:.https://doi.org/10.1111/j.1600-0447.1983.tb09716.x
34 Perneger TV , Leplège A , Etter JF , Rougemont A . Validation of a French-language version of the MOS 36-Item Short Form Health Survey (SF-36) in young healthy adults. J Clin Epidemiol. 1995;48(8):1051–60. doi:.https://doi.org/10.1016/0895-4356(94)00227-H
35Ware J, Snow K, Kosinski M, Gandek B. SF-36 health survey: manual and interpretation guide. Boston, Massachusetts: The Health Institute, New England Medical Center; 1993.
36 Kyle UG , Genton L , Karsegard L , Slosman DO , Pichard C . Single prediction equation for bioelectrical impedance analysis in adults aged 20--94 years. Nutrition. 2001;17(3):248–53. doi:.https://doi.org/10.1016/S0899-9007(00)00553-0
37 Miller RG , Jackson CE , Kasarskis EJ , England JD , Forshew D , Johnston W , et al.; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218–26. doi:.https://doi.org/10.1212/WNL.0b013e3181bc0141
38 Rubin AD , Griffin GR , Hogikyan ND , Feldman EL . A new member of the multidisciplinary ALS team: the otolaryngologist. Amyotroph Lateral Scler. 2012;13(2):229–32. doi:.https://doi.org/10.3109/17482968.2011.643898
39 Heritier Barras AC , Adler D , Iancu Ferfoglia R , Ricou B , Gasche Y , Leuchter I , et al., CeSLA group. Is tracheostomy still an option in amyotrophic lateral sclerosis? Reflections of a multidisciplinary work group. Swiss Med Wkly. 2013;143:w13830. doi:.https://doi.org/10.4414/smw.2013.13830
40 Austin PC , Lee DS , Fine JP . Introduction to the Analysis of Survival Data in the Presence of Competing Risks. Circulation. 2016;133(6):601–9. doi:.https://doi.org/10.1161/CIRCULATIONAHA.115.017719
41Richard JL, Bouzourène K, Gallant S, Ricciardi P, Sudre P, Iten A, Burnand B. Validation et normes du SF-36 dans la population du canton de Vaud. Lausanne: Institut universitaire de médecine sociale et préventive, Raisons de santé, 28; 2000
42 Pouget J . À propos des recommandations de diagnostic et de prise en charge dans les maladies neuromusculaires [About recommendations for diagnosis and management in neuromuscular diseases]. Rev Neurol (Paris). 2012;168(12):901. Article in French. doi:.https://doi.org/10.1016/j.neurol.2012.10.002
43 Aridegbe T , Kandler R , Walters SJ , Walsh T , Shaw PJ , McDermott CJ . The natural history of motor neuron disease: assessing the impact of specialist care. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(1):13–9. doi:.https://doi.org/10.3109/17482968.2012.690419
44 Schellenberg KL , Hansen G . Patient perspectives on transitioning to amyotrophic lateral sclerosis multidisciplinary clinics. J Multidiscip Healthc. 2018;11:519–24. doi:.https://doi.org/10.2147/JMDH.S177563
45 Mitchell JD , Callagher P , Gardham J , Mitchell C , Dixon M , Addison-Jones R , et al. Timelines in the diagnostic evaluation of people with suspected amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND)--a 20-year review: can we do better? Amyotroph Lateral Scler. 2010;11(6):537–41. doi:.https://doi.org/10.3109/17482968.2010.495158
46 Kano O , Iwamoto K , Ito H , Kawase Y , Cridebring D , Ikeda K , et al. Limb-onset amyotrophic lateral sclerosis patients visiting orthopedist show a longer time-to-diagnosis since symptom onset. BMC Neurol. 2013;13(1):19. doi:.https://doi.org/10.1186/1471-2377-13-19
47 Paulukonis ST , Roberts EM , Valle JP , Collins NN , English PB , Kaye WE . Survival and Cause of Death among a Cohort of Confirmed Amyotrophic Lateral Sclerosis Cases. PLoS One. 2015;10(7):e0131965. doi:.https://doi.org/10.1371/journal.pone.0131965
48 Hirose T , Kimura F , Tani H , Ota S , Tsukahara A , Sano E , et al. Clinical characteristics of long-term survival with noninvasive ventilation and factors affecting the transition to invasive ventilation in amyotrophic lateral sclerosis. Muscle Nerve. 2018;58(6):770–6. doi:.https://doi.org/10.1002/mus.26149
49 Mandrioli J , Malerba SA , Beghi E , Fini N , Fasano A , Zucchi E , et al.; ERRALS Group. Riluzole and other prognostic factors in ALS: a population-based registry study in Italy. J Neurol. 2018;265(4):817–27. doi:.https://doi.org/10.1007/s00415-018-8778-y
50 Calvo A , Moglia C , Lunetta C , Marinou K , Ticozzi N , Ferrante GD , et al. Factors predicting survival in ALS: a multicenter Italian study. J Neurol. 2017;264(1):54–63. doi:.https://doi.org/10.1007/s00415-016-8313-y
51 Govaarts R , Beeldman E , Kampelmacher MJ , van Tol MJ , van den Berg LH , van der Kooi AJ , et al. The frontotemporal syndrome of ALS is associated with poor survival. J Neurol. 2016;263(12):2476–83. doi:.https://doi.org/10.1007/s00415-016-8290-1
52 Yates E , Rafiq MK . Prognostic factors for survival in patients with amyotrophic lateral sclerosis: analysis of a multi-centre clinical trial. J Clin Neurosci. 2016;32:51–6. doi:.https://doi.org/10.1016/j.jocn.2015.12.037
53 Magnus T , Beck M , Giess R , Puls I , Naumann M , Toyka KV . Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve. 2002;25(5):709–14. doi:.https://doi.org/10.1002/mus.10090
54 Körner S , Hendricks M , Kollewe K , Zapf A , Dengler R , Silani V , et al. Weight loss, dysphagia and supplement intake in patients with amyotrophic lateral sclerosis (ALS): impact on quality of life and therapeutic options. BMC Neurol. 2013;13(1):84. doi:.https://doi.org/10.1186/1471-2377-13-84
55 Héritier AC , Janssens JP , Adler D , Ferfoglia RI , Genton L . Should patients with ALS gain weight during their follow-up? Nutrition. 2015;31(11-12):1368–71. doi:.https://doi.org/10.1016/j.nut.2015.06.005
56 Genton L , Viatte V , Janssens JP , Héritier AC , Pichard C . Nutritional state, energy intakes and energy expenditure of amyotrophic lateral sclerosis (ALS) patients. Clin Nutr. 2011;30(5):553–9. doi:.https://doi.org/10.1016/j.clnu.2011.06.004
57 Burgos R , Bretón I , Cereda E , Desport JC , Dziewas R , Genton L , et al. ESPEN guideline clinical nutrition in neurology. Clin Nutr. 2018;37(1):354–96. doi:.https://doi.org/10.1016/j.clnu.2017.09.003
58 Bourke SC , O’Neill CL , Williams TL , Peel ET , Gibson GJ , McDermott CJ , et al. The changing landscape of non-invasive ventilation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(4):368–9. doi:.https://doi.org/10.1136/jnnp-2012-302253
59 Bourke SC , Tomlinson M , Williams TL , Bullock RE , Shaw PJ , Gibson GJ . Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5(2):140–7. doi:.https://doi.org/10.1016/S1474-4422(05)70326-4
60 O’Neill CL , Williams TL , Peel ET , McDermott CJ , Shaw PJ , Gibson GJ , et al. Non-invasive ventilation in motor neuron disease: an update of current UK practice. J Neurol Neurosurg Psychiatry. 2012;83(4):371–6. doi:.https://doi.org/10.1136/jnnp-2011-300480
61 Gonzalez-Bermejo J , Morelot-Panzini C , Arnol N , Meininger V , Kraoua S , Salachas F , et al. Prognostic value of efficiently correcting nocturnal desaturations after one month of non-invasive ventilation in amyotrophic lateral sclerosis: a retrospective monocentre observational cohort study. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5-6):373–9. doi:.https://doi.org/10.3109/21678421.2013.776086
62 Georges M , Attali V , Golmard JL , Morélot-Panzini C , Crevier-Buchman L , Collet JM , et al. Reduced survival in patients with ALS with upper airway obstructive events on non-invasive ventilation. J Neurol Neurosurg Psychiatry. 2016;87(10):1045–50. doi:.https://doi.org/10.1136/jnnp-2015-312606
63 Sancho J , Servera E , Morelot-Panzini C , Salachas F , Similowski T , Gonzalez-Bermejo J . Non-invasive ventilation effectiveness and the effect of ventilatory mode on survival in ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(1-2):55–61. doi:.https://doi.org/10.3109/21678421.2013.855790
Contributed equally to the manuscript
The study was supported financially by the Ligue Pulmonaire Genevoise, a non-profit association involved in home care for patients with respiratory disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.