The cellular prion protein beyond prion diseases
DOI: https://doi.org/10.4414/smw.2020.20222
Giorgia
Mannia, Victoria
Lewisb, Matteo
Senesib, Giovanni
Spagnollic, Francesca
Fallarinoa, Steven J.
Collinsbd, Sophie
Mouillet-Richarde, Emiliano
Biasinic
aDepartment of Experimental Medicine, University of Perugia, Italy
bDepartment of Medicine (Royal Melbourne Hospital), The University of Melbourne, Parkville, Victoria, Australia
cDulbecco Telethon Laboratory of Prions and Amyloids, Department of Cellular, Computational and Integrative Biology (CIBIO), University of Trento, Italy
dFlorey Institute of Neuroscience and Mental Health, Parkville, Victoria, Australia
eCentre de Recherche des Cordeliers, INSERM, Sorbonne Université, Université de Paris, France
Summary
The cellular prion protein (PrPC), a cell surface glycoprotein originally identified for its central role in prion diseases (also called transmissible spongiform encephalopathies), has recently been implicated in the pathogenesis of other neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases, by acting as a toxicity-transducing receptor for different misfolded protein isoforms, or in some case by exerting neuroprotective effects. Interestingly, PrPC has also been reported to play unexpected functions outside the nervous system, for example by contributing to myelin homeostasis, regulating specific processes of the immune system and participating in various aspects of cancer progression. Collectively, these observations point to a much broader role for PrPC in physiological and disease processes than originally assumed. In this manuscript, we provide an overview of what is known about the role of PrPC beyond prion disorders and discuss the potential implications of targeting this protein in different diseases.
Introduction
Aging is accompanied by molecular, cellular and functional changes, which particularly affect the nervous system. Among the physiological processes known to be altered by aging is the protein folding quality control machinery, deputed to monitor and ameliorate protein misfolding. Once present, misfolded proteins typically acquire alternative conformations that can lead to their aggregation and accumulation intracellularly or extracellularly, and eventually initiate a cascade of toxic molecular events, ultimately resulting in cellular dysfunction [1]. A wide range of age-related disorders is indeed linked to protein misfolding and aggregation in the brain. Examples include highly prevalent disorders such as Parkinson’s and Alzheimer’s diseases, as well as rarer disorders such as prion diseases. Alzheimer’s disease is the most common form of dementia in the elderly population, currently affecting almost 40 million individuals worldwide. The number will increase dramatically in the coming decades as the population ages, producing challenging medical and socioeconomic consequences [2].
According to the amyloid cascade hypothesis, Alzheimer’s disease is a consequence of the accumulation in the brain of the 40−42 amino acid Aβ peptide, a cleavage product of the amyloid precursor protein (APP). The Aβ peptide spontaneously forms polymers ranging from small, soluble oligomers to large, insoluble fibrils [3]. Multiple pieces of evidence suggest that soluble Aβ oligomers, rather than fibrillar aggregates, are primarily responsible for the synaptic dysfunction underlying the cognitive decline in Alzheimer’s disease [4]. Aβ oligomers are believed to act, at least in part, by binding to cell surface receptors that transduce their detrimental effects on synapses. Recently, a novel candidate has emerged as a receptor for Aβ oligomers: the cellular form of the prion protein (PrPC) [5].
PrPC, an endogenous, cell-surface glycoprotein, plays a central role in transmissible neurodegenerative disorders commonly referred to as prion diseases. These diseases, which can be sporadic, inherited or acquired, are caused by the conformational conversion of PrPC into a misfolded isoform (called scrapie form of PrP or PrPSc) that accumulates in the central nervous system of affected individuals. PrPSc is an infectious protein (constituting “prions”) that propagates itself by binding to PrPC triggering its conformational rearrangement (“templating”) into new PrPSc molecules [6]. A great deal of evidence indicates a distinction between prion infectivity and toxicity, and suggests that a physiological function of PrPC may be altered upon binding to PrPSc, to deliver neurotoxic signals [7]. In fact, the presence of PrPC on the neuronal surface has been shown to be critical not only for supporting PrPSc propagation, but also for transducing its neurotoxicity [8–10]. This conclusion recently found unexpected support from data involving other pathogenic protein oligomers. Different studies provided evidence that PrPC could mediate the toxicity of oligomeric assemblies of Aβ, alpha-synuclein and other β-sheet-rich protein conformers [5, 11–14]. These results indicate that misfolded assemblies of several different pathogenic proteins could exert their effects by blocking, enhancing or altering the normal activity of PrPC [15]. This conclusion highlights a close connection between the role of PrPC in several neurodegenerative diseases and its physiological function. What is this function? Several activities have been attributed to PrPC in the nervous system, mostly based on subtle abnormalities detected in mice or cells depleted of PrPC [16]. These include roles in neuroprotection, synaptic integrity, neuronal excitability and memory formation. However, most of these observations have not been reproduced in subsequent studies, found little or no physiological or pathological correlates, or were later shown to arise from genetic impurities of the employed mouse models [17, 18]. In fact, a number of previous lines used to study the physiological function of PrPC were non-co-isogenic Prnp(-/-) mice, in some case leading to artifactual conclusions [18]. Curiously, some of the clearest observations regarding PrPC function have been collected by studying the protein outside prion diseases. These include roles in the regulation of myelin homeostasis [19], immune processes [20] and in the progression of cancer [21]. Although it seems unlikely that a single protein could be involved in such a wide range of physiological processes, particularly in light of the relatively small number of phenotypic changes observed in PrPC-deficient mice, the lack of a clear understanding of the activity of this protein forces us to remain open minded. Thus, in this manuscript we review the most compelling data suggesting a putative role for PrPC beyond prion diseases and discuss potential therapeutic implications arising from such observations.
A role for PrPC in other neurodegenerative disorders
Despite the lack of consensus around the normal function(s) of PrPC in the central nervous system (CNS), misfolding of PrPC with accumulation of altered conformers (PrPSc) in the brain is considered the fundamental pathogenic event in prion diseases [22, 23], with a potential role for PrPC in the development of other neurodegenerative diseases such as Alzheimer’s disease and the α-synucleinopathies increasingly described.
Alzheimer’s disease
In early studies predominantly relying on cell culture models, PrPC was described as favourably regulating the activity of β-secretase (β-site APP cleaving enzyme; BACE1), whereby production of neurotoxic Aβ peptides was reduced [24]. This reported capacity of PrPC appeared to require PrPC localisation in cholesterol-rich lipid rafts and the N-terminal polybasic region [24] thereby allowing direct interaction with Golgi-localised, immature forms of BACE1 causing trapping within the Golgi and reduced BACE1 levels at the cell surface and in endosomes [25]. Of interest, a mutant form of APP (carrying the Swedish mutation) was reported to escape this beneficial regulatory effect of PrPC, in keeping with a potential protective effect for sporadic Alzheimer’s disease but probably not for at least some types of genetic Alzheimer’s disease. Unfortunately, the translational relevance and validity of these early observations has become less clear with the passage of time. In one follow-up study from the same laboratory, Whitehouse and colleagues reported that human brains demonstrated an ~50% reduction of PrPC expression in sporadic Alzheimer’s disease frontal cortex compared with age-matched controls, with PrPC levels inversely correlated with BACE1 activity, Aβ load, soluble Aβ levels and the Braak neurofibrillary tangle stage of disease [26]. In contrast however, a more recent report from this group, primarily utilising PrPC gene ablated (PrP0/0) transgenic mice expressing wild-type human APP, the absence of PrPC appeared to have no effect on BACE1 activity, with levels of APP proteolytic fragments, cognate Aβ peptides and histopathological findings in the brains of these mice unaltered compared to controls [27]. Further potentially linking PrPC to the processing of APP and the generation of deleterious Aβ peptides, another group has reported that the genes influenced by the amyloid intracellular domain transcription regulation fragment produced through γ-secretase processing of β-APP includes the gene encoding PrPC (Prnp) through a p53-dependent pathway [28], possibly constituting a negative feedback loop. In a subsequent report employing a combination of experimental approaches, however, this putative role for the amyloid intracellular domain in influencing PrPC expression levels could not be reproduced, once again leaving uncertainty about the biological validity of the original observations [29]. Additional observations suggesting a potential neuroprotective effect of PrPC in Alzheimer’s disease have been provided by Rial and co-workers [30]. Utilising a mouse model centred on the effects of a single intracerebroventricular injection of 400 pmol of Aβ1-40 peptide on spatial learning and memory, these authors demonstrated reduced cognitive impairment in transgenic Tg-20 mice (that overexpress PrPC five-fold) compared with wild-type and transgenic PrP0/0 mice, with the Tg-20 mice also displaying less evidence of apoptosis and cell damage in the hippocampus. The mechanism of neuroprotection was not explored by the authors but other reports raise the possibility that glutamate excitotoxicity may be relevant with PrPC able to directly attenuate excessive N-methyl-D-aspartate receptor (NMDAR) activity in a copper-dependent manner, including that induced by the presence of Aβ1-42 peptide [31–33]. In contrast to any potential neuroprotective effects afforded by PrPC in Alzheimer’s disease, there is considerable evidence supporting a likely deleterious role in Alzheimer’s disease pathogenesis. PrPC, through direct binding to residues 95-113, may act to disassemble amyloid fibrils composed of Aβ peptides thereby trapping constituent peptides into an oligomeric state effectively enriching the concentration of putative neurotoxic oligomers [34], but most evidence suggests the harmful behaviour of PrPC is through acting as a receptor to transduce the toxic signal of soluble Aβ peptides. Such deleterious effects of this PrPC mediated toxic signal transduction include impairment of hippocampal long-term potentiation (LTP), dendritic spine retraction and disruption of rodent spatial memory. In their seminal report, Lauren and co-workers exploited expression cloning to determine that PrPC binds with nanomolar affinity to soluble Aβ oligomers (principally through the charge cluster residues 95−110) sub-serving blockade of hippocampal slice LTP, with synaptic function rescued by anti-PrP antibodies [5]. Despite the inability of early follow-up reports to replicate this implicated pathogenic role for PrPC [35–37], subsequent reports have re-affirmed and elaborated this apparent crucial transduction role for mediating soluble Aβ oligomer toxicity. After Aβ oligomers bind PrPC at dendritic spines (possibly also inhibiting constitutive endocytosis and causing clustering of PrPC on the cell surface [38]), the Aβ oligomer-PrPC complex associates with Fyn causing activation of this Src kinase leading to tau hyperphosphorylation [39], as well as phosphorylation of the NR2B subunit of NMDARs. The kinase activity of Fyn on NMDARs culminates in depletion of these glutamatergic ion channels at the synaptic surface in parallel with loss of dendritic spines [40–42]. In addition to deleterious synaptic changes, axonal and neuronal loss are reported as downstream pathophysiological consequences of Aβ oligomers binding to PrPC along with impairment of spatial learning and memory [43, 44]. As a sequitur to these various reports of the importance of PrPC as a key transducing mediator of soluble Aβ neurotoxicity, anti-PrP antibodies primarily directed against an epitope within the oligomer binding site have been described as ameliorating or rescuing rodent hippocampal LTP and cognitive function [5, 45–48].
Alpha-synucleinopathies
Beyond a likely participation of PrPC in Alzheimer’s disease pathogenesis, the normal form of the prion protein has also recently been suggested to contribute to the pathogenesis of α-synucleinopathies, such as Parkinson’s disease and diffuse Lewy body disease, although discrepancies in findings across reports is noteworthy. Harnessing in vivo and in vitro models, Ferreira and colleagues reported a deleterious interaction of α-synuclein oligomers (but not α-synuclein monomers or fibrils) with PrPC at the NMDAR causing a failure of LTP in wild-type mouse hippocampal slices; this putative role for PrPC was supported by the abrogation of LTP impairment when utilising PrP0/0 hippocampal slices [12]. Moreover, attempts to block the interaction of α-synuclein oligomers with PrPC using antibodies targeting specific PrPC amino acid segments revealed that the integrity of the 93−109 (charge cluster) region was necessary to observe such LTP impairment. The PrPC mediated inhibition of synaptic plasticity was also prevented with the use of a specific Fyn inhibitor when co-incubated with the α-synuclein oligomers, suggesting that an interaction of the α-synuclein oligomer-PrPC complex promotes phosphorylation of the NMDAR through Fyn, thereby causing excessive Ca2+ influx at the post-synaptic terminal. The interaction between complexes of glycosyl-phosphatidyl-inositol (GPI)-anchored PrPC and α-synuclein oligomers with cytosolic Fyn appears possible through metabotropic glutamate receptor 5 (mGluR5), as using specific inhibitors of mGluR5-mediated phosphorylation of NMDAR was also able to rescue LTP and cognitive deficits in these mice to levels equivalent to controls. Interestingly, an analogous molecular pathophysiological mechanism has also been observed in ex vivo and in vivo models of Alzheimer’s disease assessing synaptic impairment driven by soluble Aβ oligomers, with blockade of the adenosine A2A receptors responsible for mGluR5 activation resulting in the inhibition of the deleterious NMDAR phosphorylation via Fyn [40, 41]. Apparently incongruous with the aforementioned study showing that PrPC selectively bound only to α-synuclein oligomers to sub-serve their detrimental effects, Aulić and co-workers reported that PrPC mediated the cellular uptake and spread of recombinant α-synuclein amyloid fibrils, with this activity attenuating the propagation of misfolded PrPSc in in vitro and in vivo scrapie infection models [11]. Although supporting a role for PrPC in mediating the movement of α-synuclein, another group suggested that although the pathological spreading of α-synuclein may be facilitated by PrPC, it is not exclusively dependent on PrPC [49]. Despite the reported inability of α-synuclein oligomers to induce LTP impairment in PrP0/0 mouse hippocampal slices, the role of PrPC in directly mediating neurotoxicity and any direct interaction between α-synuclein oligomers and PrPC are still a matter of controversy. Employing a range of biophysical techniques to assess an intimate interaction between α-synuclein and PrPC, including surface plasmon resonance, La Vitola and colleagues were unable to confirm any direct association of PrPC with recombinant α-synuclein oligomers [50], as well as α-synuclein monomers and fibrils, although α-synuclein monomers appeared to suppress PrPC concatenation through inhibiting nucleation. In addition, La Vitola and colleagues observed that primary neuronal cultures derived from wild-type and PrP0/0 mice were equally susceptible to α-synuclein oligomer neurotoxicity in a dose-dependent manner [46]. Finally, employing an in vivo model using intracerebroventricular injection of α-synuclein oligomers, they also reported that PrP0/0 mice displayed similar memory deficits and hippocampal gliosis to wild-type controls. Although the influence of differences in methodology cannot be ruled out, these findings support the likelihood of α-synuclein oligomer mediated neurotoxicity independent from PrPC. Clearly, whereas any role of PrPC in non-prion neurodegenerative diseases remains incompletely understood and a subject of contention (especially in α-synucleinopathies), it appears likely that the normal form of the prion protein may play some part in these other diseases, which for Alzheimer’s disease may involve both protective and pathogenic contributions.
PrPC in the immune system and related diseases
Over the last few years the interest of immunologists in PrPC and immune diseases has vastly increased. Two main pieces of evidence may justify such interest: firstly, PrPC has extensively been studied in the central nervous system but is also widely expressed in cells of the immune system [51]; secondly, immune tolerance to PrPSc has been documented [52–54]. Indeed, immune tolerance may prevent robust immune responses to prions; accordingly, PrP-specific antibodies have not been detected in animals infected with prions. In addition, other studies reported that the immune system may also actively contribute to prion disease pathogenesis, by amplifying prion load in lymphoid compartments, transferring the pathogenic PrPSc to cells and facilitating efficient neuroinvasion [20, 52]. Although it is clear that components of the immune system can contribute to the spread of prions, none of these pieces of evidence have been extensively validated at the molecular level, and conflicting results have often been reported [20, 55]. Overall, based on these observations, two specific roles of the immune system in prion diseases can be identified: immune cells may perform, when properly activated, as a protective shield against prions but, at the same time, they may be involved in the accumulation and spreading of pathogenic PrPSc [56]. For these reasons, manipulation of the immune system has been envisioned as a potential therapeutic option for prion diseases [57]. Immunotherapy strategies have reported promising results in vitro and in vivo. In particular, three main approaches have been undertaken so far: (i) treatment with antibodies targeting PrPSc [58–61]; (ii) vaccines with antigen-loaded dendritic cells [62, 63]; and (iii) adoptive transfer of PrP-specific CD4+ T lymphocytes [64]. Although more research into mechanism and safety of these approaches is still required, these immunotherapies may offer potential novel tools to clear the pathological form of PrP. However, because the function of cellular PrPC in the lymphoid system and in the CNS remains to be fully elucidated, it is not yet clear how therapies targeting PrPSc, which shows similarities with PrPC, will affect immune or other specific endogenous functions. For this reason, uncovering the role of PrPC in cells of the immune system may provide novel insights both into its role in the pathogenesis of prion diseases and in specific functions of immune cells in general.
PrPC in immune cells: expression and functions
Its high evolutionary conservation suggests that PrPC fulfils ancient and still essential biological functions [65–67]. Notably, PrPC is abundantly expressed in neural cells, including neurons and glia [68], as well as in subsets of cells of haematopoietic origin (e.g., myeloid dendritic cells, DCs, and T cells) [69]. In particular, data suggest that PrPC is involved in specific immune functions, including T cell development, DC activation, inhibition of macrophage phagocytosis and immunological quiescence [70, 71]. In addition to DCs and T cells, PrPC has been detected also in B lymphocytes, natural killer cells, platelets, monocytes and in follicular DCs [72–75]. Within lymphoid cells, B cells express lower levels of PrPC compared with T cells and natural killer cells [76]. In addition, it has long been known that PrPC is present on the surface of lymphocytes and it is rapidly upregulated upon their activation [77]. Following T cell activation, PrPC is redistributed in specific structures such as lipid rafts, together with signalling molecules, leading to immunomodulation [78]. It has been shown that GPI-anchored PrPC is enriched at the immunological synapse and can interact with components of the T cell receptor , such as the Fyn tyrosine kinase and the zeta chain-associated protein kinase 70 (ZAP-70), leading to the modulation of T cell receptor signalling cascade [75, 77, 79]. Moreover, PrPC expression was reported to be higher in T cells than in B lymphocytes, with CD8+ cell subsets expressing slightly more PrPC than CD4+ cells [76, 80]. PrPC expression is also higher in CD45RO+ memory compared with CD45RA+ naive T lymphocytes [75, 81]. Interestingly, data from gene arrays have revealed the murine Prnp gene to be up-regulated in T cell [82], via a Stat6-dependent mechanism, during interleukin (IL)-4 driven Th2 differentiation [83] and in CD8+ memory T cells [84]. In addition, it has been reported that regulatory CD4+ CD25+ T cells (Tregs) expressed 4.5 fold higher levels of PrP messenger RNA and showed a 10-fold higher intensity of surface PrPC than effector CD4+ CD25− T cells, despite no loss-of-function phenotypes could be recognised in Treg cells from PrP0/0 mice [85]. Hence, PrPC may be more important in certain types of functionally differentiated lymphocytes that operate in particular immune environments. Outside the nervous system, the antigen-presenting cells, DCs display the highest expression levels of PrPC, in both humans and mice [69, 86]. Studies on myeloid DCs showed that PrPC levels particularly increase during differentiation and maturation of these cells, in parallel with molecules involved in antigen presentation, such as major histocompatibility complex type II (MHC-II) and costimulatory molecules [69]. Interestingly, it has been demonstrated that important differences of PrPC expression exist between different DC subpopulations either analysed after ex vivo isolation or differentiated in vitro. DCs can be classified in two major categories: conventional DCs (cDCs), which include at least two different DC subsets (e.g., cDC1 and cDC2) and plasmacytoid DCs (pDCs) [87, 88]. PrPC was found on the surface of bone marrow-derived human and mouse conventional DCs generated in vitro or isolated from the spleen, but not in pDCs [88]. PrPC expression in these cDCs was strongly up-regulated after maturation by TLR ligands, such as bacterial lipopolysaccharide and CpG. Interestingly, a study from Ballerini et al. showed that membrane PrPC on DCs enhanced the stimulation of specific naïve T cells both in vitro and in vivo [89]. High expression of PrPC was also found on the surface of CD8+ cDC subset, both in the spleen and the lymph-nodes [88]. Although the different PrPC expression between cDCs and pDCs could be related to the specific developmental programme of these two cell types, the specific role of PrPC in cDC functions still remains to be explored. Related to these issues, specific evidence suggests that absence of PrPC in T cells and DCs had different outcomes in T-cell proliferation. Specifically, T cells devoid of PrPC exhibited a normal allogenic antigen response, while DCs lacking PrPC significantly reduced proliferation of interacting T cells, suggesting that PrPC might serve different signalling roles in the two cell types [90]. Another class of dendritic cells, the follicular dendritic cells (FDCs), express high levels of the PrPC, although its function in these cells is still uncertain. In fact, it has been shown that PrPC is dispensable for the maturation of FDCs and for maintaining antigen-specific antibody responses [91]. PrPC has also been found in macrophages and its expression is associated with both the inflammatory M1 phenotypes and with the immunosuppressive M2 types. Interestingly, recent studies have demonstrated that PrP0/0 mice produce reduced amounts of the anti-inflammatory cytokine IL-10 in response to systemic lipopolysaccharide, potentially suggesting a role for PrPC in promoting IL-10 production in M2 macrophages [92]. Moreover, again in macrophages, it has been demonstrated that PrPC plays important role in phagocytosis [93, 94]. In particular, Wang and colleagues demonstrated that mouse bone marrow-derived macrophages infected with Escherichia coli express high levels of Prnp mRNA, leading to inefficient phagocytosis. Conversely, macrophages devoid of PrPC internalised bacteria and increased the expression of cytokines such as interleukin-1β, decreasing bacterial proliferation [95]. These data reveal a potentially important role of PrPC as a negative regulator of phagocytosis, phagosome maturation, cytokine expression, and macrophage microbicidal activity. Further studies are required to determine how PrPC regulates vesicular trafficking associated with phagocytosis and cytokine secretion.
PrPC in immune disorders
The role of PrPC in health and homeostatic cell functions is still obscure, but several potential roles have been attributed to this protein in the immune system. Specifically, several studies suggest that PrPC may act as a modulator of innate immune responses in pathologies beyond prion diseases [96–98]. The detailed molecular means by which PrPC modulates immune signalling pathways contributing to immune modulation are not yet clarified and stand out as a necessary area of future research. Interestingly, PrPC is expressed in various organs that, by multiple mechanisms, are relatively protected from inflammation (i.e., immuno-privileged sites) such as the brain, eye, placenta, the pregnant uterus and testes [99, 100]. This high expression in immuno-privileged organs suggest that PrPC has an important protective role under inflammatory stress and/or tissue damage [90, 101]. Accordingly, specific reports have shown that the absence of PrPC increases inflammatory damage in different models of inflammation such as experimental brain ischaemia, brain trauma and experimental autoimmune encephalomyelitis [102]. For example, experimental autoimmune encephalomyelitis, the animal model for human multiple sclerosis, is worsened in mice lacking PrPC. In particular, in the acute stage, the spinal cords, cerebellums and forebrains of Prn-p-deficient mice were shown to be more heavily infiltrated with leucocytes and exhibited stronger proinflammatory cytokine gene expression, as compared with those seen in wild-type mice. Remarkably, the persistence of leucocyte infiltration in the forebrain and cerebellum was accompanied by increased pathogenic cytokines, such as interferon (IFN)-γ and IL-17 [103]. In this particular model, disease exacerbation has been attributed to T cells that would differentiate into more inflammatory (i.e., Th1 and Th17) and behave more aggressively against the CNS effectors, when deprived of PrPC [104]. Thus, based on these results, attenuation of T cell-dependent neuroinflammation may represent a potential novel function of PrPC. In addition to experimental autoimmune encephalomyelitis, PrPC also appears to be protective in autoimmune colitis. Inflammatory bowel disease, induced by dextran sodium sulphate (DSS), is more severe in PrP0/0 mice than in wild-type mice. Accordingly, overexpression of PrPC greatly attenuates DSS-induced colitis [105]. Again, depletion of PrPC was able to skew T cells toward more pronounced Th1 and Th17 inflammatory phenotypes [79]. Based on these data, variations in the human PRNP gene or its sequence [106] might have effects on disease susceptibility or the clinical course of autoimmune diseases; however, these specific studies have not yet been performed. Another interesting observation is that PrPC may act as antimicrobial peptide. It was demonstrated that synthetic peptides derived from the N-terminal region of PrPC are cytotoxic to several bacterial species, including E. coli, Pseudomonas aeruginosa, Bacillus subtilis and Staphylococcus aureus [107]. In addition, in 2013 Ding and co-workers showed that PrPC participates in the regulation of microglial response to Mycobacterium bovis infection, through the upregulation of pro-inflammatory cytokines and the modulation of apoptosis [108]. In particular, they found a significant increase of Prnp mRNA expression upon microglial cell infection with M. bovis, and Prnp silencing did not alter the expression pattern of anti-inflammatory cytokines IL-10 and transforming growth factor (TGF)-β. PrPC was also shown to possess antiviral properties by inhibiting the replication of the human immunodeficiency virus type 1 (HIV-1) and the murine leukaemia virus [109]. In these studies, PrPC was able to bind the viral genomic RNA of HIV-1 negatively affecting its translation. Moreover, PrPC was found to co-localise with the virus assembly machinery at the plasma membrane and at the virological synapse in infected T cells. Depletion of PrPC in infected T cells and microglia favoured HIV-1 replication [109]. Within this conceptual framework, it has been suggested that PrPC may serve two principal roles in immune system: to modulate the inflammatory potential of immune cells, and to protect vulnerable parenchymal cells against noxious insults generated through inflammation. The mechanisms lying behind the role of PrPC and their significance for pathogenesis and its regulatory roles in specific immune disorders require further investigation.
PrPC and cancer
The first hint of a link between PrPC and cancer dates back to the early 2000s when PRNP was identified as one of the 30 genes most overexpressed in pancreatic cancer cell lines as compared with normal cells [110]. At the same time, PrPC was reported to be upregulated in a drug-resistant gastric cancer cell line as compared with the parental cell line [111]. That elevated PrPC may confer resistance to anticancer agents was soon confirmed by Diarra-Mehrpour and colleagues, who demonstrated a causal relationship between increased PrPC expression and resistance to tumour necrosis factor-α (TNFα) in a breast cancer cell line [112]. Thereafter, de Wit and colleagues came across PrPC when screening for cell surface molecules associated with adenoma to carcinoma transition in colon cancer [113]. Following these pioneering findings, further studies have consolidated the involvement of PrPC in four main aspects of cancer biology: proliferation; resistance to anticancer agents; cell migration and invasion; and epithelial to mesenchymal transition. More recently, links between PrPC and cancer stem cells (CSCs) (see below), as well as aneuploidy, were also uncovered [114]. Furthermore, although scarce, studies featuring patients globally point to an association between high PrPC expression and poor prognosis [115–120]. It is now well established that PrPC may sustain cancer cell proliferation in various types of cancers: gastric [121], pancreatic [120] and colon cancer [116, 122, 123], as well as glioblastoma [117, 124] and schwannoma [125]. From a mechanistic point of view, PrPC was shown to promote the recruitment of a PI3 kinase (PI3K)-AKT pathway, itself controlling the transcription of CyclinD1 in gastric cancer cells [121] and to activate the MAP kinases ERK1/2 upon interaction with the STI1 chaperone in glioblastoma [117, 126]. In pancreatic cancer cells, the pro-proliferative action of PrPC appears to involve activation of the Notch pathway [127]. Moreover, the capacity of PrPC to sustain cell proliferation in colon cancer cells may relate to enhanced glucose uptake, as PrPC-dependent signalling leads to transcription of the GLUT1 gene [123]. Overall, the contribution of PrPC to cancer cell proliferation fully fits with a gain of its physiological function in normal cells where it controls the activation of several effectors associated with cell growth [128, 129].
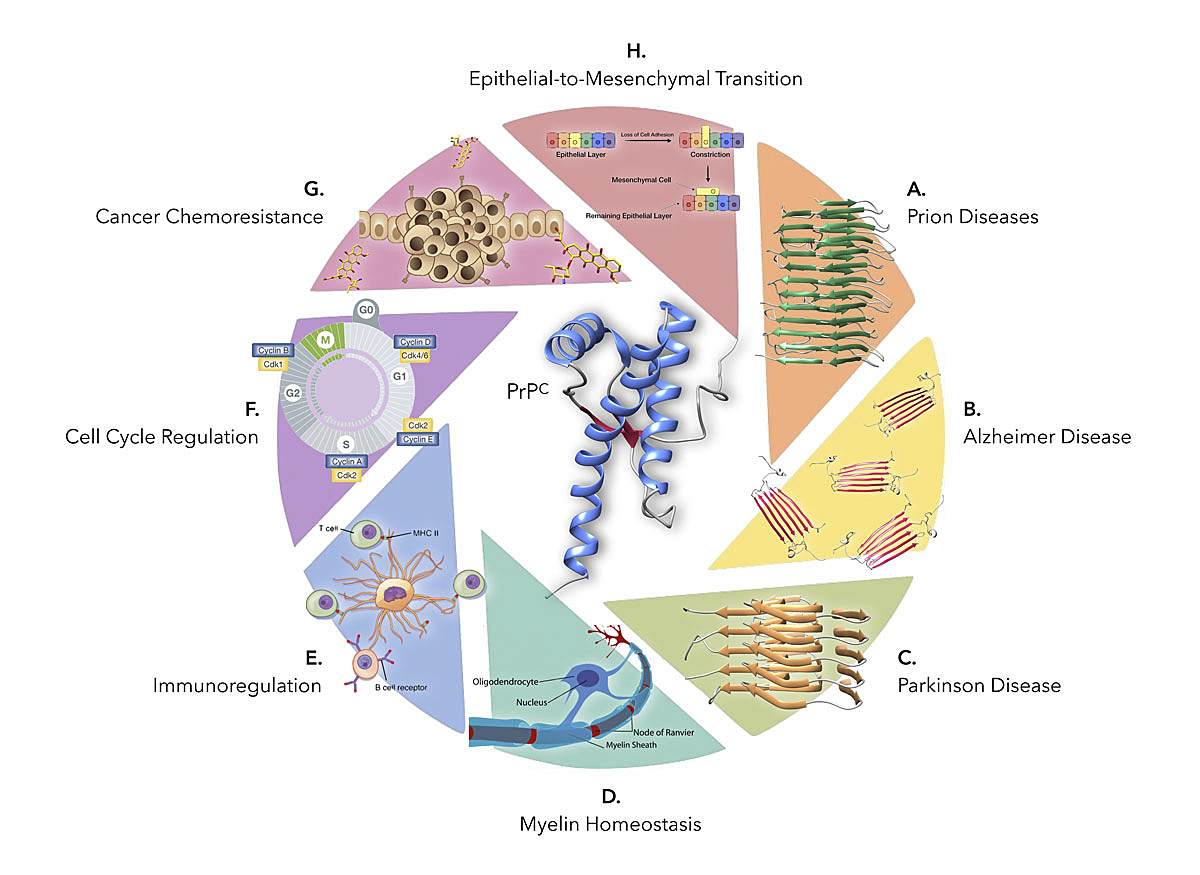
Figure 1
The physiological and pathological processes in which PrPC (PDB 2LFT) may play a role. In addition to serving as a substrate for PrPSc replication (model depicted in A from [149]), the protein has also been reported to act as a receptor for Aβ oligomers (B, PDB 6RHY) and misfolded α-synuclein (C, PDB 2N0A). Surprisingly, PrPC has also been involved in myelin homeostasis (D), immunoregulatory processes (E) and cancer, mainly at the levels of cell cycle regulation (F), drug resistance (G) and epithelial-to-mesenchymal transition (H), among others.
A second field of investigation focuses on the correlation between PrPC and chemo-resistance. High PrPC expression levels is indeed associated with increased resistance to various types of agents in glioblastoma [130], gastric [111, 121, 131, 132], breast [112, 133–135], and colon cancer [122, 136, 137]. According to several studies, the PrPC-PI3K-AKT pathway could contribute to drug resistance by enhancing the expression of MDR1 (multidrug-resistance protein 1) [138]. Very recently, PrPC was found to confer resistance to doxorubicin in breast cancer cells by directly binding and sequestering the drug via its N-terminal domain [119]. Consistently, the authors found a significant correlation between PRNP gene expression levels and resistance to treatment in breast cancer patients, arguing that PRNP monitoring could help stratify patients for adequate therapy. A third process to which PrPC takes part in cancer cells is invasion/migration. Elevated PrPC was shown to confer enhanced migratory and/or invasive properties to glioblastoma [126], gastric [118], breast [133, 139], pancreas [127], colon [140] lung [141] and melanoma [142] cell lines. In pancreatic cancer and melanoma, PrPC, which is present as pro-PrP (an isoform retaining its C-terminus instead of a GPI anchor), appears to exert its pro-migratory action by interacting with filamin A, itself connected with the actin cytoskeleton [142, 143]. In colon cancer cells, this is triggered by the binding of PrPC with its ligand STI1 [140]. Of note, the pro-invasive and pro-migratory role of PrPC extends to the in vivo situation in animal models. Indeed, Du and colleagues found that among colon primary tumour cells, only those positive for PrPC were able to promote liver metastasis after injection in the caecal wall of immunocompromised mice [115]. Whether this holds true for other types of cancer remains to be investigated.
Metastatic dissemination is highly correlated with epithelial‐to‐mesenchymal transition (EMT), a process whereby cells lose epithelial markers and cell-cell and cell-matrix contacts, remodel their actin cytoskeleton and acquire mesenchymal hallmarks, favouring cell migration [144]. At a molecular level, EMT induction is controlled by various transcription factors, including ZEB1, ZEB2, SNAIL, SLUG and TWIST [144]. The expression of PRNP is highly associated with and EMT signature in colon cancer patients, and PrPC controls the expression of ZEB1 in colon cancer cells [116]. Interestingly, EMT appears to be intimately connected with CSC properties [145]. Accordingly, Du and colleagues documented that PrPC-positive primary colon cancer cells express high levels of the EMT-associated markers TWIST and N-cadherin and low levels of the epithelial marker E-cadherin and exhibit CSC properties such as expression of the CSC marker CD44 and tumour-initiating capacity [115]. In line with this, PrPC was shown to interact with CD44 in multi-resistant breast cancer cells [133]. Furthermore, in primary glioblastoma cells, PrPC silencing reduces the expression of the CSC markers SOX2 and NANOG, as well as self-renewal and tumorigenic potential [124]. Similar findings were obtained by Iglesia et al working on glioblastoma cell lines grown as neurospheres [126]. As with proliferation, the contribution of PrPC to CSC self-renewal may be envisioned as a diversion of its physiological role in normal stem cell maintenance [146]. Collectively, the involvement of PrPC in various aspects of cancer progression may be viewed as directly related to its physiological role in normal cells. From a therapeutic perspective, reducing PrPC expression through antisense oligonucleotide-based strategies [147] may prove beneficial, as documented for glioblastoma [148] or colon cancer [115]. Besides, alternative opportunities may ensue from a better knowledge of the signals upregulating PrPC expression in cancer cells.
Conclusions
After more than three decades of intense research across numerous research laboratories around the planet there is still much to learn about the biology of PrPC. A large amount of data provides solid experimental support for the notion that the simple accumulation of PrPSc in nerve tissues may not explain the whole spectrum of neurotoxic events occurring in prion diseases, which instead is likely to require some poorly understood subversion of PrPC function upon binding to PrPSc. Such a role for corruption of PrPC as a mediator of prion toxicity has received unexpected support from research in other neurodegenerative disorders, showing that PrPC can bind disease-associated misfolded proteins, such as oligomers of Aβ and alpha-synuclein. Research in even more distant fields of biology supports expanded and surprising roles for PrPC in several physiological and disease contexts outside the brain, such as myelin homeostasis, immunoregulatory processes and cancer (figure 1). These approaches, which might not appear directly relevant to prion biology and patho-biology, are nevertheless laying the groundwork for a more comprehensive understanding of the physiological function(s) of PrPC, and the likelihood of achieving novel insights that could elucidate some fundamental cytotoxic mechanisms potentially shared by prion disorders and several other diseases.
References
1
Chiti
F
,
Dobson
CM
. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem. 2017;86(1):27–68. doi:.https://doi.org/10.1146/annurev-biochem-061516-045115
2
Powell
T
. Health Policy and Dementia. Curr Psychiatry Rep. 2018;20(1):4. doi:.https://doi.org/10.1007/s11920-018-0868-0
3
Selkoe
DJ
,
Hardy
J
. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi:.https://doi.org/10.15252/emmm.201606210
4
Walsh
DM
,
Klyubin
I
,
Fadeeva
JV
,
Cullen
WK
,
Anwyl
R
,
Wolfe
MS
, et al.
Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9. doi:.https://doi.org/10.1038/416535a
5
Laurén
J
,
Gimbel
DA
,
Nygaard
HB
,
Gilbert
JW
,
Strittmatter
SM
. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457(7233):1128–32. doi:.https://doi.org/10.1038/nature07761
6
Prusiner
SB
. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–83. doi:.https://doi.org/10.1073/pnas.95.23.13363
7
Aguzzi
A
,
Heikenwalder
M
,
Polymenidou
M
. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8(7):552–61. doi:.https://doi.org/10.1038/nrm2204
8
Brandner
S
,
Isenmann
S
,
Raeber
A
,
Fischer
M
,
Sailer
A
,
Kobayashi
Y
, et al.
Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379(6563):339–43. doi:.https://doi.org/10.1038/379339a0
9
Brandner
S
,
Raeber
A
,
Sailer
A
,
Blättler
T
,
Fischer
M
,
Weissmann
C
, et al.
Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci USA. 1996;93(23):13148–51. doi:.https://doi.org/10.1073/pnas.93.23.13148
10
Mallucci
G
,
Dickinson
A
,
Linehan
J
,
Klöhn
PC
,
Brandner
S
,
Collinge
J
. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–4. doi:.https://doi.org/10.1126/science.1090187
11
Aulić
S
,
Masperone
L
,
Narkiewicz
J
,
Isopi
E
,
Bistaffa
E
,
Ambrosetti
E
, et al.
α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci Rep. 2017;7(1):10050. doi:.https://doi.org/10.1038/s41598-017-10236-x
12
Ferreira
DG
,
Temido-Ferreira
M
,
Vicente Miranda
H
,
Batalha
VL
,
Coelho
JE
,
Szegö
ÉM
, et al.
α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017;20(11):1569–79. doi:.https://doi.org/10.1038/nn.4648
13
Resenberger
UK
,
Winklhofer
KF
,
Tatzelt
J
. Cellular prion protein mediates toxic signaling of amyloid beta. Neurodegener Dis. 2012;10(1-4):298–300. doi:.https://doi.org/10.1159/000332596
14
Corbett
GT
,
Wang
Z
,
Hong
W
,
Colom-Cadena
M
,
Rose
J
,
Liao
M
, et al.
PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020;139(3):503–26. doi:.https://doi.org/10.1007/s00401-019-02114-9
15
Biasini
E
,
Turnbaugh
JA
,
Unterberger
U
,
Harris
DA
. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012;35(2):92–103. doi:.https://doi.org/10.1016/j.tins.2011.10.002
16
Linden
R
. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front Mol Neurosci. 2017;10:77. doi:.https://doi.org/10.3389/fnmol.2017.00077
17
Nuvolone
M
,
Hermann
M
,
Sorce
S
,
Russo
G
,
Tiberi
C
,
Schwarz
P
, et al.
Strictly co-isogenic C57BL/6J-Prnp-/- mice: A rigorous resource for prion science. J Exp Med. 2016;213(3):313–27. doi:.https://doi.org/10.1084/jem.20151610
18
Wulf
MA
,
Senatore
A
,
Aguzzi
A
. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15(1):34. doi:.https://doi.org/10.1186/s12915-017-0375-5
19
Küffer
A
,
Lakkaraju
AK
,
Mogha
A
,
Petersen
SC
,
Airich
K
,
Doucerain
C
, et al.
The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536(7617):464–8. doi:.https://doi.org/10.1038/nature19312
20
Mabbott
NA
. Immunology of Prion Protein and Prions. Prog Mol Biol Transl Sci. 2017;150:203–40. doi:.https://doi.org/10.1016/bs.pmbts.2017.06.004
21
Hirsch
TZ
,
Martin-Lannerée
S
,
Mouillet-Richard
S
. Functions of the Prion Protein. Prog Mol Biol Transl Sci. 2017;150:1–34. doi:.https://doi.org/10.1016/bs.pmbts.2017.06.001
22
Chesebro
B
,
Race
R
,
Wehrly
K
,
Nishio
J
,
Bloom
M
,
Lechner
D
, et al.
Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature. 1985;315(6017):331–3. doi:.https://doi.org/10.1038/315331a0
23
Prusiner
SB
. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–44. doi:.https://doi.org/10.1126/science.6801762
24
Parkin
ET
,
Watt
NT
,
Hussain
I
,
Eckman
EA
,
Eckman
CB
,
Manson
JC
, et al.
Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc Natl Acad Sci USA. 2007;104(26):11062–7. doi:.https://doi.org/10.1073/pnas.0609621104
25
Griffiths
HH
,
Whitehouse
IJ
,
Baybutt
H
,
Brown
D
,
Kellett
KA
,
Jackson
CD
, et al.
Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J Biol Chem. 2011;286(38):33489–500. doi:.https://doi.org/10.1074/jbc.M111.278556
26
Whitehouse
IJ
,
Miners
JS
,
Glennon
EB
,
Kehoe
PG
,
Love
S
,
Kellett
KA
, et al.
Prion protein is decreased in Alzheimer’s brain and inversely correlates with BACE1 activity, amyloid-β levels and Braak stage. PLoS One. 2013;8(4):e59554. doi:.https://doi.org/10.1371/journal.pone.0059554
27
Whitehouse
IJ
,
Brown
D
,
Baybutt
H
,
Diack
AB
,
Kellett
KA
,
Piccardo
P
, et al.
Ablation of Prion Protein in Wild Type Human Amyloid Precursor Protein (APP) Transgenic Mice Does Not Alter The Proteolysis of APP, Levels of Amyloid-β or Pathologic Phenotype. PLoS One. 2016;11(7):e0159119. doi:.https://doi.org/10.1371/journal.pone.0159119
28
Vincent
B
,
Sunyach
C
,
Orzechowski
HD
,
St George-Hyslop
P
,
Checler
F
. p53-Dependent transcriptional control of cellular prion by presenilins. J Neurosci. 2009;29(20):6752–60. doi:.https://doi.org/10.1523/JNEUROSCI.0789-09.2009
29
Lewis
V
,
Whitehouse
IJ
,
Baybutt
H
,
Manson
JC
,
Collins
SJ
,
Hooper
NM
. Cellular prion protein expression is not regulated by the Alzheimer’s amyloid precursor protein intracellular domain. PLoS One. 2012;7(2):e31754. doi:.https://doi.org/10.1371/journal.pone.0031754
30
Rial
D
,
Piermartiri
TC
,
Duarte
FS
,
Tasca
CI
,
Walz
R
,
Prediger
RD
. Overexpression of cellular prion protein (PrPC) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-β (Aβ1–40) administration in mice. Neuroscience. 2012;215:79–89. doi:.https://doi.org/10.1016/j.neuroscience.2012.04.034
31
Khosravani
H
,
Zhang
Y
,
Tsutsui
S
,
Hameed
S
,
Altier
C
,
Hamid
J
, et al.
Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Gen Physiol. 2008;131(6):i5. doi:.https://doi.org/10.1085/JGP1316OIA5
32
Khosravani
H
,
Zhang
Y
,
Zamponi
GW
. Cellular prion protein null mice display normal AMPA receptor mediated long term depression. Prion. 2008;2(2):48–50. doi:.https://doi.org/10.4161/pri.2.2.6628
33
You
H
,
Tsutsui
S
,
Hameed
S
,
Kannanayakal
TJ
,
Chen
L
,
Xia
P
, et al.
Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci USA. 2012;109(5):1737–42. doi:.https://doi.org/10.1073/pnas.1110789109
34
Younan
ND
,
Sarell
CJ
,
Davies
P
,
Brown
DR
,
Viles
JH
. The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013;27(5):1847–58. doi:.https://doi.org/10.1096/fj.12-222588
35
Balducci
C
,
Beeg
M
,
Stravalaci
M
,
Bastone
A
,
Sclip
A
,
Biasini
E
, et al.
Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA. 2010;107(5):2295–300. doi:.https://doi.org/10.1073/pnas.0911829107
36
Kessels
HW
,
Nguyen
LN
,
Nabavi
S
,
Malinow
R
. The prion protein as a receptor for amyloid-beta. Nature. 2010;466(7308):E3–4, discussion E4–5. doi:.https://doi.org/10.1038/nature09217
37
Calella
AM
,
Farinelli
M
,
Nuvolone
M
,
Mirante
O
,
Moos
R
,
Falsig
J
, et al.
Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2(8):306–14. doi:.https://doi.org/10.1002/emmm.201000082
38
Caetano
FA
,
Beraldo
FH
,
Hajj
GN
,
Guimaraes
AL
,
Jürgensen
S
,
Wasilewska-Sampaio
AP
, et al.
Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem. 2011;117(3):538–53. doi:.https://doi.org/10.1111/j.1471-4159.2011.07225.x
39
Larson
M
,
Sherman
MA
,
Amar
F
,
Nuvolone
M
,
Schneider
JA
,
Bennett
DA
, et al.
The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci. 2012;32(47):16857–71. doi:.https://doi.org/10.1523/JNEUROSCI.1858-12.2012
40
Um
JW
,
Nygaard
HB
,
Heiss
JK
,
Kostylev
MA
,
Stagi
M
,
Vortmeyer
A
, et al.
Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15(9):1227–35. doi:.https://doi.org/10.1038/nn.3178
41
Um
JW
,
Strittmatter
SM
. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013;7(1):37–41. doi:.https://doi.org/10.4161/pri.22212
42
Fluharty
BR
,
Biasini
E
,
Stravalaci
M
,
Sclip
A
,
Diomede
L
,
Balducci
C
, et al.
An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J Biol Chem. 2013;288(11):7857–66. doi:.https://doi.org/10.1074/jbc.M112.423954
43
Gimbel
DA
,
Nygaard
HB
,
Coffey
EE
,
Gunther
EC
,
Laurén
J
,
Gimbel
ZA
, et al.
Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30(18):6367–74. doi:.https://doi.org/10.1523/JNEUROSCI.0395-10.2010
44
Kudo
W
,
Lee
HP
,
Zou
WQ
,
Wang
X
,
Perry
G
,
Zhu
X
, et al.
Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet. 2012;21(5):1138–44. doi:.https://doi.org/10.1093/hmg/ddr542
45
Barry
AE
,
Klyubin
I
,
Mc Donald
JM
,
Mably
AJ
,
Farrell
MA
,
Scott
M
, et al.
Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31(20):7259–63. doi:.https://doi.org/10.1523/JNEUROSCI.6500-10.2011
46
Chung
E
,
Ji
Y
,
Sun
Y
,
Kascsak
RJ
,
Kascsak
RB
,
Mehta
PD
, et al.
Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010;11(1):130. doi:.https://doi.org/10.1186/1471-2202-11-130
47
Cox
TO
,
Gunther
EC
,
Brody
AH
,
Chiasseu
MT
,
Stoner
A
,
Smith
LM
, et al.
Anti-PrPC antibody rescues cognition and synapses in transgenic alzheimer mice. Ann Clin Transl Neurol. 2019;6(3):554–74. doi:.https://doi.org/10.1002/acn3.730
48
Freir
DB
,
Nicoll
AJ
,
Klyubin
I
,
Panico
S
,
Mc Donald
JM
,
Risse
E
, et al.
Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2(1):336. doi:.https://doi.org/10.1038/ncomms1341
49
Urrea
L
,
Segura-Feliu
M
,
Masuda-Suzukake
M
,
Hervera
A
,
Pedraz
L
,
García-Aznar
JM
, et al.
Involvement of Cellular Prion Protein in α-Synuclein Transport in Neurons. Mol Neurobiol. 2018;55(3):1847–60. doi:.https://doi.org/10.1007/s12035-017-0451-4
50
La Vitola
P
,
Beeg
M
,
Balducci
C
,
Santamaria
G
,
Restelli
E
,
Colombo
L
, et al.
Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain. 2019;142(2):249–54. doi:.https://doi.org/10.1093/brain/awy318
51
Linden
R
,
Martins
VR
,
Prado
MA
,
Cammarota
M
,
Izquierdo
I
,
Brentani
RR
. Physiology of the prion protein. Physiol Rev. 2008;88(2):673–728. doi:.https://doi.org/10.1152/physrev.00007.2007
52
Aguzzi
A
. Prions and the immune system: a journey through gut, spleen, and nerves. Adv Immunol. 2003;81:123–71. doi:.https://doi.org/10.1016/S0065-2776(03)81004-0
53
McKinley
MP
,
Bolton
DC
,
Prusiner
SB
. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35(1):57–62. doi:.https://doi.org/10.1016/0092-8674(83)90207-6
54
Stahl
N
,
Baldwin
MA
,
Teplow
DB
,
Hood
L
,
Gibson
BW
,
Burlingame
AL
, et al.
Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32(8):1991–2002. doi:.https://doi.org/10.1021/bi00059a016
55
Mabbott
NA
,
Alibhai
JD
,
Manson
J
. The role of the immune system in prion infection. Handb Clin Neurol. 2018;153:85–107. doi:.https://doi.org/10.1016/B978-0-444-63945-5.00005-2
56
Elhelaly
AE
,
Inoshima
Y
,
Ishiguro
N
. Characterization of early transient accumulation of PrP(Sc) in immune cells. Biochem Biophys Res Commun. 2013;439(3):340–5. doi:.https://doi.org/10.1016/j.bbrc.2013.08.085
57
Aguzzi
A
,
Sigurdson
CJ
. Antiprion immunotherapy: to suppress or to stimulate?
Nat Rev Immunol. 2004;4(9):725–36. doi:.https://doi.org/10.1038/nri1437
58
Buchholz
CJ
,
Bach
P
,
Nikles
D
,
Kalinke
U
. Prion protein-specific antibodies for therapeutic intervention of transmissible spongiform encephalopathies. Expert Opin Biol Ther. 2006;6(3):293–300. doi:.https://doi.org/10.1517/14712598.6.3.293
59
Wisniewski
T
,
Chabalgoity
JA
,
Goni
F
. ¿Es factible la vacunación contra la encefalopatía espongiforme transmisible? [Is vaccination against transmissible spongiform encephalopathy feasible?]
Rev Sci Tech. 2007;26(1):243–51. doi:.https://doi.org/10.20506/rst.26.1.1739
60
Wisniewski
T
,
Goñi
F
. Immunomodulation for prion and prion-related diseases. Expert Rev Vaccines. 2010;9(12):1441–52. doi:.https://doi.org/10.1586/erv.10.131
61
Wisniewski
T
,
Goñi
F
. Could immunomodulation be used to prevent prion diseases?
Expert Rev Anti Infect Ther. 2012;10(3):307–17. doi:.https://doi.org/10.1586/eri.11.177
62
Bachy
V
,
Ballerini
C
,
Gourdain
P
,
Prignon
A
,
Iken
S
,
Antoine
N
, et al.
Mouse vaccination with dendritic cells loaded with prion protein peptides overcomes tolerance and delays scrapie. J Gen Virol. 2010;91(Pt 3):809–20. doi:.https://doi.org/10.1099/vir.0.013417-0
63
Gregoire
S
,
Logre
C
,
Metharom
P
,
Loing
E
,
Chomilier
J
,
Rosset
MB
, et al.
Identification of two immunogenic domains of the prion protein--PrP--which activate class II-restricted T cells and elicit antibody responses against the native molecule. J Leukoc Biol. 2004;76(1):125–34. doi:.https://doi.org/10.1189/jlb.1203656
64
Carnaud
C
,
Bachy
V
. Cell-based immunotherapy of prion diseases by adoptive transfer of antigen-loaded dendritic cells or antigen-primed CD(4+) T lymphocytes. Prion. 2010;4(2):66–71. doi:.https://doi.org/10.4161/pri.4.2.12597
65
Málaga-Trillo
E
,
Sempou
E
. PrPs: Proteins with a purpose: Lessons from the zebrafish. Prion. 2009;3(3):129–33. doi:.https://doi.org/10.4161/pri.3.3.9651
66
Málaga-Trillo
E
,
Solis
GP
,
Schrock
Y
,
Geiss
C
,
Luncz
L
,
Thomanetz
V
, et al.
Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7(3):e55. doi:.https://doi.org/10.1371/journal.pbio.1000055
67
Solis
GP
,
Malaga-Trillo
E
,
Plattner
H
,
Stuermer
CA
. Cellular roles of the prion protein in association with reggie/flotillin microdomains. Front Biosci. 2010;15(1):1075–85. doi:.https://doi.org/10.2741/3662
68
Brown
HR
,
Goller
NL
,
Rudelli
RD
,
Merz
GS
,
Wolfe
GC
,
Wisniewski
HM
, et al.
The mRNA encoding the scrapie agent protein is present in a variety of non-neuronal cells. Acta Neuropathol. 1990;80(1):1–6. doi:.https://doi.org/10.1007/BF00294214
69
Burthem
J
,
Urban
B
,
Pain
A
,
Roberts
DJ
. The normal cellular prion protein is strongly expressed by myeloid dendritic cells. Blood. 2001;98(13):3733–8. doi:.https://doi.org/10.1182/blood.V98.13.3733
70
Aguzzi
A
,
Nuvolone
M
,
Zhu
C
. The immunobiology of prion diseases. Nat Rev Immunol. 2013;13(12):888–902. doi:.https://doi.org/10.1038/nri3553
71
Brown
KL
,
Stewart
K
,
Ritchie
DL
,
Mabbott
NA
,
Williams
A
,
Fraser
H
, et al.
Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat Med. 1999;5(11):1308–12. doi:.https://doi.org/10.1038/15264
72
Dodelet
VC
,
Cashman
NR
. Prion protein expression in human leukocyte differentiation. Blood. 1998;91(5):1556–61. doi:.https://doi.org/10.1182/blood.V91.5.1556
73
Holada
K
,
Vostal
JG
. Different levels of prion protein (PrPc) expression on hamster, mouse and human blood cells. Br J Haematol. 2000;110(2):472–80. doi:.https://doi.org/10.1046/j.1365-2141.2000.02158.x
74
Kubosaki
A
,
Yusa
S
,
Nasu
Y
,
Nishimura
T
,
Nakamura
Y
,
Saeki
K
, et al.
Distribution of cellular isoform of prion protein in T lymphocytes and bone marrow, analyzed by wild-type and prion protein gene-deficient mice. Biochem Biophys Res Commun. 2001;282(1):103–7. doi:.https://doi.org/10.1006/bbrc.2001.4538
75
Li
R
,
Liu
D
,
Zanusso
G
,
Liu
T
,
Fayen
JD
,
Huang
JH
, et al.
The expression and potential function of cellular prion protein in human lymphocytes. Cell Immunol. 2001;207(1):49–58. doi:.https://doi.org/10.1006/cimm.2000.1751
76
Dürig
J
,
Giese
A
,
Schulz-Schaeffer
W
,
Rosenthal
C
,
Schmücker
U
,
Bieschke
J
, et al.
Differential constitutive and activation-dependent expression of prion protein in human peripheral blood leucocytes. Br J Haematol. 2000;108(3):488–95. doi:.https://doi.org/10.1046/j.1365-2141.2000.01881.x
77
Isaacs
JD
,
Jackson
GS
,
Altmann
DM
. The role of the cellular prion protein in the immune system. Clin Exp Immunol. 2006;146(1):1–8. doi:.https://doi.org/10.1111/j.1365-2249.2006.03194.x
78
Hugel
B
,
Martínez
MC
,
Kunzelmann
C
,
Blättler
T
,
Aguzzi
A
,
Freyssinet
JM
. Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell Mol Life Sci. 2004;61(23):2998–3007. doi:.https://doi.org/10.1007/s00018-004-4318-2
79
Hu
W
,
Nessler
S
,
Hemmer
B
,
Eagar
TN
,
Kane
LP
,
Leliveld
SR
, et al.
Pharmacological prion protein silencing accelerates central nervous system autoimmune disease via T cell receptor signalling. Brain. 2010;133(Pt 2):375–88. doi:.https://doi.org/10.1093/brain/awp298
80
Politopoulou
G
,
Seebach
JD
,
Schmugge
M
,
Schwarz
HP
,
Aguzzi
A
. Age-related expression of the cellular prion protein in human peripheral blood leukocytes. Haematologica. 2000;85(6):580–7.
81
Cashman
NR
,
Loertscher
R
,
Nalbantoglu
J
,
Shaw
I
,
Kascsak
RJ
,
Bolton
DC
, et al.
Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell. 1990;61(1):185–92. doi:.https://doi.org/10.1016/0092-8674(90)90225-4
82
Huehn
J
,
Siegmund
K
,
Lehmann
JC
,
Siewert
C
,
Haubold
U
,
Feuerer
M
, et al.
Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199(3):303–13. doi:.https://doi.org/10.1084/jem.20031562
83
Chen
Z
,
Lund
R
,
Aittokallio
T
,
Kosonen
M
,
Nevalainen
O
,
Lahesmaa
R
. Identification of novel IL-4/Stat6-regulated genes in T lymphocytes. J Immunol. 2003;171(7):3627–35. doi:.https://doi.org/10.4049/jimmunol.171.7.3627
84
Goldrath
AW
,
Luckey
CJ
,
Park
R
,
Benoist
C
,
Mathis
D
. The molecular program induced in T cells undergoing homeostatic proliferation. Proc Natl Acad Sci USA. 2004;101(48):16885–90. doi:.https://doi.org/10.1073/pnas.0407417101
85
Isaacs
JD
,
Garden
OA
,
Kaur
G
,
Collinge
J
,
Jackson
GS
,
Altmann
DM
. The cellular prion protein is preferentially expressed by CD4+ CD25+ Foxp3+ regulatory T cells. Immunology. 2008;125(3):313–9. doi:.https://doi.org/10.1111/j.1365-2567.2008.02853.x
86
Ford
MJ
,
Burton
LJ
,
Morris
RJ
,
Hall
SM
. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience. 2002;113(1):177–92. doi:.https://doi.org/10.1016/S0306-4522(02)00155-0
87
Durai
V
,
Murphy
KM
. Functions of Murine Dendritic Cells. Immunity. 2016;45(4):719–36. doi:.https://doi.org/10.1016/j.immuni.2016.10.010
88
del Hoyo
GM
,
Martín
P
,
Vargas
HH
,
Ruiz
S
,
Arias
CF
,
Ardavín
C
. Characterization of a common precursor population for dendritic cells. Nature. 2002;415(6875):1043–7. doi:.https://doi.org/10.1038/4151043a
89
Ballerini
C
,
Gourdain
P
,
Bachy
V
,
Blanchard
N
,
Levavasseur
E
,
Grégoire
S
, et al.
Functional implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J Immunol. 2006;176(12):7254–62. doi:.https://doi.org/10.4049/jimmunol.176.12.7254
90
Bakkebø
MK
,
Mouillet-Richard
S
,
Espenes
A
,
Goldmann
W
,
Tatzelt
J
,
Tranulis
MA
. The Cellular Prion Protein: A Player in Immunological Quiescence. Front Immunol. 2015;6:450. doi:.https://doi.org/10.3389/fimmu.2015.00450
91
McCulloch
L
,
Brown
KL
,
Mabbott
NA
. Ablation of the cellular prion protein, PrPC, specifically on follicular dendritic cells has no effect on their maturation or function. Immunology. 2013;138(3):246–57. doi:.https://doi.org/10.1111/imm.12031
92
Liu
J
,
Zhao
D
,
Liu
C
,
Ding
T
,
Yang
L
,
Yin
X
, et al.
Prion protein participates in the protection of mice from lipopolysaccharide infection by regulating the inflammatory process. J Mol Neurosci. 2015;55(1):279–87. doi:.https://doi.org/10.1007/s12031-014-0319-2
93
de Almeida
CJ
,
Chiarini
LB
,
da Silva
JP
,
E Silva
PM
,
Martins
MA
,
Linden
R
. The cellular prion protein modulates phagocytosis and inflammatory response. J Leukoc Biol. 2005;77(2):238–46. doi:.https://doi.org/10.1189/jlb.1103531
94
Uraki
R
,
Sakudo
A
,
Ando
S
,
Kitani
H
,
Onodera
T
. Enhancement of phagocytotic activity by prion protein in PrP-deficient macrophage cells. Int J Mol Med. 2010;26(4):527–32.
95
Wang
M
,
Zhao
D
,
Yang
Y
,
Liu
J
,
Wang
J
,
Yin
X
, et al.
The cellular prion protein negatively regulates phagocytosis and cytokine expression in murine bone marrow-derived macrophages. PLoS One. 2014;9(7):e102785. doi:.https://doi.org/10.1371/journal.pone.0102785
96
McBride
SM
. Prion protein: a pattern recognition receptor for viral components and uric acid responsible for the induction of innate and adaptive immunity. Med Hypotheses. 2005;65(3):570–7. doi:.https://doi.org/10.1016/j.mehy.2005.02.038
97
Obst
J
,
Simon
E
,
Mancuso
R
,
Gomez-Nicola
D
. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front Aging Neurosci. 2017;9:207. doi:.https://doi.org/10.3389/fnagi.2017.00207
98
Dervishi
E
,
Lam
TH
,
Dunn
SM
,
Zwierzchowski
G
,
Saleem
F
,
Wishart
DS
, et al.
Recombinant mouse prion protein alone or in combination with lipopolysaccharide alters expression of innate immunity genes in the colon of mice. Prion. 2015;9(1):59–73. doi:.https://doi.org/10.1080/19336896.2015.1019694
99
Johnson
ML
,
Grazul-Bilska
AT
,
Reynolds
LP
,
Redmer
DA
. Prion (PrPC) expression in ovine uteroplacental tissues increases after estrogen treatment of ovariectomized ewes and during early pregnancy. Reproduction. 2014;148(1):1–10. doi:.https://doi.org/10.1530/REP-13-0548
100
Tanji
K
,
Saekia
K
,
Matsumoto
Y
,
Takeda
M
,
Hirasawa
K
,
Doi
K
, et al.
Analysis of PrPc mRNA by in situ hybridization in brain, placenta, uterus and testis of rats. Intervirology. 1995;38(6):309–15. doi:.https://doi.org/10.1159/000150457
101
Salvesen
Ø
,
Reiten
MR
,
Espenes
A
,
Bakkebø
MK
,
Tranulis
MA
,
Ersdal
C
. LPS-induced systemic inflammation reveals an immunomodulatory role for the prion protein at the blood-brain interface. J Neuroinflammation. 2017;14(1):106. doi:.https://doi.org/10.1186/s12974-017-0879-5
102
Onodera
T
,
Sakudo
A
,
Tsubone
H
,
Itohara
S
. Review of studies that have used knockout mice to assess normal function of prion protein under immunological or pathophysiological stress. Microbiol Immunol. 2014;58(7):361–74. doi:.https://doi.org/10.1111/1348-0421.12162
103
Tsutsui
S
,
Hahn
JN
,
Johnson
TA
,
Ali
Z
,
Jirik
FR
. Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am J Pathol. 2008;173(4):1029–41. doi:.https://doi.org/10.2353/ajpath.2008.071062
104
Gourdain
P
,
Ballerini
C
,
Nicot
AB
,
Carnaud
C
. Exacerbation of experimental autoimmune encephalomyelitis in prion protein (PrPc)-null mice: evidence for a critical role of the central nervous system. J Neuroinflammation. 2012;9(1):25. doi:.https://doi.org/10.1186/1742-2094-9-25
105
Martin
GR
,
Keenan
CM
,
Sharkey
KA
,
Jirik
FR
. Endogenous prion protein attenuates experimentally induced colitis. Am J Pathol. 2011;179(5):2290–301. doi:.https://doi.org/10.1016/j.ajpath.2011.07.025
106
Bishop
MT
,
Pennington
C
,
Heath
CA
,
Will
RG
,
Knight
RS
. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet. 2009;10(1):146. doi:.https://doi.org/10.1186/1471-2350-10-146
107
Pasupuleti
M
,
Roupe
M
,
Rydengård
V
,
Surewicz
K
,
Surewicz
WK
,
Chalupka
A
, et al.
Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. 2009;4(10):e7358. doi:.https://doi.org/10.1371/journal.pone.0007358
108
Ding
T
,
Zhou
X
,
Kouadir
M
,
Shi
F
,
Yang
Y
,
Liu
J
, et al.
Cellular prion protein participates in the regulation of inflammatory response and apoptosis in BV2 microglia during infection with Mycobacterium bovis. J Mol Neurosci. 2013;51(1):118–26. doi:.https://doi.org/10.1007/s12031-013-9962-2
109
Alais
S
,
Soto-Rifo
R
,
Balter
V
,
Gruffat
H
,
Manet
E
,
Schaeffer
L
, et al.
Functional mechanisms of the cellular prion protein (PrP(C)) associated anti-HIV-1 properties. Cell Mol Life Sci. 2012;69(8):1331–52. doi:.https://doi.org/10.1007/s00018-011-0879-z
110
Han
H
,
Bearss
DJ
,
Browne
LW
,
Calaluce
R
,
Nagle
RB
,
Von Hoff
DD
. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002;62(10):2890–6.
111
Zhao
Y
,
You
H
,
Liu
F
,
An
H
,
Shi
Y
,
Yu
Q
, et al.
Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett. 2002;185(2):211–8. doi:.https://doi.org/10.1016/S0304-3835(02)00264-1
112
Diarra-Mehrpour
M
,
Arrabal
S
,
Jalil
A
,
Pinson
X
,
Gaudin
C
,
Piétu
G
, et al.
Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64(2):719–27. doi:.https://doi.org/10.1158/0008-5472.CAN-03-1735
113
de Wit
M
,
Jimenez
CR
,
Carvalho
B
,
Belien
JA
,
Delis-van Diemen
PM
,
Mongera
S
, et al.
Cell surface proteomics identifies glucose transporter type 1 and prion protein as candidate biomarkers for colorectal adenoma-to-carcinoma progression. Gut. 2012;61(6):855–64. doi:.https://doi.org/10.1136/gutjnl-2011-300511
114
Domingues
PH
,
Nanduri
LSY
,
Seget
K
,
Venkateswaran
SV
,
Agorku
D
,
Viganó
C
, et al.
Cellular Prion Protein PrPC and Ecto-5′-Nucleotidase Are Markers of the Cellular Stress Response to Aneuploidy. Cancer Res. 2017;77(11):2914–26. doi:.https://doi.org/10.1158/0008-5472.CAN-16-3052
115
Du
L
,
Rao
G
,
Wang
H
,
Li
B
,
Tian
W
,
Cui
J
, et al.
CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013;73(8):2682–94. doi:.https://doi.org/10.1158/0008-5472.CAN-12-3759
116
Le Corre
D
,
Ghazi
A
,
Balogoun
R
,
Pilati
C
,
Aparicio
T
,
Martin-Lannerée
S
, et al.
The cellular prion protein controls the mesenchymal-like molecular subtype and predicts disease outcome in colorectal cancer. EBioMedicine. 2019;46:94–104. doi:.https://doi.org/10.1016/j.ebiom.2019.07.036
117
Lopes
MH
,
Santos
TG
,
Rodrigues
BR
,
Queiroz-Hazarbassanov
N
,
Cunha
IW
,
Wasilewska-Sampaio
AP
, et al.
Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene. 2015;34(25):3305–14. doi:.https://doi.org/10.1038/onc.2014.261
118
Pan
Y
,
Zhao
L
,
Liang
J
,
Liu
J
,
Shi
Y
,
Liu
N
, et al.
Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006;20(11):1886–8. doi:.https://doi.org/10.1096/fj.06-6138fje
119
Wiegmans
AP
,
Saunus
JM
,
Ham
S
,
Lobb
R
,
Kutasovic
JR
,
Dalley
AJ
, et al.
Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight. 2019;5:5. doi:.https://doi.org/10.1172/jci.insight.124092
120
Li
QQ
,
Cao
XX
,
Xu
JD
,
Chen
Q
,
Wang
WJ
,
Tang
F
, et al.
The role of P-glycoprotein/cellular prion protein interaction in multidrug-resistant breast cancer cells treated with paclitaxel. Cell Mol Life Sci. 2009;66(3):504–15. doi:.https://doi.org/10.1007/s00018-008-8548-6
121
Liang
J
,
Luo
G
,
Ning
X
,
Shi
Y
,
Zhai
H
,
Sun
S
, et al.
Differential expression of calcium-related genes in gastric cancer cells transfected with cellular prion protein. Biochem Cell Biol. 2007;85(3):375–83. doi:.https://doi.org/10.1139/O07-052
122
Chieng
CK
,
Say
YH
. Cellular prion protein contributes to LS 174T colon cancer cell carcinogenesis by increasing invasiveness and resistance against doxorubicin-induced apoptosis. Tumour Biol. 2015;36(10):8107–20. doi:.https://doi.org/10.1007/s13277-015-3530-z
123
Li
QQ
,
Sun
YP
,
Ruan
CP
,
Xu
XY
,
Ge
JH
,
He
J
, et al.
Cellular prion protein promotes glucose uptake through the Fyn-HIF-2α-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci. 2011;102(2):400–6. doi:.https://doi.org/10.1111/j.1349-7006.2010.01811.x
124
Corsaro
A
,
Bajetto
A
,
Thellung
S
,
Begani
G
,
Villa
V
,
Nizzari
M
, et al.
Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget. 2016;7(25):38638–57. doi:.https://doi.org/10.18632/oncotarget.9575
125
Provenzano
L
,
Ryan
Y
,
Hilton
DA
,
Lyons-Rimmer
J
,
Dave
F
,
Maze
EA
, et al.
Cellular prion protein (PrPC) in the development of Merlin-deficient tumours. Oncogene. 2017;36(44):6132–42. doi:.https://doi.org/10.1038/onc.2017.200
126
Iglesia
RP
,
Prado
MB
,
Cruz
L
,
Martins
VR
,
Santos
TG
,
Lopes
MH
. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res Ther. 2017;8(1):76. doi:.https://doi.org/10.1186/s13287-017-0518-1
127
Wang
Y
,
Yu
S
,
Huang
D
,
Cui
M
,
Hu
H
,
Zhang
L
, et al.
Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am J Pathol. 2016;186(11):2945–56. doi:.. Correction in: Am J Pathol. 2016;187(3):689
https://doi.org/10.1016/j.ajpath.2016.07.010
128
Halliez
S
,
Martin-Lannerée
S
,
Passet
B
,
Hernandez-Rapp
J
,
Castille
J
,
Urien
C
, et al.
Prion protein localizes at the ciliary base during neural and cardiovascular development, and its depletion affects α-tubulin post-translational modifications. Sci Rep. 2015;5(1):17146. doi:.https://doi.org/10.1038/srep17146
129
Santos
TG
,
Silva
IR
,
Costa-Silva
B
,
Lepique
AP
,
Martins
VR
,
Lopes
MH
. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells. 2011;29(7):1126–36. doi:.https://doi.org/10.1002/stem.664
130
Zhuang
D
,
Liu
Y
,
Mao
Y
,
Gao
L
,
Zhang
H
,
Luan
S
, et al.
TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. Int J Cancer. 2012;130(2):309–18. doi:.https://doi.org/10.1002/ijc.25985
131
Du
J
,
Pan
Y
,
Shi
Y
,
Guo
C
,
Jin
X
,
Sun
L
, et al.
Overexpression and significance of prion protein in gastric cancer and multidrug-resistant gastric carcinoma cell line SGC7901/ADR. Int J Cancer. 2005;113(2):213–20. doi:.https://doi.org/10.1002/ijc.20570
132
Luo
G
,
Wang
W
,
Wu
Q
,
Lu
Y
,
Su
T
,
Gu
N
, et al.
MGr1-Antigen/37 kDa laminin receptor precursor promotes cellular prion protein induced multi-drug-resistance of gastric cancer. Oncotarget. 2017;8(42):71630–41. doi:.https://doi.org/10.18632/oncotarget.17795
133
Cheng
Y
,
Tao
L
,
Xu
J
,
Li
Q
,
Yu
J
,
Jin
Y
, et al.
CD44/cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol Carcinog. 2014;53(9):686–97. doi:.https://doi.org/10.1002/mc.22021
134
Meslin
F
,
Conforti
R
,
Mazouni
C
,
Morel
N
,
Tomasic
G
,
Drusch
F
, et al.
Efficacy of adjuvant chemotherapy according to Prion protein expression in patients with estrogen receptor-negative breast cancer. Ann Oncol. 2007;18(11):1793–8. doi:.https://doi.org/10.1093/annonc/mdm406
135
Meslin
F
,
Hamaï
A
,
Gao
P
,
Jalil
A
,
Cahuzac
N
,
Chouaib
S
, et al.
Silencing of prion protein sensitizes breast adriamycin-resistant carcinoma cells to TRAIL-mediated cell death. Cancer Res. 2007;67(22):10910–9. doi:.https://doi.org/10.1158/0008-5472.CAN-07-0512
136
Atkinson
CJ
,
Kawamata
F
,
Liu
C
,
Ham
S
,
Győrffy
B
,
Munn
AL
, et al.
EGFR and Prion protein promote signaling via FOXO3a-KLF5 resulting in clinical resistance to platinum agents in colorectal cancer. Mol Oncol. 2019;13(4):725–37. doi:.https://doi.org/10.1002/1878-0261.12411
137
Park
JY
,
Jeong
JK
,
Lee
JH
,
Moon
JH
,
Kim
SW
,
Lee
YJ
, et al.
Induction of cellular prion protein (PrPc) under hypoxia inhibits apoptosis caused by TRAIL treatment. Oncotarget. 2015;6(7):5342–53. doi:.https://doi.org/10.18632/oncotarget.3028
138
Liang
J
,
Ge
F
,
Guo
C
,
Luo
G
,
Wang
X
,
Han
G
, et al.
Inhibition of PI3K/Akt partially leads to the inhibition of PrP(C)-induced drug resistance in gastric cancer cells. FEBS J. 2009;276(3):685–94. doi:.https://doi.org/10.1111/j.1742-4658.2008.06816.x
139
Gil
M
,
Kim
YK
,
Kim
KE
,
Kim
W
,
Park
CS
,
Lee
KJ
. Cellular prion protein regulates invasion and migration of breast cancer cells through MMP-9 activity. Biochem Biophys Res Commun. 2016;470(1):213–9. doi:.https://doi.org/10.1016/j.bbrc.2016.01.038
140
de Lacerda
TC
,
Costa-Silva
B
,
Giudice
FS
,
Dias
MV
,
de Oliveira
GP
,
Teixeira
BL
, et al.
Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin Exp Metastasis. 2016;33(5):441–51. doi:.https://doi.org/10.1007/s10585-016-9788-8
141
Lin
SC
,
Lin
CH
,
Shih
NC
,
Liu
HL
,
Wang
WC
,
Lin
KY
, et al.
Cellular prion protein transcriptionally regulated by NFIL3 enhances lung cancer cell lamellipodium formation and migration through JNK signaling. Oncogene. 2020;39(2):385–98. doi:.https://doi.org/10.1038/s41388-019-0994-0
142
Li
C
,
Yu
S
,
Nakamura
F
,
Pentikäinen
OT
,
Singh
N
,
Yin
S
, et al.
Pro-prion binds filamin A, facilitating its interaction with integrin beta1, and contributes to melanomagenesis. J Biol Chem. 2010;285(39):30328–39. doi:.https://doi.org/10.1074/jbc.M110.147413
143
Yang
L
,
Gao
Z
,
Hu
L
,
Wu
G
,
Yang
X
,
Zhang
L
, et al.
Glycosylphosphatidylinositol anchor modification machinery deficiency is responsible for the formation of pro-prion protein (PrP) in BxPC-3 cells and increases cancer cell motility. J Biol Chem. 2016;291(13):6785. doi:.https://doi.org/10.1074/jbc.A115.705830
144
Lu
W
,
Kang
Y
. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49(3):361–74. doi:.https://doi.org/10.1016/j.devcel.2019.04.010
145
Shibue
T
,
Weinberg
RA
. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29. doi:.https://doi.org/10.1038/nrclinonc.2017.44
146
Martin-Lannerée
S
,
Hirsch
TZ
,
Hernandez-Rapp
J
,
Halliez
S
,
Vilotte
JL
,
Launay
JM
, et al.
PrP(C) from stem cells to cancer. Front Cell Dev Biol. 2014;2:55.
147
Raymond
GJ
,
Zhao
HT
,
Race
B
,
Raymond
LD
,
Williams
K
,
Swayze
EE
, et al.
Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019;4(16):e131175. doi:.https://doi.org/10.1172/jci.insight.131175
148
Barbieri
G
,
Palumbo
S
,
Gabrusiewicz
K
,
Azzalin
A
,
Marchesi
N
,
Spedito
A
, et al.
Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy. 2011;7(8):840–53. doi:.https://doi.org/10.4161/auto.7.8.15615
149
Spagnolli
G
,
Rigoli
M
,
Orioli
S
,
Sevillano
AM
,
Faccioli
P
,
Wille
H
, et al.
Full atomistic model of prion structure and conversion. PLoS Pathog. 2019;15(7):e1007864. doi:.https://doi.org/10.1371/journal.ppat.1007864