A review of HLA allele and SNP associations with highly prevalent infectious diseases in human populations
DOI: https://doi.org/10.4414/smw.2020.20214
Department of Genetics and Evolution, Anthropology Unit, University of Geneva, Switzerland
Summary
Human leucocyte antigen (HLA) alleles and single nucleotide polymorphisms (SNPs) lying in the HLA region are known to be associated with several infectious diseases among which acquired immunodeficiency syndrome, hepatitis B, hepatitis C, tuberculosis, leprosy and malaria are highly prevalent in many human populations worldwide. Distinct approaches such as case-control comparisons, immunogenetic analyses, bioinformatic peptide-binding predictions, ancient DNA and genome-wide association studies (GWAS) have contributed to improving this knowledge during the last decade, although many results still need stronger statistical and/or functional support. The present review updates the information regarding the main HLA allele and SNP associations observed to date for six of the most widespread and some other infectious diseases, and provides a synthetic illustration of these findings on a schematic HLA genomic map. It then discusses these results by stressing the importance of integrating information on HLA population diversity in disease-association studies.
Introduction
Human leucocyte antigen (HLA) molecules are encoded by a set of 21 protein-coding loci lying amongst many other genes and pseudogenes at region 6p21 of our genome [1, 2]. The HLA transmembrane proteins encoded by the classical (A, B, C, DR, DQ and DP) HLA genes are principally involved in the presentation, at the cell surface, of small pathogen-derived peptides to T cells, which triggers an immune response [3]. HLA class I (A, B, C) molecules are composed of a single α chain non-covalently bound to a small β2-microglobulin polypeptide encoded by another chromosome (15q21). Their α1 and α2 domains form the peptide-binding site where small peptides of 8 to 10 amino acids derived from viral proteins produced by an infected cell are presented to cytotoxic CD8+ lymphocytes, with the effect of destroying that cell. HLA class II (DR, DQ, DP) molecules are heterodimers composed of one α (encoded by DRA, DQA and DPA genes) and one β (encoded by DRB, DQB and DPB genes) chain, the α1 and β1 domains of which form the peptide binding site. These proteins present longer peptides of 13 to 25 amino acids derived from endocytosed antigens to helper CD4+ lymphocytes. In this case, the recognition of the HLA-peptide complex by the lymphocytes, via their T-cell receptors, induces the release of cytokines that will orchestrate a tailored immune response against the pathogen, for instance by helping B cells to secrete high affinity antibodies or by inducing macrophage activation.
All classical HLA genes except DRA and DPA are highly polymorphic at their peptide binding site-encoding exons (exons 2 and 3 for class I and exon 2 for class II genes), which translates into several thousands of currently known HLA alleles and high levels of heterozygosity (often around 90%) in most human populations [1, 4, 5]. This huge diversity is usually explained by the advantage it may confer to heterozygous individuals, which would be capable of presenting a larger variety of peptides than in homozygous individuals, therefore enhancing their T-cell repertoire and protection against pathogens. This heterozygous advantage model, first described by Doherty and Zinkernagel [6], was further refined by the consideration that heterozygotes carrying functionally divergent alleles would have a higher fitness (ability to survive and reproduce) than heterozygotes carrying more similar alleles, hence the naming of divergent allele advantage [7–9], also recently extended, under the name of joint divergent asymmetric selection, to the simultaneous action of multiple HLA loci [10].
The extreme genetic variation observed at classical HLA genes in human populations is thus principally considered as the result of an adaptation to pathogen-rich environments during evolution, although other mechanisms have been proposed as well [11]. This population heterozygote advantage is a global effect of HLA allelic variation that does not necessarily indicate allele-specific overdominance whereby a heterozygote would be more protected than the homozygotes for the same allele [12]. On the other hand, protective or harmful effects of specific HLA alleles or single nucleotide polymorphisms (SNPs) in the HLA region have been suggested through several epidemiological approaches, a number of them relating to infectious diseases, as documented below. Such diseases are characterised by the fact that they can be passed directly or indirectly from one individual to another through several categories of infectious agents (viruses, bacteria, parasites and fungi), with the dramatic consequences that they may quickly affect and kill hundred thousands or even millions of individuals each year (table 1). This is why there is an urgent need to understand better how each individual reacts against different kinds of infections, taking into account their HLA genotype. From an evolutionary genetics perspective, a fascinating issue is also to understand how specific HLA markers evolved in relation to the spread of infectious diseases worldwide. For example, in the case of a severe epidemic leading to a reduction in size of a given population (i.e., a population bottleneck), we may expect highly protective alleles to be maintained in the population through positive selection and susceptibility alleles to be eliminated through purifying selection or selective sweep, as revealed in the major histocompatibility complex (MHC) genes of chimpanzees following putative simian immunodeficiency virus epidemics [13–15]. This partly explains why genetic differences are observed at HLA genes between populations (if the latter underwent distinct disease outbreaks during evolution) despite a general effect of population heterozygous advantage [16]. In the present paper, we first review the associations that have been best documented between markers at classical HLA genes and six of the most prevalent infectious diseases in human populations, and we then discuss these findings in a population genetics and evolutionary perspective.
|
Category
|
Disease
|
Infecting agent
|
Number of cases worldwide*
|
Number of deaths worldwide*
|
| Viral |
Acquired immunodeficiency syndrome |
Human immunodeficiency virus |
37.9 million in 2018 |
770,000 in 2018 |
| Viral |
Hepatitis B |
Hepatitis B virus |
257 million in 2015 |
887,000 in 2015 |
| Viral |
Hepatitis C |
Hepatitis C virus |
71 million in 2015 |
399,000 in 2016 |
| Bacterial |
Tuberculosis |
Mycobacterium tuberculosis
|
10 million in 2018 |
1.5 million in 2018 |
| Bacterial |
Leprosy |
Mycobacterium leprae
|
>200,000 new cases in 2018 |
− |
| Parasitic |
Malaria |
Plasmodium falciparum and vivax
|
228 million in 2018 |
405,000 in 2018 |
HLA allele and SNP associations with infectious diseases
Causal relationships between host genetic variation and infectious disease susceptibility have been found at different genes across the genome, one of the best documented examples being CCR5 Δ32, which impairs the penetration of human immunodeficiency virus (HIV) into the cells [17]. In addition, although precise causal determinants have not been identified, for about half a century HLA genes have been reported to be associated with diseases including many different autoimmune disorders and infections, as well as several kinds of cancers (reviewed in [2, 18–25]). Genome-wide association studies (GWAS), which explore putative associations between genetic markers and particular phenotypes across the whole genome, have identified significant signals of positive selection in that region (e.g., [26]) and a large number of SNPs within the HLA region have been found to be associated with viral infections, as recently reported in several reviews [27–29]. To date, HLA associations have been more particularly documented for acquired immune deficiency syndrome (AIDS), hepatitis B, hepatitis C, tuberculosis, leprosy and malaria, as described below (note that regarding HLA alleles, we focused this review on alleles defined at the second-field level of resolution that corresponds to specific HLA proteins [1] except when presenting the historical backgrounds of some findings).
Acquired immune deficiency syndrome
One of the best documented associations of markers in the HLA region with viral infections is for the acquired immune deficiency syndrome (AIDS) due to infection by the human immunodeficiency virus (HIV) [30], which is one of the major global public health issues today (table 1). Earliest studies suggested that AIDS progression was accelerated in homozygotes at HLA class I genes compared with heterozygotes [31, 32] and in HLA-B*35 [32, 33] and/or HLA-C*04 carriers [32]. B*35 subtypes differing in their peptide-binding specificity at position 9 of the peptide binding site were also considered, and it was shown that changes at a single amino acid position of the HLA molecules could lead to differences in terms of HIV disease progression [34]. A significant negative correlation was also observed between the rapidity of AIDS progression and the number of viral epitopes recognised by HLA class I types, suggesting a direct relationship between HLA alleles and the progression of the disease [35]. More recently, Pereyra et al. [36] performed GWAS in a multi-ethnic cohort of HIV-1 controllers (infected people who show ability to control HIV replication without any therapy and who do not develop clinical disease) and progressors (used as controls): in the largest European group, they identified 313 SNPs reaching genome-wide significance in HLA and none in other genomic regions. Four independent markers lie in proximity to HLA class I, among which are rs4418214 close to MICA and rs2395029 related to the HLA Complex P5 gene (HCP5) coding for a long non coding regulatory RNA associated with several diseases [37]. The G variant of rs2395029 is itself in linkage disequilibrium with allele HLA-B*57:01 [38–40] protecting against HIV progression to AIDS in populations of European ancestry [41], although also found to confer hypersensitivity to abacavir, an antiretroviral drug used to treat HIV-infected patients [42–45]. However, as reported by Kulski et al. [37], the association of HCP5-rs2395029 with HIV viral load set point has not been confirmed in African populations, where HLA-B*57:03 is found in place of B*57:01. Actually, the closely related HLA-B*57:02, HLA-B*57:03 and HLA-B*58:01 alleles are all found to be protective against HIV disease progression by presenting several p24 Gag-specific epitopes [46], although Env protein-derived epitopes also likely play an important role in HLA-HIV restriction involving HLA-B*57:01 and HLA-B*58:01 [47]. As reviewed by Bardeskar and Mania-Pramanik [48], another SNP located 35 kb upstream of HLA-C, rs9264942 (termed −35Kb), was putatively associated with both HLA-C expression levels and HIV control (variant C being associated with higher expression and slower disease progression) [39, 49], the causal variant being however an in/del located at position 263 of HLA-C 3’-UTR (rs67384697, in linkage disequilibrium with rs9264942) regulating the binding of a micro-RNA (miR-148a) [50–55]. In the African American group, Pereyra et al. [36] identified 33 significant SNPs (none in common with Europeans) among which rs2523590 2kb upsteam of HLA-B in common with the (so-called by the authors) Hispanic group, as well as the intronic rs2523608, also observed by Pelak et al. [56] to be associated with HLA-B*57:03, which was previously suggested to be involved in slower HIV-1 disease progression in populations of African descent [57]. Pereyra et al. [36] also identified (through amino acid imputation) six significant amino acid positions among which five within the PBR, more particularly position 97 (C-pocket) in HLA-B that would mediate the protection or susceptibility to HIV progression. Finally, besides HLA-B*27:05 [36, 38], HLA-B*27:02 was recently identified as another protective factor in HIV-1 (slow disease progression) [58], which confirms the implication of the formerly proposed HLA-B*27 allele [59]. More recently, bioinformatic peptide-binding predictions have again stressed the putative advantage of HLA heterozygotes in HIV-1 control [60].
Hepatitis B
Viral hepatitis includes a set a diseases due to different viruses (A, B, C, D, E) [61], among which HBV and HCV cause 96% of hepatitis mortality in humans [62] (table 1). Former association studies focusing on the HLA region (partly reviewed in [63–65]) suggested that homozygosity, as well as specific alleles at HLA class II loci (DQA1*05:01, DQB1*03:01, DPB1*09:01, DRB1*11:02), increased the risk of HBV infection or progression [66–68], whereas both HLA class I (A*03:01) and class II (DRB1*13:01, DRB1*13:02, DQB1*02:01, DPB1*02:01) alleles were associated with protection or viral clearance [68–72]. More recently, GWAS studies identified HLA-DP as the locus most strongly associated with chronic HBV infection [73, 74]. In the 3′-UTR region of both DPA1 and DPB1, 11 SNPs were found to be associated with chronic hepatitis B in Asian populations, the strongest associations being found for rs3077 and rs9277535, respectively (A alleles conferring protection in both cases) [73].
Based on these results, two protective (DPA1*01:03~DPB1*04:02 and DPA1*01:03~DPB1*04:01) and two risk (DPA1*02:02~DPB1*05:01 and DPA1*02:02~DPB1*03:01) HLA haplotypes were proposed. Further fine-mapping of the MHC region in East Chinese HBV carriers and controls also revealed a strong association with chronic hepatitis B infection of HLA-DPβ1 amino acid positions 84-87 lying in the PBR, themselves in high linkage disequilibrium with SNP rs9277535 at HLA-DPB1 and medium linkage disequilibrium with SNP rs3077 at HLA-DPA1, making these positions possible causal variants involved in peptide presentation [75]. However, decreased expression of HLA-DPA1 and HLA-DPB1 has also been associated with rs3077 and rs9277535, respectively, suggesting that low gene expression may be a risk factor [76], in line with the more general idea that associations between HLA markers and diseases are partly driven by differential expression of HLA alleles [77]. A different SNP located in the 3′-UTR of HLA-DPB1 − rs9277534 (or 496A/G), in complete linkage disequilibrium with rs9277530, rs9277531, rs9277533 and rs9277536 − was found to be associated with either recovery (variant A, in linkage disequilibrium with allele HLA-DPB1*04:01) or persistence (variant G, in linkage disequilibrium with allele HLA-DPB1*01:01) of HBV in both Europeans and African-Americans [78]. Here again, the associations would be due to differences in HLA-DPB1 expression levels rather than in peptide binding properties, but in this case higher levels of HLA-DP expression (496 GG genotype) would confer HBV persistence [78]. A novel SNP near HLA-DPA3 (rs9366816) was also found to be significantly associated with persistent HBV infection in Han-Taiwanese [79] and a SNP near HLA-DPB1 (rs9277542) with protection against chronic HBV and viral clearance in Japanese and Koreans [80] as well as in Argentinians [81]. Mbarek et al. [74] also showed that two SNPs located within the HLA-DQ locus, namely rs2856718 located in the intergenic region between HLA-DQA2 and HLA-DQB1, and rs7453920 located in intron 1 of HLA-DQB2, exerted effects on hepatitis B susceptibility (A conferring protection and G susceptibility in both cases) independently from the effects of the SNPs found at locus HLA-DP in the studied Japanese cohort. Based on this study, HLA haplotypes DQA1*01:02~DQB1*03:03 and DQA1*03:01~DQB1*06:01 were associated with risk and DQA1*01:01~DQB1*05:01 and DQA1*01:02~DQB1*06:04 to protective effects. Two significant SNPs were also identified near the HLA-DQA2 (rs9276370) and HLA-DQB2 (rs7756516) loci [79] and one SNP (rs9268652) in HLA-DRA [29]. Whereas the involvement of HLA-DP SNPs rs3077, rs9277534 and rs9277535 and HLA-DQ SNPs rs2856718 and rs7453920 were confirmed in other studies (e.g., [63, 79–88]), although sometimes contradicted in cohorts of different origins (e.g., [89]), significant associations of SNPs in the HLA-DP/HLA-DQ regions were further expanded by Tao et al. [90], the most significant ones among a total of 76 being again found in HLA-DPB1 3′-UTR. Together with a large meta-analysis confirming the significant associations of SNPs rs3077 and rs9277535 [91], this genomic region thus appears to play a leading functional role in HBV immunity. Some studies also observed significant associations between HBV and SNPs in the HLA class I region, namely rs3130542 near HLA-C (the A allele being associated with a low HLA-C expression and increased risk of developing chronic infection) [85], further found to be in linkage disequilibrium with HLAC*07:02 [88], as well as rs2853953, in linkage disequilibrium with HLA-C*06:02 [88].
Hepatitis C
Some studies also suggested associations between HLA alleles and hepatitis C virus (HCV) persistence, such as HLA-DRB1*03:01, or spontaneous clearance, such as HLA-A*02:01, A*11:01, B*57:01, B*57:03, C*01:02, DQB1*03:01, DRB1*01:01, DRB1*04:01 and DRB1*11:01, among others [92–99]. However, these associations depend on population origin, as stressed in a recent review and meta-analysis of HCV clearance [100] that reported highly significant associations for alleles HLA-DQB1*02:01, DQB1*03:01, DRB1*07:01, DRB1*11:01 and/or DRB1*12:01, depending on the ethnicity. SNP rs4273729 (C allele) associated with HLA-DQB1*03:01 (which would be expressed at higher levels compared to other DQB alleles) was proposed to be involved in spontaneous resolution of HCV infection in individuals of both European and African ancestry [101]. SNP rs9275572, located in the HLA-DQB1 region, showed a significant association with chronic HCV in Japanese, however giving the same signal as SNP rs1130380, a variant specific to HLA-DQB1*03 that causes an amino acid substitution in the peptide-binding pocket of HLA-DQB1 (position 55) [102]. Note also that alleles HLA-B*57:01 and B*57:03, associated with HCV clearance [92, 95] have also been associated with slow HIV disease progression [41, 57], that SNP rs3077 of the HLA-DPA1 region, mentioned above as associated with HBV infection, has been suggested to increase the risk of chronic HCV infection in a Chinese population [103] and that SNP rs9264942 putatively linked to HIV control (see above) has been found to be associated with persistent HCV seronegativity among C/C genotyped people who inject drugs [104].
Other associations within the HLA region with HBV or HCV progression towards different diseases have recently been documented by Sawai et al. [105] and Omae and Tokunaga [106].
Tuberculosis
Tuberculosis is due to infection by the bacteria Mycobacterium tuberculosis. Although curable, it is still the major cause of death from infectious diseases on the global scale (table 1). A number of studies have reported significant associations between HLA alleles and either protection or susceptibility to several respiratory diseases [107], including tuberculosis (review and meta-analysis in [108]), but with many inconsistencies [107, 109–111]. GWAS studies have revealed several significant markers in the HLA class II region: according to Sveinbjornsson et al. [112], two markers, rs557011 [T] located between HLA-DQA1 and HLA–DRB1 and rs9272785 located in exon 4 of HLA-DQA1 (a missense mutation p.Ala210Thr which defines HLA-DQA1*03 alleles) would confer susceptibility to M. tuberculosis infection in European populations, the former SNP being also a risk factor for the development of the disease in infected individuals (note that about one third of the global population is latently infected by the bacteria, some 10% of them developing the disease); by contrast, rs9271378 [G], also located between HLA-DQA1 and HLA–DRB1, would protect against the development of the disease. The same authors confirmed the susceptibility of HLA-DQA1*03:01 to tuberculosis after imputation of HLA alleles. They suggest that HLA-DQA1*03 molecules would have a reduced stability and would be less able to present M. tuberculosis antigens. Other missense mutations classified as probably damaging were found in the Han Chinese population, namely rs41553512 and rs1136744 in HLA-DRB5 and rs41553512 in HLA-DQB1, in addition to a significant association of TB with HLA-DQB1*02:01 [113]. Because six different M. tuberculosis strains are unevenly distributed geographically [114], putative associations between HLA polymorphisms and tuberculosis caused by specific strains were investigated, which revealed higher susceptibility conferred by alleles HLA-DRB1*09:01 and HLA-DQB1*03:03 (both in linkage disequilibrium) to modern strains in Southeast Asia [115]. The association of HLA-DQB1*03 with tuberculosis was supported by a recent study [116] that also suggested a protective effect of HLA-B*58:01 through M. tuberculosis antigens epitopes CFP-10 (ESAT-6-like protein) binding and a susceptibility effect of HLA-B*58:02, which differs by 3 amino acids from the former.
Leprosy
Leprosy is due to infection by the bacteria Mycobacterium leprae. Although the disease is decreasing at the global level, a high number of new cases are still reported per year (table 1), India, Brazil, Indonesia, the western Pacific and some parts of Africa being the most affected. As reviewed by Jarduli et al. [117], many studies identified both protective and risk class I and class II HLA alleles associated with leprosy. A case-control study in India revealed two SNPs, one in the HLA-DRB1 region (rs9270650) and one within the HLA-DQA1 gene (rs1071630), as major susceptibility markers [118]. Based on genome-wide studies, HLA-DRB1 further appeared as a key region for leprosy susceptibility, with SNPs rs9271366, rs602875 [119], rs9271011 and rs9271100 [120] identified in Chinese Han, the two latter being in strong linkage disequilibrium with HLA-DRB1*15:01, another significant variant being HLA-DQB1*04:01. SNP rs9271011 showed a regulatory effect, the risk allele [T] being responsible of an increased HLA-DRB1 expression. By analysing both ancient and modern DNA, an associated SNP, rs3135388 [T], was found to be significantly more frequent in a sample of 69 M. leprae-positive individuals showing lesions specific for lepromatous leprosy in a medieval population from Denmark than in both ancient and modern randomly sampled individuals from Denmark and north Germany [121]. Based on both direct identification through high-throughput sequencing of HLA-DRB1*15(01) and HLADQB1*06:02 alleles in a substantial number of medieval individuals with lepromatous leprosy and a limited M. leprae peptide-binding capacity predicted for the HLA-DRB1*15:01 allele, the same study suggested that HLA-DRB*15:01~DQB1*06:02 was a strong risk haplotype for this disease. HLA class I alleles may also be implicated in leprosy pathogenesis, as suggested by the identification of two class I intergenic SNPs - rs2394885 and rs2922997 - associated with leprosy susceptibility in Vietnamese and Indians, the former one being in strong linkage disequilibrium with HLA-C*15:05 [122].
Malaria
According to the 2019 report of the World Health Organization [123], 228 million cases of malaria were reported worldwide in 2018 (table 1), among which 213 million (93%) were in Africa, followed by South-East Asia (3.4%). Plasmodium falciparum is the most virulent species and the most widespread in Africa (99.7% of cases), South-East Asia (62.8%), the Eastern Mediterranean (69%) and the Western Pacific (71.9%), whereas Plasmodium vivax is the predominant parasite in the Americas (74.1%). Several genes across the genome have been shown to display polymorphisms conferring protection against malaria, namely markers related to haemoglobinopathies (sickle-cell trait HbS, HbE, HbC, α- and β-thalassemias), erythrocyte antigens (ovalocytosis SLC4A1, Duffy DARC), enzymopathies (glucose-6-phosphate dehydrogenase [G6PD], pyruvate kinase [PKLR]) and immunogenetic variants (complement receptor-1 CR1, HLA) (reviewed in 124]). Besides the putative role of HLA-G variation in the regulation of the response to P. falciparum infection [125, 126], several alleles of classical HLA genes have also been suggested to be protective to this form of malaria. The best known of them is HLA-B*53, first identified, in addition to the HLA class II DRB1*13:02~DQB1*05:01 haplotype, in a case-control study in Gambia [127]. HLA-B*53 was shown to recognise a conserved nonamer peptide from liver-stage-specific antigen-1 (LSA-1) of P. falciparum [128]. Based on their frequency in populations, several HLA alleles were also proposed as resistance (B*53:01, DQB1*05:01, DRB1*01:01 and DRB1*13:02) or susceptibility (A*30:01, A*33:01, DPB1*17:01 and DRB1*04:01) factors [129, 130]. The protective effect of HLA-B*53:01 was further supported by a population genetics modelling showing a clear relationship, after controlling for other genetic, geographic and environmental variables, between the frequency of this allele and the prevalence of P. falciparum in Africa [131]. The same study also identified HLA-B*78:01 as another possible candidate. Interestingly, based on peptide binding predictions both HLA-B*53:01 and B*78:01 share a high affinity for peptides with amino acid proline at position 2 (accommodated by pocket B of the peptide binding site), a property that is shared by 50% of the most frequent (frequency above 15% in at least one population) HLA-B alleles found in Africa [131]. Among these alleles, HLA-B*35:01 also revealed a significant protective effect against malaria in Ghana [132], in agreement with a previous finding that two antigenic octamer peptides of P. falciparum’s circumsporozoite protein, cp26 and cp29, bind HLAB*35 [133]. Despite the common idea that HLA class II, rather than HLA class I, molecules operate at the peptide-binding level in the case of parasitic infections (although cross-presentation of exogenous antigens on HLA class I is a well described phenomenon in professional antigen presenting cells), these findings suggest that several HLA-B alleles from the standing genetic variation increased in frequency when malaria expanded in Africa, probably with the development of agriculture [134]. This mechanism of soft selective sweep, which is likely to be a common evolutionary process in the human genome [135, 136], does not exclude, however, that pathogen-mediated hard selective sweep also operates at HLA genes, as recently suggested by the very high HLA class II allele and haplotype frequencies, presumably not resulting from rapid genetic drift, observed in a large West African population [137]. Interaction between HLA-B*53:01, carrier of the Bw4 epitope, and killer cell immunoglobulin-like receptor KIR3DL1 has also been suggested to contribute to its protective effect [138]. Genes of the HLA region would also be associated with P. vivax infection [139], although, to our knowledge, no specific HLA allele defined at the second field level of resolution nor specific SNP has been proposed so far.
Other infectious diseases
Thanks to GWAS, HLA associations with other infectious diseases have also been suggested. Regarding viral infections, numerous SNPs in the HLA-B region (among which rs114045064) have been associated with herpes zoster (shingles due to varicella-zoster virus, VZV), likely playing a role in viral suppression [140, 141]. A SNP located upstream to HLA-DQB1 (rs9357152) has been associated with human papillomavirus (HPV) seropositivity [142], while both risk (HLA-DRB1*15:01~DQB1*06:02) and protective (HLA-DRB1*03:01~DQB1*02:01) HLA class II haplotypes had previously been suggested for this disease (cited by the same authors). Regarding bacterial infections, a significant association has been found at SNP rs7765379 near HLA-DQB1 and HLA-DRB1 for enteric fever due to infection by Salmonella enterica, pointing to allele HLA-DRB1*04:05 (after imputation) as a protective factor [143]. In addition, DeLorenze et al. [144] identified three SNPs associated with Staphylococcus aureus infection in HLA-DRA (rs4321864) and close to HLA-DRB1 (rs115231074, rs35079132). Among parasitic infections, Leishmaniasis (due to infection by protozoan parasites Leishmania donovani or Leishmaniasi infantum chagasi) was significantly associated with SNP rs9271858 located between HLA-DRB1 and HLA-DQA1 in three distinct cohorts from India and Brazil [145], more recent studies also showing that HLA-DRB1*15:01 (with specific amino acid residues at positions 11 and 13) and HLA-DRB1*14:04/HLA-DRB1*13:01 were the most significant protective and risk alleles, respectively [146, 147]. By performing GWAS on a large sample of over 200,000 individuals of European ancestry recorded in the database 23andme, Tian et al. [29] also found new significant associations between several infectious diseases and SNPs located within the HLA region: chickenpox with rs9266089 within HLA-B; shingles with rs2523591 upstream to HLA-B (also reported in [141]); cold sores with rs885950 between HLA-A and HLA-C; mononucleosis caused by the Epstein-Barr virus (EBV) with SNP rs2596465 within the HCP5 complex upstream to HLA-B; mumps with rs114193679 near HLA-A; plantar warts with rs9272050 within HLA-DQA1; streptococcal sore throat with rs1055821 within HLA-B; scarlet fever with rs36205178 within HLA-DQB1; pneumonia with rs3131623 within the HCP5 complex; and childhood ear infections with rs4329147 between HLA-DRB1 and HLA-DQA1.
Interestingly, these authors identified several associations with amino acid polymorphisms in the peptide-binding region, thus involving specific HLA alleles like HLA-A*02:01 associated with chickenpox and shingles, and HLA-A*02:05 with mumps.
A schematic HLA genomic map showing the positions of the main HLA alleles and SNPs (according to the e!Ensembl [148] and NCBI dbSNP [149] databases accessed on 9 December 2019 at https://www.ensembl.org and https://www.ncbi.nlm.nih.gov/snp, respectively) associated with the diseases mentioned in this review is provided in figure 1. Interestingly, this figure indicates that, in the current state of knowledge, specific HLA regions have been found to be involved in associations with AIDS (B-C region), tuberculosis (DRB-DQA1), hepatitis C (DQB1-DQA1) and (to a lesser extent) leprosy (mostly DRB-DQA1), whereas putative associations with hepatitis B and malaria are more widespread across the whole HLA region.
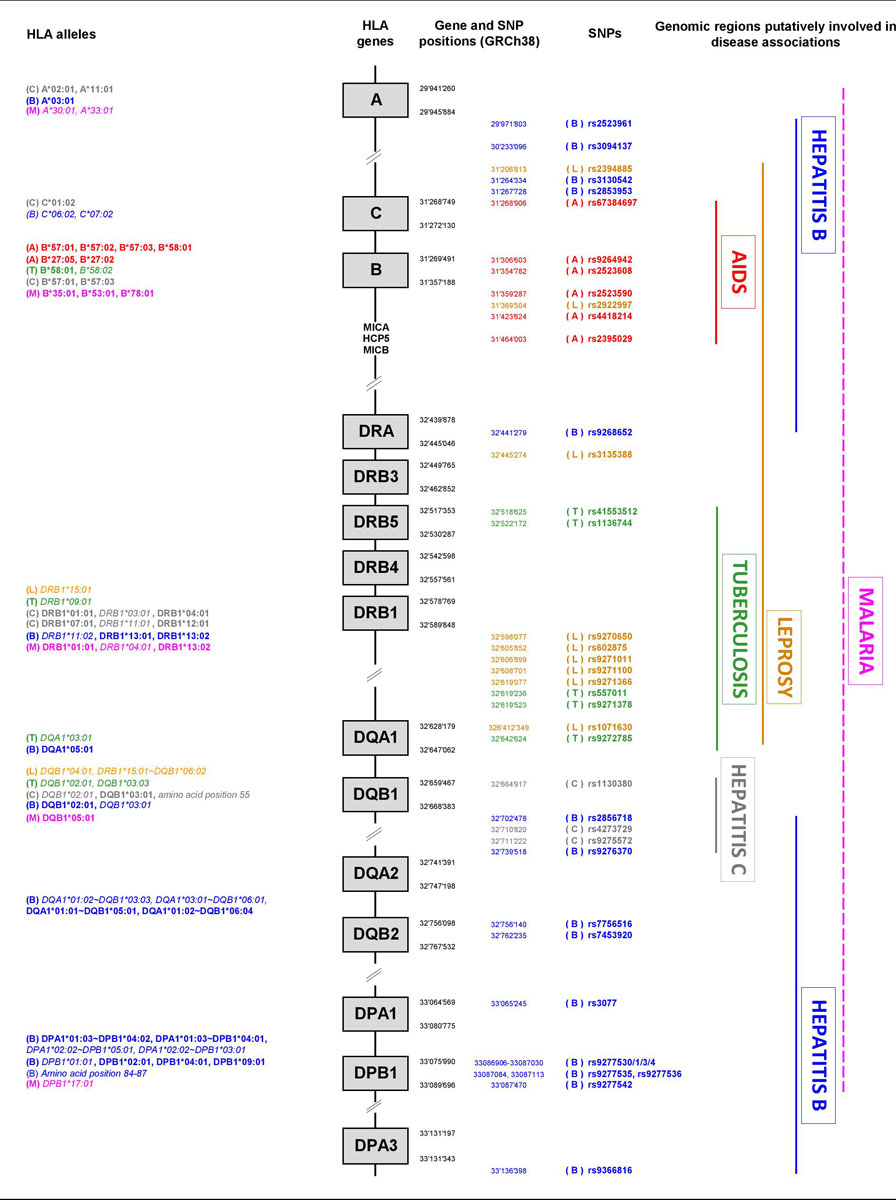
Figure 1 Schematic map of the HLA genomic region showing the positions of the HLA alleles and SNPs associated to six of the most prevalent infectious diseases in human populations.
(A): AIDS, in red; (B): Hepatitis B, in blue; (C): Hepatitis C, in grey; (L): Leprosy, in orange; (M): Malaria, in pink; (T): Tuberculosis, in green. Protective HLA alleles are in bold, susceptibility HLA alleles are in italics.
The genomic regions shown at the right as putatively involved in disease associations are only based on SNP information, except for malaria (dashed line).
The genomic positions (GRCh38) of the HLA genes and SNPs have been determined by using the e!Ensembl [148] and NCBI dbSNP [149] databases accessed on 9 December 2019 at https://www.ensembl.org/index.html and https://www.ncbi.nlm.nih.gov/snp/, respectively.
Population genetics in HLA and infectious disease association studies
This review shows that, in addition to case-control comparisons and immunogenetic analyses, new kinds of approaches such as GWAS, bioinformatic peptide-binding predictions and ancient DNA studies have helped to improve our knowledge on putative associations between HLA molecular variation and infectious diseases in the last decades. Of course, precise identification of causal markers remains complicated not only because the mechanisms by which the immune responses to infectious diseases is modulated are very diverse, as reviewed by Crux et al. [150] for HIV and HCV infections, but also because the analysis of the HLA polymorphism itself raises specific methodological problems and is especially challenging in a population genetics an evolutionary context [28].
First, HLA genes are particularly polymorphic. SNPs in the HLA region are often characterised by multiple non-synonymous substitutions at peptide-binding regions [151], such as rs1130380 located within HLA-DQB1 and associated with hepatitis C, and many SNPs are also multi-allelic in non-coding or intergenic HLA regions, such as rs9271100, rs557011, rs9275572 and rs7453920 associated with leprosy, tuberculosis, hepatitis C and hepatitis B, respectively (major and minor alleles of each SNP can be found at https://www.ncbi.nlm.nih.gov/snp/). As a consequence, HLA variation is still difficult to characterise in particularly dense polymorphic regions (e.g., SNP array probes may be difficult to design), unless long-read sequencing techniques are applied, which represents a strong limiting factor for disease-association studies. Although imputation methods (i.e., methods that infer HLA variation from SNPs genotyped at sites flanking the highly polymorphic regions) are currently used [152], their accuracy varies greatly among studies because of numerous factors such as SNP density in the studied HLA region, as well as which reference panels and bioinformatics methods are used for imputation [153]. Moreover, the heterogeneous recombination patterns prevailing in the HLA genomic region [154–157] are a source of confounding effects, as many genetic markers in linkage disequilibrium may cosegregate in association to the same disease phenotypes without all being instrumental as protective or susceptibility factors. As an example, the rs9264942 (or -35Kb) SNP was first considered as modulating HLA-C expression levels with direct consequences on HIV control before the linked rs67384697 in/del SNP was identified as a micro-RNA binder acting as a real regulatory factor. Also, nonadditive effects such as interactions among loci within the HLA region, recently investigated for a number of autoimmune diseases (e.g., [158–160]) may complicate the identification of one among several alternative models explaining the effects of HLA variation on disease phenotypes. Finally, the extreme HLA molecular variation observed within populations requires very large sample sizes to be satisfactorily characterised and for the statistical tests to reach sufficient power, avoid false positives and allow replication of the results, in particular because the large number of comparisons needed to consider all possible alleles and SNPs may fail to reach significance after correction for multiple tests (e.g., odds ratios are often close to 1 and/or p-values borderline, e.g., in [66]).
Besides the specificity of the HLA region in terms of high molecular variation within populations, HLA allele or SNP frequencies, as well as linkage disequilibrium across the HLA region, greatly differ between populations from different geographic regions [4, 5, 161]. Examples are given in figure 2, which displays global frequencies of the main HIV and malaria protective alleles, as well as those of two DQB1*03 subtypes that have been associated with hepatitis B, hepatitis C and/or tuberculosis (all data were taken from published articles and the allelefrequencies.net database [162]). This partly explains why given disease associations are often not observed in populations of distinct origins (e.g., for HBV-HCV [163]). A dramatic example is that of African populations, which are known to exhibit much higher levels of genetic diversity and much lower levels of linkage disequilibrium than populations from other continents both at the genome level [164] and across the HLA region [161]. This necessarily handicaps the discovery of significant disease associations, whereas Africa is one of the world regions where the greatest burden of infectious disease exists today (e.g., this region accounted for 68% of the world’s people living with HIV in 2018) while being still largely underrepresented in genetic variation studies [165–168]. A better characterisation of HLA molecular diversity in populations worldwide and the integration of population genetics data in disease-association studies are clearly essential.
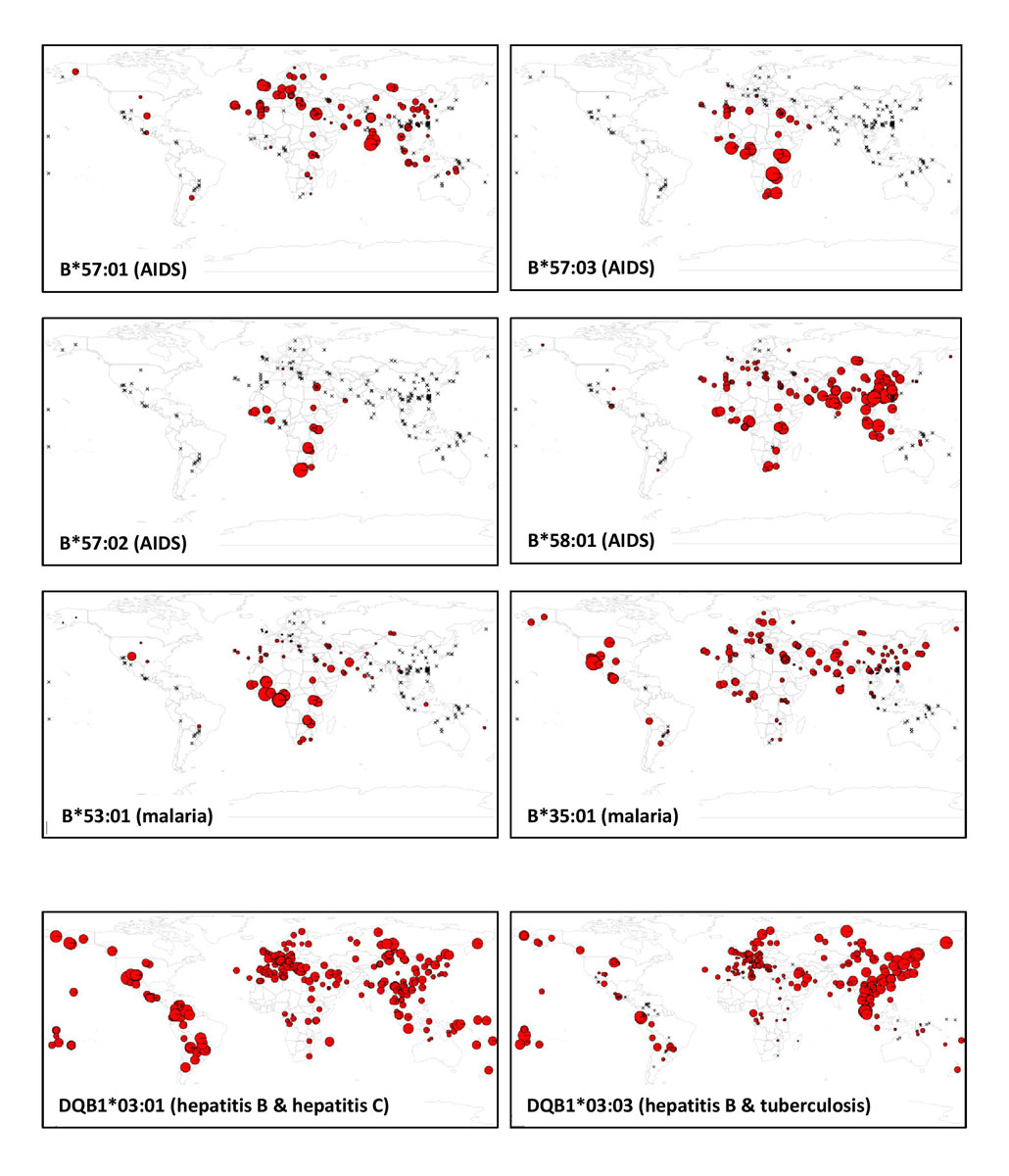
Figure 2 Worldwide population frequency maps of some of the HLA-B alleles suggested to confer protection against AIDS (B*57:01, B*57:02, B*57:03, B*58:01) and malaria (B*53:01, B*35:01) as well as of two HLA-DQB1*03 alleles suggested to be associated to hepatitis B susceptibility (DQB1*03:01 and DQB1*03:03), to hepatitis C protection (DQB1*03:01) and to tuberculosis susceptibility (DQB1*03:03) (see text for references).
In each map, the sizes of the pie charts are proportional to the frequencies of the allele in the corresponding populations, but pie charts are not at the same scale in the different maps. The maximum size of the pie charts in each map correspond to the following frequencies: B*57:01 = 0.082 (Tamil from South India; South Asia); B*57:02 = 0.042 (Xhosa from South Africa, sub-Saharan Africa); B*57:03 = 0.057 (Lusaka from Zambia, sub-Saharan Africa); B*58:01 = 0.170 (Han, China, East Asia); B*53:01 = 0.244 (Angolan, Sao Tome Island, sub-Saharan Africa); B*35:01 = 0.485 (Seri, Mexico, North America); DQB1*03:01 = 0.896 (Gila River Amerindian from Arizona, North America); DQB1*03:03 = 0.313 (Jehai from Malaysia, Southeast Asia).
In each map, the crosses represent HLA-tested populations where the allele was not observed.
All HLA population frequency data were taken from both published articles and the allelefrequency.net database [162].
HLA genetic differences between populations result from both selective effects due to the prominent role of HLA genes in immunity and to human history of migration since the emergence of modern humans from Africa, that likely happened between 300,000 and 200,000 years ago [169, 170]. Disease-association studies thus need to be controlled for population structure, for example by comparing HLA with genome-wide neutral variation in the same populations [16, 171]. If selective and demographic factors are correctly disentangled, differences in HLA frequencies between populations living in distinct environments may be more easily interpreted in terms of local adaptations to pathogens. However, selective signals may also be weak and difficult to identify because multiple alleles at a given HLA locus (e.g., through soft selective sweep) and/or several (HLA and non-HLA) loci across the genome may jointly confer susceptibility or protection to the same diseases, malaria resistance probably being the best example of polyallelic and polygenic adaptation [127, 131, 172–174]. Moreover, host-pathogen interaction is a dynamic process where host molecules may drive changes in pathogens molecules and vice versa, leading to nonpermanent protective or susceptibility effects as a result of fluctuating or frequencydependent selection [175]. Simultaneous analyses of host genome versus pathogen genome diversity in relation to specific diseases (e.g., [176]) may thus help to better understand the evolution of the HLA polymorphism in relation to its pathogenic environment.
Of course, the evolutionary timeframe is not the same for all disease. Some infectious diseases are known to be much more recent than others, making more difficult the exploration of host-pathogen co-evolution through time. For example, it has been suggested that the last common ancestor of HIV-1 pandemic strains dates back to the beginning of the 20th century [177], with a likely transmission of the disease through zoonotic infections from simian reservoirs and an early spread among humans from the region of Kinshasa in the Democratic Republic of Congo [178]. This corresponds to about four or five human generations, a very short time for protective factors to be positively selected and for susceptibility variants to be eliminated under selective pressure, in contrast to the long timeframes for diseases that disseminated in much earlier times (e.g., before or during the Neolithic), such as tuberculosis, leprosy or malaria [179]. This probably explains, for example, why AIDS protective alleles are found at low frequencies (<10%), whereas some malaria protective alleles reach higher frequencies (>20%) in regions where these diseases are highly prevalent (fig. 2), even though soft rather than hard selective sweep mechanisms were at work.
Actually, distinct signals of selection resulting from different evolutionary processes and corresponding to either recent or ancient selective pressures are expected to be observed through population genetic studies. For example, long-term balancing selection likely decreased population differentiation, such as at the HLA-B locus [131, 180], whereas local adaptations to specific diseases probably drove adaptive HLA markers to high frequencies and led to marked genetic differences among populations, as at some HLA class II loci [137]. Such signatures are detectable through comparisons of HLA with neutral genomic variation [16, 181], computer simulations mimicking the evolution of the HLA polymorphism during human migration history with variable intensities of selection [182, 183] and other population genetics approaches taking into account the functional properties of the HLA molecules [184]. This is what makes population genetics approaches powerful to decipher the signatures of pathogen-driven selection on HLA molecular variability. Clearly, the identification of genetic markers involved in disease protection or susceptibility in the HLA region would largely benefit from a better integration of population genetics, with its demographic and evolutionary perspectives, in disease association studies.
Acknowledgments
This work was supported by the Swiss National Science Foundation, grant #310030_188820 (HLA-AFRICA). We also thank Da Di for the allele frequency maps and his very helpful comments, and José Manuel Nunes for stimulating discussions and two anonymous reviewers for their constructive comments.
References
1
Robinson
J
,
Barker
DJ
,
Georgiou
X
,
Cooper
MA
,
Flicek
P
,
Marsh
SGE
. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020;48(D1):D948–55. doi:.https://doi.org/10.1093/nar/gkz950
2
Shiina
T
,
Hosomichi
K
,
Inoko
H
,
Kulski
JK
. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54(1):15–39. doi:.https://doi.org/10.1038/jhg.2008.5
3Parham P, Janeway C. The immune system. 4th edition. New York, NY: Garland Science, Taylor & Francis Group; 2015
4
Buhler
S
,
Sanchez-Mazas
A
. HLA DNA sequence variation among human populations: molecular signatures of demographic and selective events. PLoS One. 2011;6(2):e14643. doi:.https://doi.org/10.1371/journal.pone.0014643
5
Solberg
OD
,
Mack
SJ
,
Lancaster
AK
,
Single
RM
,
Tsai
Y
,
Sanchez-Mazas
A
, et al.
Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum Immunol. 2008;69(7):443–64. doi:.https://doi.org/10.1016/j.humimm.2008.05.001
6
Doherty
PC
,
Zinkernagel
RM
. A biological role for the major histocompatibility antigens. Lancet. 1975;305(7922):1406–9. doi:.https://doi.org/10.1016/S0140-6736(75)92610-0
7
Lenz
TL
. Computational prediction of MHC II-antigen binding supports divergent allele advantage and explains trans-species polymorphism. Evolution. 2011;65(8):2380–90. doi:.https://doi.org/10.1111/j.1558-5646.2011.01288.x
8
Bitarello
BD
,
Francisco
RS
,
Meyer
D
. Heterogeneity of dN/dS ratios at the classical HLA Class I genes over divergence time and across the allelic phylogeny. J Mol Evol. 2016;82(1):38–50. doi:.https://doi.org/10.1007/s00239-015-9713-9
9
Wakeland
EK
,
Boehme
S
,
She
JX
,
Lu
CC
,
McIndoe
RA
,
Cheng
I
, et al.
Ancestral polymorphisms of MHC class II genes: divergent allele advantage. Immunol Res. 1990;9(2):115–22. doi:.https://doi.org/10.1007/BF02918202
10
Buhler
S
,
Nunes
JM
,
Sanchez-Mazas
A
. HLA class I molecular variation and peptide-binding properties suggest a model of joint divergent asymmetric selection. Immunogenetics. 2016;68(6-7):401–16. doi:.https://doi.org/10.1007/s00251-016-0918-x
11
Meyer
D
,
Thomson
G
. How selection shapes variation of the human major histocompatibility complex: a review. Ann Hum Genet. 2001;65(Pt 1):1–26. doi:.https://doi.org/10.1046/j.1469-1809.2001.6510001.x
12
Lipsitch
M
,
Bergstrom
CT
,
Antia
R
. Effect of human leukocyte antigen heterozygosity on infectious disease outcome: the need for allele-specific measures. BMC Med Genet. 2003;4(1):2. doi:.https://doi.org/10.1186/1471-2350-4-2
13
de Groot
NG
,
Otting
N
,
Doxiadis
GG
,
Balla-Jhagjhoorsingh
SS
,
Heeney
JL
,
van Rood
JJ
, et al.
Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc Natl Acad Sci USA. 2002;99(18):11748–53. doi:.https://doi.org/10.1073/pnas.182420799
14
de Groot
NG
,
Heijmans
CM
,
de Groot
N
,
Otting
N
,
de Vos-Rouweller
AJ
,
Remarque
EJ
, et al.
Pinpointing a selective sweep to the chimpanzee MHC class I region by comparative genomics. Mol Ecol. 2008;17(8):2074–88. doi:.https://doi.org/10.1111/j.1365-294X.2008.03716.x
15
de Groot
NG
,
Heijmans
CMC
,
Helsen
P
,
Otting
N
,
Pereboom
Z
,
Stevens
JMG
, et al.
Limited MHC class I intron 2 repertoire variation in bonobos. Immunogenetics. 2017;69(10):677–88. doi:.https://doi.org/10.1007/s00251-017-1010-x
16
Meyer
D
,
C Aguiar
VR
,
Bitarello
BD
,
C Brandt
DY
,
Nunes
K
. A genomic perspective on HLA evolution. Immunogenetics. 2018;70(1):5–27. doi:.https://doi.org/10.1007/s00251-017-1017-3
17
Klebanov
N
. Genetic predisposition to infectious disease. Cureus. 2018;10(8):e3210. doi:.https://doi.org/10.7759/cureus.3210
18
Blackwell
JM
,
Jamieson
SE
,
Burgner
D
. HLA and infectious diseases. Clin Microbiol Rev. 2009;22(2):370–85. doi:.https://doi.org/10.1128/CMR.00048-08
19Alper CA. Major Histocompatibility Complex: Disease Associations, in Encyclopedia of Life Sciences. Chichester, UK: John Wiley & Sons, Ltd; 2009. pp. 1−7.
20
Trowsdale
J
. The MHC, disease and selection. Immunol Lett. 2011;137(1-2):1–8. doi:.https://doi.org/10.1016/j.imlet.2011.01.002
21
Dendrou
CA
,
Petersen
J
,
Rossjohn
J
,
Fugger
L
. HLA variation and disease. Nat Rev Immunol. 2018;18(5):325–39. doi:.https://doi.org/10.1038/nri.2017.143
22
Trowsdale
J
,
Knight
JC
. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. 2013;14(1):301–23. doi:.https://doi.org/10.1146/annurev-genom-091212-153455
23
Abel
L
,
Alcaïs
A
,
Schurr
E
. The dissection of complex susceptibility to infectious disease: bacterial, viral and parasitic infections. Curr Opin Immunol. 2014;30:72–8. doi:.https://doi.org/10.1016/j.coi.2014.07.002
24Cardoso DM, Marangon AV, Sell AM, Visentainer JEL, De Souza CA. HLA and infectious diseases. In: HLA and associated important diseases. Xi Y, editor. Rijeka, Croatia: InTech; 2014. pp. 259−300.
25
Shiina
T
,
Inoko
H
,
Kulski
JK
. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens. 2004;64(6):631–49. doi:.https://doi.org/10.1111/j.1399-0039.2004.00327.x
26
Sabeti
PC
,
Schaffner
SF
,
Fry
B
,
Lohmueller
J
,
Varilly
P
,
Shamovsky
O
, et al.
Positive natural selection in the human lineage. Science. 2006;312(5780):1614–20. doi:.https://doi.org/10.1126/science.1124309
27
Matzaraki
V
,
Kumar
V
,
Wijmenga
C
,
Zhernakova
A
. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017;18(1):76. doi:.https://doi.org/10.1186/s13059-017-1207-1
28
Mozzi
A
,
Pontremoli
C
,
Sironi
M
. Genetic susceptibility to infectious diseases: Current status and future perspectives from genome-wide approaches. Infect Genet Evol. 2018;66:286–307. doi:.https://doi.org/10.1016/j.meegid.2017.09.028
29
Tian
C
,
Hromatka
BS
,
Kiefer
AK
,
Eriksson
N
,
Noble
SM
,
Tung
JY
, et al.
Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. 2017;8(1):599. doi:.https://doi.org/10.1038/s41467-017-00257-5
30
McLaren
PJ
,
Carrington
M
. The impact of host genetic variation on infection with HIV-1. Nat Immunol. 2015;16(6):577–83. doi:.https://doi.org/10.1038/ni.3147
31
Tang
J
,
Costello
C
,
Keet
IP
,
Rivers
C
,
Leblanc
S
,
Karita
E
, et al.
HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 1999;15(4):317–24. doi:.https://doi.org/10.1089/088922299311277
32
Carrington
M
,
Nelson
GW
,
Martin
MP
,
Kissner
T
,
Vlahov
D
,
Goedert
JJ
, et al.
HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283(5408):1748–52. doi:.https://doi.org/10.1126/science.283.5408.1748
33
Itescu
S
,
Mathur-Wagh
U
,
Skovron
ML
,
Brancato
LJ
,
Marmor
M
,
Zeleniuch-Jacquotte
A
, et al.
HLA-B35 is associated with accelerated progression to AIDS. J Acquir Immune Defic Syndr (1988). 1992;5(1):37–45.
34
Gao
X
,
Nelson
GW
,
Karacki
P
,
Martin
MP
,
Phair
J
,
Kaslow
R
, et al.
Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344(22):1668–75. doi:.https://doi.org/10.1056/NEJM200105313442203
35
Scherer
A
,
Frater
J
,
Oxenius
A
,
Agudelo
J
,
Price
DA
,
Günthard
HF
, et al.; Swiss HIV Cohort Study. Quantifiable cytotoxic T lymphocyte responses and HLA-related risk of progression to AIDS. Proc Natl Acad Sci USA. 2004;101(33):12266–70. doi:.https://doi.org/10.1073/pnas.0404091101
36
Pereyra
F
,
Jia
X
,
McLaren
PJ
,
Telenti
A
,
de Bakker
PI
,
Walker
BD
, et al., International HIV Controllers Study. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330(6010):1551–7. doi:.https://doi.org/10.1126/science.1195271
37
Kulski
JK
. Long noncoding RNA HCP5, a hybrid HLA Class I endogenous retroviral gene: structure, expression, and disease associations. Cells. 2019;8(5):480. doi:.https://doi.org/10.3390/cells8050480
38
Fellay
J
,
Ge
D
,
Shianna
KV
,
Colombo
S
,
Ledergerber
B
,
Cirulli
ET
, et al.; NIAID Center for HIV/AIDS Vaccine Immunology (CHAVI). Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5(12):e1000791. doi:.https://doi.org/10.1371/journal.pgen.1000791
39
Fellay
J
,
Shianna
KV
,
Ge
D
,
Colombo
S
,
Ledergerber
B
,
Weale
M
, et al.
A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317(5840):944–7. doi:.https://doi.org/10.1126/science.1143767
40
Limou
S
,
Le Clerc
S
,
Coulonges
C
,
Carpentier
W
,
Dina
C
,
Delaneau
O
, et al.; ANRS Genomic Group. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02). J Infect Dis. 2009;199(3):419–26. doi:.https://doi.org/10.1086/596067
41
Migueles
SA
,
Sabbaghian
MS
,
Shupert
WL
,
Bettinotti
MP
,
Marincola
FM
,
Martino
L
, et al.
HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA. 2000;97(6):2709–14. doi:.https://doi.org/10.1073/pnas.050567397
42
Hetherington
S
,
Hughes
AR
,
Mosteller
M
,
Shortino
D
,
Baker
KL
,
Spreen
W
, et al.
Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359(9312):1121–2. doi:.https://doi.org/10.1016/S0140-6736(02)08158-8
43
Mallal
S
,
Nolan
D
,
Witt
C
,
Masel
G
,
Martin
AM
,
Moore
C
, et al.
Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–32. doi:.https://doi.org/10.1016/S0140-6736(02)07873-X
44
Mallal
S
,
Phillips
E
,
Carosi
G
,
Molina
JM
,
Workman
C
,
Tomazic
J
, et al.; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358(6):568–79. doi:.https://doi.org/10.1056/NEJMoa0706135
45
Saag
M
,
Balu
R
,
Phillips
E
,
Brachman
P
,
Martorell
C
,
Burman
W
, et al.; Study of Hypersensitivity to Abacavir and Pharmacogenetic Evaluation Study Team. High sensitivity of human leukocyte antigen-b*5701 as a marker for immunologically confirmed abacavir hypersensitivity in white and black patients. Clin Infect Dis. 2008;46(7):1111–8. doi:.https://doi.org/10.1086/529382
46
Kløverpris
HN
,
Leslie
A
,
Goulder
P
. Role of HLA Adaptation in HIV Evolution. Front Immunol. 2016;6:665. doi:.https://doi.org/10.3389/fimmu.2015.00665
47
Arora
J
,
McLaren
PJ
,
Chaturvedi
N
,
Carrington
M
,
Fellay
J
,
Lenz
TL
. HIV peptidome-wide association study reveals patient-specific epitope repertoires associated with HIV control. Proc Natl Acad Sci USA. 2019;116(3):944–9. doi:.https://doi.org/10.1073/pnas.1812548116
48
Bardeskar
NS
,
Mania-Pramanik
J
. HIV and host immunogenetics: unraveling the role of HLA-C. HLA. 2016;88(5):221–31. doi:.https://doi.org/10.1111/tan.12882
49
Apps
R
,
Qi
Y
,
Carlson
JM
,
Chen
H
,
Gao
X
,
Thomas
R
, et al.
Influence of HLA-C expression level on HIV control. Science. 2013;340(6128):87–91. doi:.https://doi.org/10.1126/science.1232685
50
Blais
ME
,
Zhang
Y
,
Rostron
T
,
Griffin
H
,
Taylor
S
,
Xu
K
, et al.
High frequency of HIV mutations associated with HLA-C suggests enhanced HLA-C-restricted CTL selective pressure associated with an AIDS-protective polymorphism. J Immunol. 2012;188(9):4663–70. doi:.https://doi.org/10.4049/jimmunol.1103472
51
Kulkarni
S
,
Qi
Y
,
O’hUigin
C
,
Pereyra
F
,
Ramsuran
V
,
McLaren
P
, et al.
Genetic interplay between HLA-C and MIR148A in HIV control and Crohn disease. Proc Natl Acad Sci USA. 2013;110(51):20705–10. doi:.https://doi.org/10.1073/pnas.1312237110
52
Kulkarni
S
,
Savan
R
,
Qi
Y
,
Gao
X
,
Yuki
Y
,
Bass
SE
, et al.
Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472(7344):495–8. doi:.https://doi.org/10.1038/nature09914
53
Kulpa
DA
,
Collins
KL
. The emerging role of HLA-C in HIV-1 infection. Immunology. 2011;134(2):116–22. doi:.https://doi.org/10.1111/j.1365-2567.2011.03474.x
54
O’hUigin
C
,
Kulkarni
S
,
Xu
Y
,
Deng
Z
,
Kidd
J
,
Kidd
K
, et al.
The molecular origin and consequences of escape from miRNA regulation by HLA-C alleles. Am J Hum Genet. 2011;89(3):424–31. doi:.https://doi.org/10.1016/j.ajhg.2011.07.024
55
Zipeto
D
,
Beretta
A
. HLA-C and HIV-1: friends or foes?
Retrovirology. 2012;9(1):39. doi:.https://doi.org/10.1186/1742-4690-9-39
56
Pelak
K
,
Goldstein
DB
,
Walley
NM
,
Fellay
J
,
Ge
D
,
Shianna
KV
, et al.; Infectious Disease Clinical Research Program HIV Working Group; National Institute of Allergy and Infectious Diseases Center for HIV/AIDS Vaccine Immunology (CHAVI). Host determinants of HIV-1 control in African Americans. J Infect Dis. 2010;201(8):1141–9. doi:.https://doi.org/10.1086/651382
57
Costello
C
,
Tang
J
,
Rivers
C
,
Karita
E
,
Meizen-Derr
J
,
Allen
S
, et al.
HLA-B*5703 independently associated with slower HIV-1 disease progression in Rwandan women. AIDS. 1999;13(14):1990–1. doi:.https://doi.org/10.1097/00002030-199910010-00031
58
Adland
E
,
Hill
M
,
Lavandier
N
,
Csala
A
,
Edwards
A
,
Chen
F
, et al.
Differential immunodominance hierarchy of CD8+ T-Cell responses in HLA-B*27:05- and -B*27:02-mediated control of HIV-1 infection. J Virol. 2018;92(4):e01685-17. doi:.https://doi.org/10.1128/JVI.01685-17
59
Kaslow
RA
,
Carrington
M
,
Apple
R
,
Park
L
,
Muñoz
A
,
Saah
AJ
, et al.
Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med. 1996;2(4):405–11. doi:.https://doi.org/10.1038/nm0496-405
60
Arora
J
,
Pierini
F
,
McLaren
PJ
,
Carrington
M
,
Fellay
J
,
Lenz
TL
. HLA heterozygote advantage against HIV-1 is driven by quantitative and qualitative differences in HLA allele-specific peptide presentation. Mol Biol Evol. 2020;37(3):639–50. doi:.https://doi.org/10.1093/molbev/msz249
61Zuckerman AJ. Hepatitis viruses. In: Medical Microbiology. Baron S, editor. Galveston, TX: University of Texas Medical Branch at Galveston; 1996.
62WHO. Global hepatitis report 2017. Geneva, Switzerland: World Health Organization; 2017; p. 83.
63
Wang
L
,
Wu
XP
,
Zhang
W
,
Zhu
DH
,
Wang
Y
,
Li
YP
, et al.
Evaluation of genetic susceptibility loci for chronic hepatitis B in Chinese: two independent case-control studies. PLoS One. 2011;6(3):e17608. doi:.https://doi.org/10.1371/journal.pone.0017608
64
Matsuura
K
,
Isogawa
M
,
Tanaka
Y
. Host genetic variants influencing the clinical course of hepatitis B virus infection. J Med Virol. 2016;88(3):371–9. doi:.https://doi.org/10.1002/jmv.24350
65
Zhang
Z
,
Wang
C
,
Liu
Z
,
Zou
G
,
Li
J
,
Lu
M
. Host genetic determinants of hepatitis B virus infection. Front Genet. 2019;10:696. doi:.https://doi.org/10.3389/fgene.2019.00696
66
Thio
CL
,
Carrington
M
,
Marti
D
,
O’Brien
SJ
,
Vlahov
D
,
Nelson
KE
, et al.
Class II HLA alleles and hepatitis B virus persistence in African Americans. J Infect Dis. 1999;179(4):1004–6. doi:.https://doi.org/10.1086/314684
67
Thursz
MR
,
Thomas
HC
,
Greenwood
BM
,
Hill
AV
. Heterozygote advantage for HLA class-II type in hepatitis B virus infection. Nat Genet. 1997;17(1):11–2. doi:.https://doi.org/10.1038/ng0997-11
68
Zhang
Y
,
Zhao
F
,
Lan
L
,
Qin
Z
,
Jun
L
. Correlation of HLA-DQB1 gene polymorphism of Xinjiang Uygur with outcome of HBV infection. Int J Clin Exp Med. 2015;8(4):6067–72.
69
Thursz
MR
,
Kwiatkowski
D
,
Allsopp
CE
,
Greenwood
BM
,
Thomas
HC
,
Hill
AV
. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N Engl J Med. 1995;332(16):1065–9. doi:.https://doi.org/10.1056/NEJM199504203321604
70
Thio
CL
,
Thomas
DL
,
Karacki
P
,
Gao
X
,
Marti
D
,
Kaslow
RA
, et al.
Comprehensive analysis of class I and class II HLA antigens and chronic hepatitis B virus infection. J Virol. 2003;77(22):12083–7. doi:.https://doi.org/10.1128/JVI.77.22.12083-12087.2003
71
Nishida
N
,
Sawai
H
,
Kashiwase
K
,
Minami
M
,
Sugiyama
M
,
Seto
WK
, et al.
New susceptibility and resistance HLA-DP alleles to HBV-related diseases identified by a trans-ethnic association study in Asia. PLoS One. 2014;9(2):e86449. doi:.https://doi.org/10.1371/journal.pone.0086449
72
Höhler
T
,
Gerken
G
,
Notghi
A
,
Lubjuhn
R
,
Taheri
H
,
Protzer
U
, et al.
HLA-DRB1*1301 and *1302 protect against chronic hepatitis B. J Hepatol. 1997;26(3):503–7. doi:.https://doi.org/10.1016/S0168-8278(97)80414-X
73
Kamatani
Y
,
Wattanapokayakit
S
,
Ochi
H
,
Kawaguchi
T
,
Takahashi
A
,
Hosono
N
, et al.
A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41(5):591–5. doi:.https://doi.org/10.1038/ng.348
74
Mbarek
H
,
Ochi
H
,
Urabe
Y
,
Kumar
V
,
Kubo
M
,
Hosono
N
, et al.
A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum Mol Genet. 2011;20(19):3884–92. doi:.https://doi.org/10.1093/hmg/ddr301
75
Zhu
M
,
Dai
J
,
Wang
C
,
Wang
Y
,
Qin
N
,
Ma
H
, et al.
Fine mapping the MHC region identified four independent variants modifying susceptibility to chronic hepatitis B in Han Chinese. Hum Mol Genet. 2016;25(6):1225–32. doi:.https://doi.org/10.1093/hmg/ddw003
76
O’Brien
TR
,
Kohaar
I
,
Pfeiffer
RM
,
Maeder
D
,
Yeager
M
,
Schadt
EE
, et al.
Risk alleles for chronic hepatitis B are associated with decreased mRNA expression of HLA-DPA1 and HLA-DPB1 in normal human liver. Genes Immun. 2011;12(6):428–33. doi:.https://doi.org/10.1038/gene.2011.11
77
D’Antonio
M
,
Reyna
J
,
Jakubosky
D
,
Donovan
MK
,
Bonder
M-J
,
Matsui
H
, et al.
Systematic genetic analysis of the MHC region reveals mechanistic underpinnings of HLA type associations with disease. eLife. 2019;8:e48476. doi:.https://doi.org/10.7554/eLife.48476
78
Thomas
R
,
Thio
CL
,
Apps
R
,
Qi
Y
,
Gao
X
,
Marti
D
, et al.
A novel variant marking HLA-DP expression levels predicts recovery from hepatitis B virus infection. J Virol. 2012;86(12):6979–85. doi:.https://doi.org/10.1128/JVI.00406-12
79
Chang
SW
,
Fann
CS
,
Su
WH
,
Wang
YC
,
Weng
CC
,
Yu
CJ
, et al.
A genome-wide association study on chronic HBV infection and its clinical progression in male Han-Taiwanese. PLoS One. 2014;9(6):e99724. doi:.https://doi.org/10.1371/journal.pone.0099724
80
Nishida
N
,
Sawai
H
,
Matsuura
K
,
Sugiyama
M
,
Ahn
SH
,
Park
JY
, et al.
Genome-wide association study confirming association of HLA-DP with protection against chronic hepatitis B and viral clearance in Japanese and Korean. PLoS One. 2012;7(6):e39175. doi:.https://doi.org/10.1371/journal.pone.0039175
81
Trinks
J
,
Nishida
N
,
Hulaniuk
ML
,
Caputo
M
,
Tsuchiura
T
,
Marciano
S
, et al.
Role of HLA-DP and HLA-DQ on the clearance of hepatitis B virus and the risk of chronic infection in a multiethnic population. Liver Int. 2017;37(10):1476–87. doi:.https://doi.org/10.1111/liv.13405
82
Al-Qahtani
AA
,
Al-Anazi
MR
,
Abdo
AA
,
Sanai
FM
,
Al-Hamoudi
W
,
Alswat
KA
, et al.
Association between HLA variations and chronic hepatitis B virus infection in Saudi Arabian patients. PLoS One. 2014;9(1):e80445. doi:.https://doi.org/10.1371/journal.pone.0080445
83
Lau
KC
,
Lam
CW
,
Law
CY
,
Lai
ST
,
Tsang
TY
,
Siu
CW
, et al.
Non-invasive screening of HLA-DPA1 and HLA-DPB1 alleles for persistent hepatitis B virus infection: susceptibility for vertical transmission and toward a personalized approach for vaccination and treatment. Clin Chim Acta. 2011;412(11-12):952–7. doi:.https://doi.org/10.1016/j.cca.2011.01.030
84
Hu
L
,
Zhai
X
,
Liu
J
,
Chu
M
,
Pan
S
,
Jiang
J
, et al.
Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology. 2012;55(5):1426–31. doi:.https://doi.org/10.1002/hep.24799
85
Hu
Z
,
Liu
Y
,
Zhai
X
,
Dai
J
,
Jin
G
,
Wang
L
, et al.
New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat Genet. 2013;45(12):1499–503. doi:.https://doi.org/10.1038/ng.2809
86
Hu
Z
,
Yang
J
,
Xiong
G
,
Shi
H
,
Yuan
Y
,
Fan
L
, et al.
HLA-DPB1 variant effect on hepatitis b virus clearance and liver cirrhosis development among southwest Chinese population. Hepat Mon. 2014;14(8):e19747. doi:.https://doi.org/10.5812/hepatmon.19747
87
Kim
YJ
,
Kim
HY
,
Lee
JH
,
Yu
SJ
,
Yoon
JH
,
Lee
HS
, et al.
A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Hum Mol Genet. 2013;22(20):4233–8. doi:.https://doi.org/10.1093/hmg/ddt266
88
Jiang
DK
,
Ma
XP
,
Yu
H
,
Cao
G
,
Ding
DL
,
Chen
H
, et al.
Genetic variants in five novel loci including CFB and CD40 predispose to chronic hepatitis B. Hepatology. 2015;62(1):118–28. doi:.https://doi.org/10.1002/hep.27794
89
Vermehren
J
,
Lötsch
J
,
Susser
S
,
Wicker
S
,
Berger
A
,
Zeuzem
S
, et al.
A common HLA-DPA1 variant is associated with hepatitis B virus infection but fails to distinguish active from inactive Caucasian carriers. PLoS One. 2012;7(3):e32605. doi:.https://doi.org/10.1371/journal.pone.0032605
90
Tao
J
,
Su
K
,
Yu
C
,
Liu
X
,
Wu
W
,
Xu
W
, et al.
Fine mapping analysis of HLA-DP/DQ gene clusters on chromosome 6 reveals multiple susceptibility loci for HBV infection. Amino Acids. 2015;47(12):2623–34. doi:.https://doi.org/10.1007/s00726-015-2054-6
91
Yu
L
,
Cheng
YJ
,
Cheng
ML
,
Yao
YM
,
Zhang
Q
,
Zhao
XK
, et al.
Quantitative assessment of common genetic variations in HLA-DP with hepatitis B virus infection, clearance and hepatocellular carcinoma development. Sci Rep. 2015;5(1):14933. doi:.https://doi.org/10.1038/srep14933
92
Thio
CL
,
Gao
X
,
Goedert
JJ
,
Vlahov
D
,
Nelson
KE
,
Hilgartner
MW
, et al.
HLA-Cw*04 and hepatitis C virus persistence. J Virol. 2002;76(10):4792–7. doi:.https://doi.org/10.1128/JVI.76.10.4792-4797.2002
93
McKiernan
SM
,
Hagan
R
,
Curry
M
,
McDonald
GS
,
Kelly
A
,
Nolan
N
, et al.
Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology. 2004;40(1):108–14. doi:.https://doi.org/10.1002/hep.20261
94
Amini
M
,
Poustchi
H
. Hepatitis C virus spontaneous clearance: immunology and genetic variance. Viral Immunol. 2012;25(4):241–8. doi:.https://doi.org/10.1089/vim.2011.0052
95
Kuniholm
MH
,
Kovacs
A
,
Gao
X
,
Xue
X
,
Marti
D
,
Thio
CL
, et al.
Specific human leukocyte antigen class I and II alleles associated with hepatitis C virus viremia. Hepatology. 2010;51(5):1514–22. doi:.https://doi.org/10.1002/hep.23515
96
Thio
CL
,
Thomas
DL
,
Goedert
JJ
,
Vlahov
D
,
Nelson
KE
,
Hilgartner
MW
, et al.
Racial differences in HLA class II associations with hepatitis C virus outcomes. J Infect Dis. 2001;184(1):16–21. doi:.https://doi.org/10.1086/321005
97
Hong
Z
,
Smart
G
,
Dawood
M
,
Kaita
K
,
Wen
SW
,
Gomes
J
, et al.
Hepatitis C infection and survivals of liver transplant patients in Canada, 1997-2003. Transplant Proc. 2008;40(5):1466–70. doi:.https://doi.org/10.1016/j.transproceed.2008.03.089
98
Harris
RA
,
Sugimoto
K
,
Kaplan
DE
,
Ikeda
F
,
Kamoun
M
,
Chang
KM
. Human leukocyte antigen class II associations with hepatitis C virus clearance and virus-specific CD4 T cell response among Caucasians and African Americans. Hepatology. 2008;48(1):70–9. doi:.https://doi.org/10.1002/hep.22287
99
Huang
J
,
Huang
K
,
Xu
R
,
Wang
M
,
Liao
Q
,
Xiong
H
, et al.
The Associations of HLA-A*02:01 and DRB1*11:01 with Hepatitis C Virus Spontaneous Clearance Are Independent of IL28B in the Chinese Population. Sci Rep. 2016;6(1):31485. doi:.https://doi.org/10.1038/srep31485
100
Gauthiez
E
,
Habfast-Robertson
I
,
Rüeger
S
,
Kutalik
Z
,
Aubert
V
,
Berg
T
, et al.; Swiss Hepatitis C Cohort Study. A systematic review and meta-analysis of HCV clearance. Liver Int. 2017;37(10):1431–45. doi:.https://doi.org/10.1111/liv.13401
101
Duggal
P
,
Thio
CL
,
Wojcik
GL
,
Goedert
JJ
,
Mangia
A
,
Latanich
R
, et al.
Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann Intern Med. 2013;158(4):235–45. doi:.https://doi.org/10.7326/0003-4819-158-4-201302190-00003
102
Miki
D
,
Ochi
H
,
Takahashi
A
,
Hayes
CN
,
Urabe
Y
,
Abe
H
, et al.
HLA-DQB1*03 confers susceptibility to chronic hepatitis C in Japanese: a genome-wide association study. PLoS One. 2013;8(12):e84226. doi:.https://doi.org/10.1371/journal.pone.0084226
103
Xu
X
,
Yue
M
,
Jiang
L
,
Deng
X
,
Zhang
Y
,
Zhang
Y
, et al.
Genetic variants in human leukocyte antigen-DP influence both hepatitis C virus persistence and hepatitis C virus F protein generation in the Chinese Han population. Int J Mol Sci. 2014;15(6):9826–43. doi:.https://doi.org/10.3390/ijms15069826
104
Thoens
C
,
Heinold
A
,
Lindemann
M
,
Horn
PA
,
Chang
DI
,
Scherbaum
N
, et al.
A single-nucleotide polymorphism upstream of the HLA-C locus is associated with an anti-hepatitis C virus-seronegative state in a high-risk exposed cohort. J Infect Dis. 2018;218(12):2016–9. doi:.https://doi.org/10.1093/infdis/jiy492
105
Sawai
H
,
Nishida
N
,
Khor
SS
,
Honda
M
,
Sugiyama
M
,
Baba
N
, et al.
Genome-wide association study identified new susceptible genetic variants in HLA class I region for hepatitis B virus-related hepatocellular carcinoma. Sci Rep. 2018;8(1):7958. doi:.https://doi.org/10.1038/s41598-018-26217-7
106Omae Y, Tokunaga K. Genetics of infectious diseases, in Genome-wide association studies. Tsunoda T, Tanaka T, and Nakamura Y, editors. Singapore: Springer; 2019. pp. 145−74.
107
Spinola
H
. HLA loci and respiratory infectious diseases. Journal of Respiratory Research. 2016;2(3):56–66. doi:.https://doi.org/10.17554/j.issn.2412-2424.2016.02.15
108
Oliveira-Cortez
A
,
Melo
AC
,
Chaves
VE
,
Condino-Neto
A
,
Camargos
P
. Do HLA class II genes protect against pulmonary tuberculosis? A systematic review and meta-analysis. Eur J Clin Microbiol Infect Dis. 2016;35(10):1567–80. doi:.https://doi.org/10.1007/s10096-016-2713-x
109
Harishankar
M
,
Selvaraj
P
,
Bethunaickan
R
. Influence of genetic polymorphism towards pulmonary tuberculosis susceptibility. Front Med (Lausanne). 2018;5:213. doi:.https://doi.org/10.3389/fmed.2018.00213
110
Yim
JJ
,
Selvaraj
P
. Genetic susceptibility in tuberculosis. Respirology. 2010;15(2):241–56. doi:.https://doi.org/10.1111/j.1440-1843.2009.01690.x
111
Kone
A
,
Diarra
B
,
Cohen
K
,
Diabate
S
,
Kone
B
,
Diakite
MT
, et al.
Differential HLA allele frequency in Mycobacterium africanum vs Mycobacterium tuberculosis in Mali. HLA. 2019;93(1):24–31. doi:.https://doi.org/10.1111/tan.13448
112
Sveinbjornsson
G
,
Gudbjartsson
DF
,
Halldorsson
BV
,
Kristinsson
KG
,
Gottfredsson
M
,
Barrett
JC
, et al.
HLA class II sequence variants influence tuberculosis risk in populations of European ancestry. Nat Genet. 2016;48(3):318–22. doi:.https://doi.org/10.1038/ng.3498
113
Qi
H
,
Zhang
YB
,
Sun
L
,
Chen
C
,
Xu
B
,
Xu
F
, et al.
Discovery of susceptibility loci associated with tuberculosis in Han Chinese. Hum Mol Genet. 2017;26(23):4752–63. doi:.https://doi.org/10.1093/hmg/ddx365
114
Gagneux
S
,
DeRiemer
K
,
Van
T
,
Kato-Maeda
M
,
de Jong
BC
,
Narayanan
S
, et al.
Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103(8):2869–73. doi:.https://doi.org/10.1073/pnas.0511240103
115
Toyo-Oka
L
,
Mahasirimongkol
S
,
Yanai
H
,
Mushiroda
T
,
Wattanapokayakit
S
,
Wichukchinda
N
, et al.
Strain-based HLA association analysis identified HLA-DRB1*09:01 associated with modern strain tuberculosis. HLA. 2017;90(3):149–56. doi:.https://doi.org/10.1111/tan.13070
116
Ndzi
EN
,
Nkenfou
CN
,
Pefura
EWY
,
Mekue
LCM
,
Guiedem
E
,
Nguefeu
CN
, et al.
Tuberculosis diagnosis: algorithm that May discriminate latent from active tuberculosis. Heliyon. 2019;5(10):e02559. doi:.https://doi.org/10.1016/j.heliyon.2019.e02559
117
Jarduli
LR
,
Sell
AM
,
Reis
PG
,
Sippert
EA
,
Ayo
CM
,
Mazini
PS
, et al.
Role of HLA, KIR, MICA, and cytokines genes in leprosy. BioMed Res Int. 2013;2013:989837. doi:.https://doi.org/10.1155/2013/989837
118
Wong
SH
,
Gochhait
S
,
Malhotra
D
,
Pettersson
FH
,
Teo
YY
,
Khor
CC
, et al.
Leprosy and the adaptation of human toll-like receptor 1. PLoS Pathog. 2010;6(7):e1000979. doi:.https://doi.org/10.1371/journal.ppat.1000979
119
Zhang
FR
,
Huang
W
,
Chen
SM
,
Sun
LD
,
Liu
H
,
Li
Y
, et al.
Genomewide association study of leprosy. N Engl J Med. 2009;361(27):2609–18. doi:.https://doi.org/10.1056/NEJMoa0903753
120
Liu
H
,
Irwanto
A
,
Fu
X
,
Yu
G
,
Yu
Y
,
Sun
Y
, et al.
Discovery of six new susceptibility loci and analysis of pleiotropic effects in leprosy. Nat Genet. 2015;47(3):267–71. doi:.https://doi.org/10.1038/ng.3212
121
Krause-Kyora
B
,
Nutsua
M
,
Boehme
L
,
Pierini
F
,
Pedersen
DD
,
Kornell
SC
, et al.
Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans. Nat Commun. 2018;9(1):1569. doi:.https://doi.org/10.1038/s41467-018-03857-x
122
Alter
A
,
Huong
NT
,
Singh
M
,
Orlova
M
,
Van Thuc
N
,
Katoch
K
, et al.
Human leukocyte antigen class I region single-nucleotide polymorphisms are associated with leprosy susceptibility in Vietnam and India. J Infect Dis. 2011;203(9):1274–81. doi:.https://doi.org/10.1093/infdis/jir024
123WHO. World Malaria Report 2019. Geneva, Switzerland: World Health Organization; 2019.
124
Verra
F
,
Mangano
VD
,
Modiano
D
. Genetics of susceptibility to Plasmodium falciparum: from classical malaria resistance genes towards genome-wide association studies. Parasite Immunol. 2009;31(5):234–53. doi:.https://doi.org/10.1111/j.1365-3024.2009.01106.x
125
Garcia
A
,
Milet
J
,
Courtin
D
,
Sabbagh
A
,
Massaro
JD
,
Castelli
EC
, et al.
Association of HLA-G 3'UTR polymorphisms with response to malaria infection: a first insight. Infect Genet Evol. 2013;16:263–9. doi:.https://doi.org/10.1016/j.meegid.2013.02.021
126
Sabbagh
A
,
Courtin
D
,
Milet
J
,
Massaro
JD
,
Castelli
EC
,
Migot-Nabias
F
, et al.
Association of HLA-G 3′ untranslated region polymorphisms with antibody response against Plasmodium falciparum antigens: preliminary results. Tissue Antigens. 2013;82(1):53–8. doi:.https://doi.org/10.1111/tan.12140
127
Hill
AV
,
Allsopp
CE
,
Kwiatkowski
D
,
Anstey
NM
,
Twumasi
P
,
Rowe
PA
, et al.
Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991;352(6336):595–600. doi:.https://doi.org/10.1038/352595a0
128
Hill
AV
,
Elvin
J
,
Willis
AC
,
Aidoo
M
,
Allsopp
CE
,
Gotch
FM
, et al.
Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature. 1992;360(6403):434–9. doi:.https://doi.org/10.1038/360434a0
129
Lyke
KE
,
Fernández-Viňa
MA
,
Cao
K
,
Hollenbach
J
,
Coulibaly
D
,
Kone
AK
, et al.
Association of HLA alleles with Plasmodium falciparum severity in Malian children. Tissue Antigens. 2011;77(6):562–71. doi:.https://doi.org/10.1111/j.1399-0039.2011.01661.x
130
Garamszegi
LZ
. Global distribution of malaria-resistant MHC-HLA alleles: the number and frequencies of alleles and malaria risk. Malar J. 2014;13(1):349. doi:.https://doi.org/10.1186/1475-2875-13-349
131
Sanchez-Mazas
A
,
Černý
V
,
Di
D
,
Buhler
S
,
Podgorná
E
,
Chevallier
E
, et al.
The HLA-B landscape of Africa: Signatures of pathogen-driven selection and molecular identification of candidate alleles to malaria protection. Mol Ecol. 2017;26(22):6238–52. doi:.https://doi.org/10.1111/mec.14366
132
Yamazaki
A
,
Yasunami
M
,
Ofori
M
,
Horie
H
,
Kikuchi
M
,
Helegbe
G
, et al.
Human leukocyte antigen class I polymorphisms influence the mild clinical manifestation of Plasmodium falciparum infection in Ghanaian children. Hum Immunol. 2011;72(10):881–8. doi:.https://doi.org/10.1016/j.humimm.2011.06.007
133
Gilbert
SC
,
Plebanski
M
,
Gupta
S
,
Morris
J
,
Cox
M
,
Aidoo
M
, et al.
Association of malaria parasite population structure, HLA, and immunological antagonism. Science. 1998;279(5354):1173–7. doi:.https://doi.org/10.1126/science.279.5354.1173
134
Laval
G
,
Peyrégne
S
,
Zidane
N
,
Harmant
C
,
Renaud
F
,
Patin
E
, et al.
Recent adaptive acquisition by African rainforest hunter-gatherers of the late pleistocene sickle-cell mutation suggests past differences in malaria exposure. Am J Hum Genet. 2019;104(3):553–61. doi:.https://doi.org/10.1016/j.ajhg.2019.02.007
135
Schrider
DR
,
Kern
AD
. Soft sweeps are the dominant mode of adaptation in the human genome. Mol Biol Evol. 2017;34(8):1863–77. doi:.https://doi.org/10.1093/molbev/msx154
136
Pritchard
JK
,
Pickrell
JK
,
Coop
G
. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010;20(4):R208–15. doi:.https://doi.org/10.1016/j.cub.2009.11.055
137
Goeury
T
,
Creary
LE
,
Brunet
L
,
Galan
M
,
Pasquier
M
,
Kervaire
B
, et al.
Deciphering the fine nucleotide diversity of full HLA class I and class II genes in a well-documented population from sub-Saharan Africa. HLA. 2018;91(1):36–51. doi:.https://doi.org/10.1111/tan.13180
138
Norman
PJ
,
Hollenbach
JA
,
Nemat-Gorgani
N
,
Guethlein
LA
,
Hilton
HG
,
Pando
MJ
, et al.
Co-evolution of human leukocyte antigen (HLA) class I ligands with killer-cell immunoglobulin-like receptors (KIR) in a genetically diverse population of sub-Saharan Africans. PLoS Genet. 2013;9(10):e1003938. doi:.https://doi.org/10.1371/journal.pgen.1003938
139
Lima-Junior
JC
,
Pratt-Riccio
LR
. Major Histocompatibility Complex and malaria: focus on Plasmodium vivax infection. Front Immunol. 2016;7:13. doi:.https://doi.org/10.3389/fimmu.2016.00013
140
Crosslin
DR
,
Carrell
DS
,
Burt
A
,
Kim
DS
,
Underwood
JG
,
Hanna
DS
, et al.
Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015;16(1):1–7. doi:.https://doi.org/10.1038/gene.2014.51
141
Stanaway
IB
,
Hall
TO
,
Rosenthal
EA
,
Palmer
M
,
Naranbhai
V
,
Knevel
R
, et al.; eMERGE Network. The eMERGE genotype set of 83,717 subjects imputed to ~40 million variants genome wide and association with the herpes zoster medical record phenotype. Genet Epidemiol. 2019;43(1):63–81.
142
Chen
D
,
McKay
JD
,
Clifford
G
,
Gaborieau
V
,
Chabrier
A
,
Waterboer
T
, et al.
Genome-wide association study of HPV seropositivity. Hum Mol Genet. 2011;20(23):4714–23. doi:.https://doi.org/10.1093/hmg/ddr383
143
Dunstan
SJ
,
Hue
NT
,
Han
B
,
Li
Z
,
Tram
TT
,
Sim
KS
, et al.
Variation at HLA-DRB1 is associated with resistance to enteric fever. Nat Genet. 2014;46(12):1333–6. doi:.https://doi.org/10.1038/ng.3143
144
DeLorenze
GN
,
Nelson
CL
,
Scott
WK
,
Allen
AS
,
Ray
GT
,
Tsai
AL
, et al.
Polymorphisms in HLA Class II genes are associated with susceptibility to Staphylococcus aureus infection in a white population. J Infect Dis. 2016;213(5):816–23. doi:.https://doi.org/10.1093/infdis/jiv483
145
Fakiola
M
,
Strange
A
,
Cordell
HJ
,
Miller
EN
,
Pirinen
M
,
Su
Z
, et al.; LeishGEN Consortium; Wellcome Trust Case Control Consortium 2. Common variants in the HLA-DRB1-HLA-DQA1 HLA class II region are associated with susceptibility to visceral leishmaniasis. Nat Genet. 2013;45(2):208–13. doi:.https://doi.org/10.1038/ng.2518
146
Blackwell
JM
,
Fakiola
M
,
Castellucci
LC
. Human genetics of leishmania infections. Hum Genet. 2020. doi:.https://doi.org/10.1007/s00439-020-02130-w
147
Singh
T
,
Fakiola
M
,
Oommen
J
,
Singh
AP
,
Singh
AK
,
Smith
N
, et al.
Epitope-Binding characteristics for risk versus protective DRB1 alleles for visceral leishmaniasis. J Immunol. 2018;200(8):2727–37. doi:.https://doi.org/10.4049/jimmunol.1701764
148
Zerbino
DR
,
Achuthan
P
,
Akanni
W
,
Amode
MR
,
Barrell
D
,
Bhai
J
, et al.
Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–61. doi:.https://doi.org/10.1093/nar/gkx1098
149
Sherry
ST
,
Ward
MH
,
Kholodov
M
,
Baker
J
,
Phan
L
,
Smigielski
EM
, et al.
dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi:.https://doi.org/10.1093/nar/29.1.308
150
Crux
NB
,
Elahi
S
. Human Leukocyte Antigen (HLA) and immune regulation: how do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections?
Front Immunol. 2017;8:832. doi:.https://doi.org/10.3389/fimmu.2017.00832
151
Robinson
J
,
Guethlein
LA
,
Cereb
N
,
Yang
SY
,
Norman
PJ
,
Marsh
SGE
, et al.
Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet. 2017;13(6):e1006862. doi:.https://doi.org/10.1371/journal.pgen.1006862
152
Sanchez-Mazas
A
,
Meyer
D
. The relevance of HLA sequencing in population genetics studies. J Immunol Res. 2014;2014:971818. doi:.https://doi.org/10.1155/2014/971818
153
Meyer
D
,
Nunes
K
. HLA imputation, what is it good for?
Hum Immunol. 2017;78(3):239–41. doi:.https://doi.org/10.1016/j.humimm.2017.02.007
154
Cullen
M
,
Noble
J
,
Erlich
H
,
Thorpe
K
,
Beck
S
,
Klitz
W
, et al.
Characterization of recombination in the HLA class II region. Am J Hum Genet. 1997;60(2):397–407.
155
Cullen
M
,
Perfetto
SP
,
Klitz
W
,
Nelson
G
,
Carrington
M
. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet. 2002;71(4):759–76. doi:.https://doi.org/10.1086/342973
156
Sanchez-Mazas
A
,
Djoulah
S
,
Busson
M
,
Le Monnier de Gouville
I
,
Poirier
JC
,
Dehay
C
, et al.
A linkage disequilibrium map of the MHC region based on the analysis of 14 loci haplotypes in 50 French families. Eur J Hum Genet. 2000;8(1):33–41. doi:.https://doi.org/10.1038/sj.ejhg.5200391
157
Bugawan
TL
,
Klitz
W
,
Blair
A
,
Erlich
HA
. High-resolution HLA class I typing in the CEPH families: analysis of linkage disequilibrium among HLA loci. Tissue Antigens. 2000;56(5):392–404. doi:.https://doi.org/10.1034/j.1399-0039.2000.560502.x
158
Achour
Y
,
Ben Hamad
M
,
Chaabane
S
,
Rebai
A
,
Marzouk
S
,
Mahfoudh
N
, et al.
Analysis of two susceptibility SNPs in HLA region and evidence of interaction between rs6457617 in HLA-DQB1 and HLA-DRB1*04 locus on Tunisian rheumatoid arthritis. J Genet. 2017;96(6):911–8. doi:.https://doi.org/10.1007/s12041-017-0855-y
159
Lenz
TL
,
Deutsch
AJ
,
Han
B
,
Hu
X
,
Okada
Y
,
Eyre
S
, et al.
Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet. 2015;47(9):1085–90. doi:.https://doi.org/10.1038/ng.3379
160
Goudey
B
,
Abraham
G
,
Kikianty
E
,
Wang
Q
,
Rawlinson
D
,
Shi
F
, et al.
Interactions within the MHC contribute to the genetic architecture of celiac disease. PLoS One. 2017;12(3):e0172826. doi:.https://doi.org/10.1371/journal.pone.0172826
161
de Bakker
PI
,
McVean
G
,
Sabeti
PC
,
Miretti
MM
,
Green
T
,
Marchini
J
, et al.
A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38(10):1166–72. doi:.https://doi.org/10.1038/ng1885
162
Gonzalez-Galarza
FF
,
McCabe
A
,
Santos
EJMD
,
Jones
J
,
Takeshita
L
,
Ortega-Rivera
ND
, et al.
Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2019;gkz1029. doi:.https://doi.org/10.1093/nar/gkz1029
163
Singh
R
,
Kaul
R
,
Kaul
A
,
Khan
K
. A comparative review of HLA associations with hepatitis B and C viral infections across global populations. World J Gastroenterol. 2007;13(12):1770–87. doi:.https://doi.org/10.3748/wjg.v13.i12.1770
164
Auton
A
,
Brooks
LD
,
Durbin
RM
,
Garrison
EP
,
Kang
HM
,
Korbel
JO
, et al., 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:.https://doi.org/10.1038/nature15393
165
Tshabalala
M
,
Mellet
J
,
Pepper
MS
. Human Leukocyte Antigen Diversity: A Southern African Perspective. J Immunol Res. 2015;2015:746151. doi:.https://doi.org/10.1155/2015/746151
166Ndiaye Diallo R, Gadji M, Hennig BJ, Gueye MV, Gaye A, Diop JPD, et al. Strengthening human genetics research in Africa: report of the 9th meeting of the African Society of Human Genetics in Dakar in May 2016. Glob Health Epidemiol Genom. 2017. 2: p. e10.DOI: https://doi.org/10.1017/gheg.2017.3.
167
Sirugo
G
,
Williams
SM
,
Tishkoff
SA
. The missing diversity in human genetic studies. Cell. 2019;177(1):26–31. doi:.https://doi.org/10.1016/j.cell.2019.02.048
168
Ramsay
M
. Africa: continent of genome contrasts with implications for biomedical research and health. FEBS Lett. 2012;586(18):2813–9. doi:.https://doi.org/10.1016/j.febslet.2012.07.061
169
McDougall
I
,
Brown
FH
,
Fleagle
JG
. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature. 2005;433(7027):733–6. doi:.https://doi.org/10.1038/nature03258
170
Hublin
J-J
,
Ben-Ncer
A
,
Bailey
SE
,
Freidline
SE
,
Neubauer
S
,
Skinner
MM
, et al.
New fossils from Jebel Irhoud, Morocco and the pan-African origin of Homo sapiens. Nature. 2017;546(7657):289–92. doi:.. Correction in: Nature. 2018;70(141):986
https://doi.org/10.1038/nature22336
171
Meyer
D
,
Single
RM
,
Mack
SJ
,
Erlich
HA
,
Thomson
G
. Signatures of demographic history and natural selection in the human major histocompatibility complex Loci. Genetics. 2006;173(4):2121–42. doi:.https://doi.org/10.1534/genetics.105.052837
172
Kwiatkowski
DP
. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77(2):171–92. doi:.https://doi.org/10.1086/432519
173
Mangano
VD
,
Modiano
D
. An evolutionary perspective of how infection drives human genome diversity: the case of malaria. Curr Opin Immunol. 2014;30:39–47. doi:.https://doi.org/10.1016/j.coi.2014.06.004
174
Marquet
S
. Overview of human genetic susceptibility to malaria: From parasitemia control to severe disease. Infect Genet Evol. 2018;66:399–409. doi:.https://doi.org/10.1016/j.meegid.2017.06.001
175
Slade
RW
,
McCallum
HI
. Overdominant vs. frequency-dependent selection at MHC loci. Genetics. 1992;132(3):861–4.
176
Ansari
MA
,
Pedergnana
V
,
L C Ip
C
,
Magri
A
,
Von Delft
A
,
Bonsall
D
, et al.; STOP-HCV Consortium. Genome-to-genome analysis highlights the effect of the human innate and adaptive immune systems on the hepatitis C virus. Nat Genet. 2017;49(5):666–73. doi:.https://doi.org/10.1038/ng.3835
177
Korber
B
,
Muldoon
M
,
Theiler
J
,
Gao
F
,
Gupta
R
,
Lapedes
A
, et al.
Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288(5472):1789–96. doi:.https://doi.org/10.1126/science.288.5472.1789
178
Faria
NR
,
Rambaut
A
,
Suchard
MA
,
Baele
G
,
Bedford
T
,
Ward
MJ
, et al.
The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014;346(6205):56–61. doi:.https://doi.org/10.1126/science.1256739
179
Karlsson
EK
,
Kwiatkowski
DP
,
Sabeti
PC
. Natural selection and infectious disease in human populations. Nat Rev Genet. 2014;15(6):379–93. doi:.https://doi.org/10.1038/nrg3734
180
Andrés
AM
,
Hubisz
MJ
,
Indap
A
,
Torgerson
DG
,
Degenhardt
JD
,
Boyko
AR
, et al.
Targets of balancing selection in the human genome. Mol Biol Evol. 2009;26(12):2755–64. doi:.https://doi.org/10.1093/molbev/msp190
181
Penman
BS
,
Gupta
S
. Detecting signatures of past pathogen selection on human HLA loci: are there needles in the haystack?
Parasitology. 2018;145(6):731–9. doi:.https://doi.org/10.1017/S0031182017001159
182
Currat
M
,
Poloni
ES
,
Sanchez-Mazas
A
. Human genetic differentiation across the Strait of Gibraltar. BMC Evol Biol. 2010;10(1):237. doi:.https://doi.org/10.1186/1471-2148-10-237
183
Di
D
,
Sanchez-Mazas
A
,
Currat
M
. Computer simulation of human leukocyte antigen genes supports two main routes of colonization by human populations in East Asia. BMC Evol Biol. 2015;15(1):240. doi:.https://doi.org/10.1186/s12862-015-0512-0
184
Dos Santos Francisco
R
,
Buhler
S
,
Nunes
JM
,
Bitarello
BD
,
França
GS
,
Meyer
D
, et al.
HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms. Immunogenetics. 2015;67(11-12):651–63. doi:.https://doi.org/10.1007/s00251-015-0875-9