Holistic cancer genome profiling for every patient
DOI: https://doi.org/10.4414/smw.2020.20158
Serena
Nik-Zainal, Yasin
Memari, Helen R.
Davies
aMRC Cancer Unit, Hutchison/MRC Research Centre, University of Cambridge, UK
bAcademic Laboratory of Medical Genetics, Addenbrookes Treatment Centre, Cambridge, UK
Summary
Technological advances in the ability to read the human genome have accelerated the speed of sequencing, such that today we can perform whole genome sequencing (WGS) in one day. Until recently, genomic studies have largely been limited to seeking novel scientific discoveries. The application of new insights gained through cancer WGS into the clinical domain, have been relatively limited. Looking ahead, a vast amount of data can be generated by genomic studies. Of note, excellent organisation of genomic and clinical data permits the application of machine-learning methods which can lead to the development of clinical algorithms that could assist future clinicians and genomicists in the analysis and interpretation of individual cancer genomes. Here, we describe what can be gleaned from holistic whole cancer genome profiling and argue that we must build the infrastructure and educational frameworks to support the modern clinical genomicist to prepare for a future where WGS will be the norm.
Introduction
The development of sequencing-by-synthesis [1] catapulted the field of genomics into a new age. The ability to immobilise each DNA molecule on the surface of a chip and continuously read each nucleotide from every DNA molecule as a result of novel “reversible” terminator chemistry increased the speed and scale of sequencing by orders of magnitude [1]. The term “massively parallel sequencing” (MPS) was coined. Today, we can sequence a whole human genome in one day.
When studying cancer genomics of solid tumours, two samples are required per cancer patient; a DNA sample from the cancer (”tumour” DNA representative of the cancer clone) and a DNA sample extracted from peripheral blood lymphocytes (”normal” DNA derived from a heterogeneous cellular population representative of the germline genome). Sequencing tumour and normal DNA allows the identification of “somatic mutations”, those which are acquired and present only in the cancer and not the germline. The two DNA samples from each patient are subjected to fragmentation independently, each generating billions of DNA fragments. Size-selection of a fragment size of interest is performed, usually 400–600 base pairs for a whole genome. Around 150 nucleotides at each end of the size-selected fragments are sequenced using MPS technology. The aim is for each of the 3,000,000,000 base pairs present in the human genome to be re-sequenced at least 30 times on average. This strategy, called paired-end high-coverage sequencing, is a general principle that can be adapted (e.g., single-ended sequencing, 75 or 100 base-pair read lengths and/or variable fragment sizes). In a whole genome sequencing (WGS) experiment, the entire human genome is captured. In a whole exome sequencing (WES) experiment, protein-coding sequences are captured (~1.5% of the genome). Targeted sequencing experiments tend to encompass genes of interest and a raft of other loci that may be informative (e.g., gene fusions and copy number alterations) (~0.1% or less of the genome). In terms of sequencing costs, WGS is the most expensive and targeted experiments are cheapest. There are also associated costs of storage, computing and analysis to consider.
What we obtain in terms of genomics depends on the sequencing experiment. When a WGS is performed, one obtains “driver” mutations –causally implicated mutations of carcinogenesis, passenger variants, structural variants, copy number aberrations and many other insights into the noncoding genome [2, 3]. Other sequencing experiments may be cheaper but limited to only driver mutations [4] or selected patterns (table 1). Although there have been great efforts to enhance these panels [5], one will only see what one is looking for. Opportunities for new discoveries are limited.
Table 1 Data obtainable from various sequencing experiments.
| |
Whole genome sequencing
|
Whole exome sequencing
|
Targeted sequencing
|
| Drivers |
Substitutions and indels
Structural variation including gene fusions |
Substitutions and indels |
Substitutions and indels limited to specific genes on panel |
| Copy number drivers (amplifications and homozygous deletions) |
Comprehensive |
Semi-reliable |
Limited to genes on panel and not always reliable |
| Mutational signatures |
Comprehensive |
Limited |
Not reliable |
| Complex rearrangements |
Comprehensive |
Not reliable |
Not detectable |
| Copy number aberrations |
Comprehensive |
Semi-reliable |
Not detectable |
| Germline variation |
Comprehensive |
High-penetrance alleles |
Limited |
| Phylogenetic trees |
Possible.
Limited branching unless very high-depth |
Possible |
Limited |
In this review, we highlight what else can be seen in the whole human cancer genome and why it might be useful clinically. As we look towards a future when genomics may become a standard part of cancer diagnostics, we highlight current issues and how we should tackle them going forward.
Whole cancer genomes
Driver mutations in cancer
Decades of cancer research were focused on discovery of driver mutations, positively-selected genetic changes that occur in “cancer genes”, because these became targets for developing new therapeutic agents [6–9]. A key contribution of MPS, in the earlier part of the 21st century, was the acceleration of novel cancer gene discovery [10]. Improved sequencing affordability resulted in more cancers being sequenced per experiment. Thus, rare, low-frequency cancer genes present in common cancers [4, 10–15], as well as common cancer genes present in rare cancers [16, 17], were increasingly identified. These studies also revealed that there was an enormous amount of inter-tumour heterogeneity between patients, with most patients having different combinations of a long list of drivers even when they shared the same tumour type [4]. Hence, using individual driver mutations or cancer genes as a strategy to develop therapeutic targets is likely to be of limited success. Given that there are many hundreds of driver mutations / cancer genes and a mere handful of successfully developed targeted therapies that are clinically available after four decades of cancer research, we need to find alternative strategies to treat cancers more effectively.
Passenger mutations: a resource of historical information
Notably, cancers contain far more than the handful of drivers, estimated to be between one to ten per tumour [6, 8]. Each cancer carries thousands of “passenger” mutations that have historically been thought of as unimportant, inconsequential mutational noise [6]. However, they are in fact a mine of information, reporting the biological history of the tumour [2, 18, 19]. The catalogue of somatic mutations that is revealed through cancer sequencing is the final outcome of the mutational processes that have occurred through malignant transformation [2, 18, 19]. Each mutational process leaves its characteristic imprint or mutational signature on the cancer genome, defined by the mechanisms of DNA damage and DNA repair of which it is comprised [2, 18, 19]. Whatever the nature of the mutational process, the final set of mutations, be they substitutions, insertions/deletions or structural variation, is also determined by the strength and duration of exposure to each mutational process [2, 18, 19]. Some exposures may be weak or moderate in intensity, whereas others may be strong in their effect [19]. Similarly, some exposures might be on-going through the entire lifetime of the patient, even preceding the formation of the cancer, and some may start late or become dominant later in tumorigenesis [19].
WGS experiments permitted us to use mathematical methods to distil the mutational signatures buried within these cancers. In 2012, using WGS data from just 21 breast cancers, the first five mutational signatures were revealed [2]. Subsequently, similar approaches were used to unearth at least 21 different mutation signatures across 30 different cancer types [18]. This included signatures associated with past exposure to environmental carcinogens, such as tobacco smoke in lung cancer and ultraviolet radiation in malignant melanoma, and endogenous sources of mutagenesis including ubiquitous deamination at methyl-cytosines seen in nearly all human cancers and the activity of other dysregulated proteins such as polymerase epsilon (POLE). Numerous novel signatures have also been excavated. Today, there are many on-going efforts to experimentally validate [20, 21] and fully characterise the aetiologies of these mutational signatures in order to understand the sources of mutagenesis in human cancer.
It should be noted that these are early days in this field. It was initially focused almost exclusively on substitutions, whereas today there are mutational signatures reported for insertions/deletions (though not verified or widely taken up so far) [22], structural variation [23] and copy number aberrations [24]. Indeed, even the total tally of substitution signatures is not static. In excess of 40 signatures have been reported [22], although this is expected to change as more cancers are sequenced in the future and as methods and thinking evolve on these concepts.
Other angles
WGS also permits other findings to be revealed including gene fusion events and retrotransposition-driven genetic changes [25]. There remains much to learn about 3D compaction of the human genome [26, 27], enhancer [28] and promoter mutations [29] and the consequences of complex rearrangements in the genome [30], including the formation of micronuclei [31]. WGS data also contain information about immunogenicity of the tumour, including human leucocyte antigen (HLA) and T-cell receptor information [32, 33].
Quite apart from the somatic cancer genome, there is a germline genome, which reportedly sets our lifetime risk of cancer [34], modifiable by the variety of lifestyle factors that we are exposed to. Currently, we know very little about how the germline genome interacts with or influences somatic mutation acquisition [28, 35]. Unlike acquired mutations, there are well-known inherited predisposition genes that have significant implications for relatives if identified as a new finding in a cancer patient [23, 36]. There are also a variety of moderate-penetrance and low-penetrance genetic alleles that when combined can provide measures of cancer risk called polygenic risk scores [34, 37]. How these data from WGS can be used effectively for clinical application remains unknown. It is likely to become clearer with the increasing availability of WGS in the near future.
Beyond the genome, there is the transcriptome and methylome, presenting additional layers of data per patient. As a scientific community, we are still learning how best to integrate these data. The areas of metabolomics and proteomics are also coming to the fore. We may look forward to a future where these different modalities are available across each cancer, providing very rich datasets from which to learn.
Tumour phylogenies
Another area of particular growth has been the study of the phylogenetic evolution of cancers [3]. Tumour evolutionary histories or phylogenies can be constructed by taking multiple samples per patient, separated either by space [38, 39] (multiple primaries, or multiple sites per primary) or by time [40] (e.g., primary and metastasis). The digital nature of modern sequencing technology also permits estimation of subpopulations of cells within a single cancer sample [3].
For a sample with sequence coverage of 40-fold, this implies that sequence data from approximately 40 DNA molecules are available at any particular genomic coordinate, on average. If the sample were representative of the germline, a heterozygous variant of a diploid chromosome would be expected to be present in approximately 50% of reads (~20 reads), whereas a homozygous variant should be present in 100% of reads. For a tumour sample, a fraction of reads will come from normal cells such as lymphocytes or stromal tissue, but the remaining reads should be representative of the tumour. A heterozygous variant in a diploid chromosome in the tumour genome should be present in half of the remaining reads, whatever that may be. Thus, if there was an estimated 70% tumour cellularity, then a heterozygous mutation would be present at 35% variant allele fraction. When collections of mutations do not abide by this rule and are instead consistently present in a subset of the expected fraction of reads, this can be used to infer the presence of a subclonal populations in a cancer [3]. Numerous mathematical methods have been developed to identify such subpopulations [3, 41]. Phylogenetic trees of each primary cancer can therefore be constructed to a finite level of resolution.
What these studies have collectively shown us is that cancers may begin as a clonal outgrowth but inevitably evolve under the pressure of environmental changes, with resultant subclonal populations in due course. Intra-tumour heterogeneity is in fact the norm.
Insights from latest technology: adjustments to a biological perspective
Recent advancements in sequencing technology, including low-volume and single-cell sequencing, have led us to gain some fascinating new insights [42, 43] that may require us to reflect and adjust our understanding of cancer biology.
First, the concept of “driver mutations” requires re-thinking. In the past, we could see these drivers in cancers and assumed that they were causally implicated in tumorigenesis because of observed enrichment of these mutations in particular cancers, such as TP53 (tumour protein p53) in breast cancer, APC (adenomatous polyposis coli) in colorectal cancer and KRAS (Ki-ras2 Kirsten rat sarcoma viral oncogene homologue) in pancreatic cancer. The application of MPS to normal, noncancerous tissues, however, demonstrates that there are “drivers” in all normal tissues too [42–45]. In some instances, some “driver” mutations are more frequent in normal tissues than cancer tissues [42].
Second, not only do normal tissues have these “drivers”, they are present as clonal populations [42–45]. Having multiple clones present in what is nonmalignant, healthy tissue is now increasingly accepted as the norm. There is current debate as to whether these numerous clones in healthy tissues are arising through neutral drift or whether they arise due to selective pressure, and this remains unresolved. Perhaps what we have thought of as “drivers” in cancer are simply the genes that are most frequently mutated in certain tissues and do not confer any driver potential at all. It is possible that some of these alleged driver mutations could be relatively harmless and without effect initially, but as a tissue becomes more awry, it may assert intrinsic potential under selective pressure and become more like a true driver – in other words, their physiological effects are dynamic [42–45]. It is known that some tumour suppressor driver mutations can be reverted back to being normal during tumour evolution [46]. It is thus not inconceivable that in a complex, dynamic cell an alleged driver mutation could evolve to increase the strength of its properties too. This reflection is not simply an academic one; it has implications for clinical trials that rely on driver mutations in a binary way, to stratify patients into respective arms when they may in fact not be the sort of drivers that we had initially believed them to be.
Third, it has also been observed that the burden of mutation in normal tissues matches that of cancers [42–45]. Thousands and tens of thousands of mutations can be present in normal skin cells for example [43]. Statistical studies have suggested that the majority of mutations arise before the transformation into a frank malignancy [44]. For example, exposure to ultraviolet light damage in skin, tobacco damage in lung cells and deamination of methyl-cytosines – all of these create mutational signatures and may even result in heavy, widespread mutagenesis in normal cells.
Therefore, we arrive at a critical juncture: given what we can see in normal cells, what are the drivers and mutational signatures that are truly clinically important to recognise?
Identifying clinically useful genomic properties
We do not have all the answers at this point in time about which are the clinically meaningful genomic features. That is largely because we have not historically captured all the necessary genomic information to be able to answer these questions. The vast majority of genomics-informed clinical trials have used either single driver events or targeted panels for stratification. Separately, large genomic studies like The Cancer Genome Atlas (TCGA) [11–15] and International Cancer Genome Consortium (ICGC) [2, 23, 47, 48] endeavours have focused on either WES or WGS with some additional modalities such as transcriptomics or methylation studies, but well-annotated clinical data have been lacking. Although it has been immensely valuable to study these cohorts to gain biological understanding, what we need to do for the future is to collect as much of the most comprehensive genomic data as possible, to structure and store the data in modern, intelligent ways, and to learn and relearn from these extensive data as effectively as possible.
Quite apart from identifying driver events, mutational signatures, for example, will reveal a panoply of insults including prior exposures to environmental genotoxins such as tobacco smoke or ultraviolet light [18, 20]. Although this is valuable to understand, for an individual patient the signatures of these environmental agents report a past exposure and there is little that we can do to remedy what has already been acquired. Thus, although it is interesting, revealing and has public-health implications, identifying signatures of environmental agents is less clinically useful in terms of intervention for the patient – at least for now.
What is more useful is to identify mutational signatures from biological processes that are on-going, because they may indicate a dysfunctional pathway that is potentially targetable. Examples of defective DNA repair pathways that are clinically informative are mismatch repair (MMR) deficiency [49] and BRCA1/BRCA2 deficiency [36] (fig. 1). Tumours that have the former are treatable with checkpoint inhibitors whereas tumours that have the latter have been reported to be particularly sensitive to poly-ADP-ribose inhibitors (PARPi) through synthetic lethal mechanisms, where a tumour is fully-dependent on the normal functioning of PARP because BRCA1 and BRCA2 are no longer operational. Deficiencies in MMR and BRCA1/BRCA2 cause mutagenesis directly and their corresponding signatures can be used as biomarkers to report those deficiencies, making tumours that carry those signatures clinically targetable.
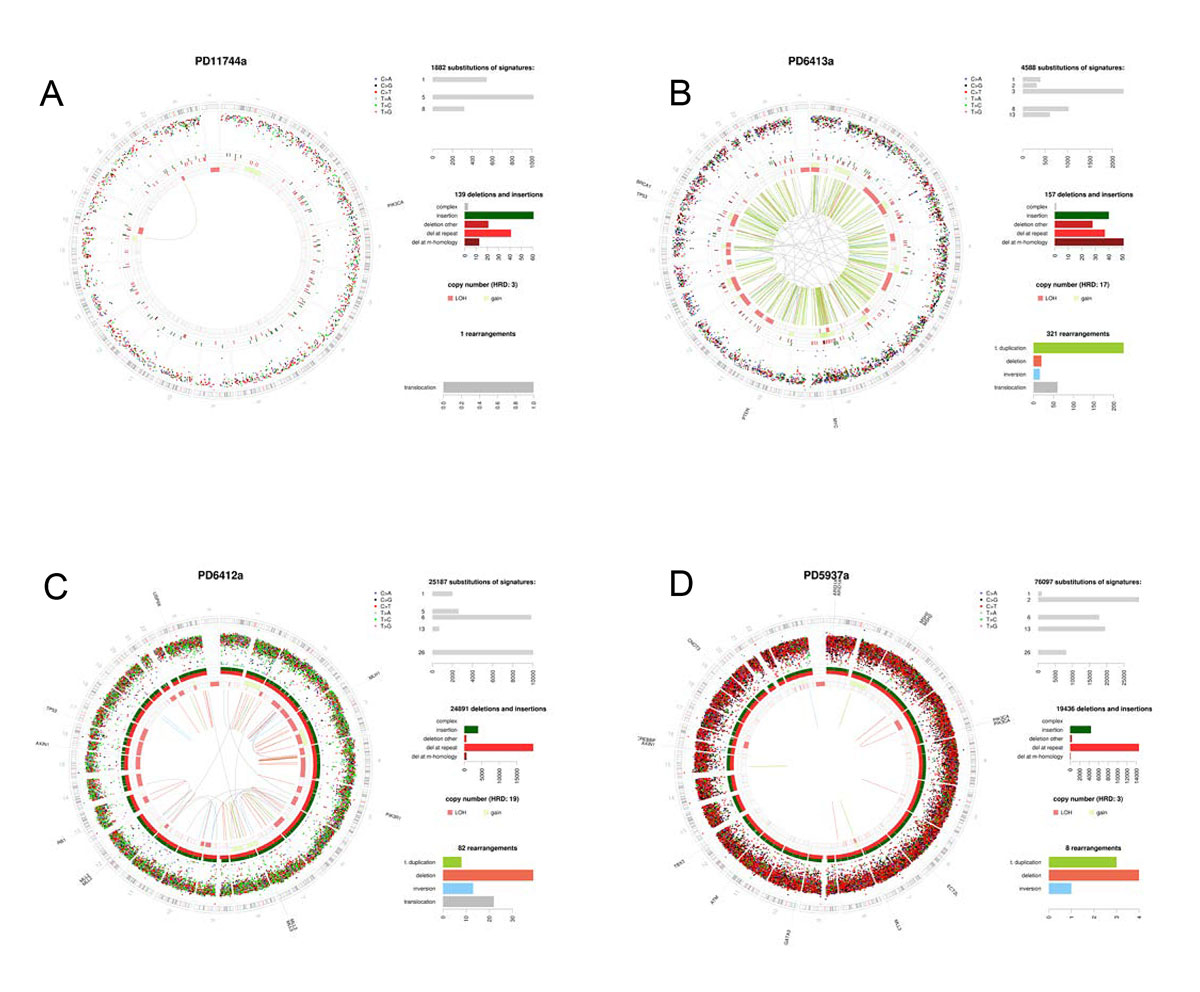
Figure 1
Holistic cancer genome profiling. Whole cancer genome profiles of four breast cancer patients. Circos plots showing the chromosomal ideogram on the outermost ring, clockwise chr1-chr22,X,Y. Subsequent circles heading inwards: Substitutions as dots plotted on log scale of intermutation distance, small insertions/deletions, copy number (pink = losses, green = gains), rearrangements = lines (green = tandem duplications, pink = large deletions, blue = inversions, grey = translocations). Top right panel depicts substitution mutational signatures, next one below showing indel classes, next one below showing rearrangements. Last panel bottom right shows curated driver mutations. (A) Oestrogen receptor positive breast cancer with good outcome: a tumour with sparse mutagenesis, low numbers of substitutions, indels and rearrangements, mainly signatures 1 and 5. 1q gain and 16q loss are common. (B) Characteristic BRCA1 null tumour with high levels of substitution signature 3, deletions with microhomology, multiple copy number losses and tandem duplications throughout the genome. (C) Mismatch repair deficient tumour with high numbers of signatures 6 and 26, and a very large number of indels at polynucleotide repeat tracts. (D) Mixed APOBEC-mutated and mismatch repair deficiency demonstrating that tumours can have multiple mutational processes.
Other endogenous mutational processes may also report biological abnormalities, even if in an indirect way. These are a little more difficult to ascertain and require extensive analytics in order to identify, to “train” using mathematical methods and to apply across cancers in order to develop into some form of biomarker. For example, the mutational signatures associated with the activity of the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of enzymes are characterised by a C>T transition (signature 2) and/or C>G transversion (signature 13) at cytosines that are almost always preceded by a thymine (otherwise described as a TpC sequence context) [2, 18]. The first mechanistic step in generating this signature is the deamination of cytosines at TpC contexts to form uracils, which can only occur when DNA is single stranded (ssDNA). The APOBEC signature tends to show many mutations happening on the same strand (strandedness) [50] for relatively long stretches in the genome (tens of kilobases) suggesting that long spools of ssDNA are available for APOBEC deamination during tumorigenesis. This may perhaps be an indicator of replication stress for the tumour. It consequently follows that tumours that have a preponderance of APOBEC mutagenesis may be tumours that are under a high level of replication stress and may be selectively sensitive to drugs that have been developed for this cause, including WEE1 G2 checkpoint kinase (WEE1) and ATR serine/shreonine kinase (ATR) inhibitors.
The value of having a total genomic picture of each cancer through WGS is that it can reveal all that is awry within each tumour. A tumour is, after all, a biological entity that rarely falls into binary categories. A tumour could have a selection of “drivers” which impair diverse pathways in different ways, as well as a selection of mutational signatures occurring in unison such as signatures of APOBEC and MMR deficiency (see fig. 1). Accordingly, to report on such a tumour, and to learn and understand why any individual tumour responds to a particular set of therapeutics or otherwise, we need to be able to interpret whole cancer genomes in a holistic way.
Moreover, to make interpretation of WGS cancers easier and assist clinicians of the future in using WGS cancer data more effectively, it is of enormous utility to develop algorithms or predictors of biological abnormalities and to test these in clinical populations. As an example, we have pioneered the development of a mutational-signature-based algorithm, capable of predicting BRCA1/BRCA2 deficiency, called HRDetect [36]. Critically, it identified many additional breast cancer patients with germline and somatic mutations of BRCA1/BRCA2, promoter hypermethylation of BRCA1, in patients not previously prioritised for BRCA1/BRCA2 screening [36]. Crucially, it also revealed a cohort of women with clear BRCA1/BRCA2-like tumours, but in whom we could not identify the underlying genetic or epigenetic driver [36]. These patients would be missed using current assays such as targeted BRCA1/BRCA2 sequencing and/or possibly the “genomic scars” test. There are thus benefits to taking the mutational signatures approach in identifying these patients.
To convince the clinical research community that these machine-learning-based algorithms are of value, algorithms such as HRDetect must be validated in alternative cohorts and their ability to relate to clinical outcome must be evaluated. Indeed, HRDetect has been applied to a population-based study in Sweden, called SCAN-B [51], in which all women in the south of Sweden with breast cancer are invited to participate in the study without exception. Focusing on a type of breast cancer associated with poor outcomes called triple negative breast cancer (TNBC), all available tumours between 2010–2015 have been whole genome sequenced [51]. Because the SCAN-B project has extraordinary supporting registries of clinical information, it is possible to make a true assessment of the frequency of BRCA1/BRCA2-deficient cancers in TNBC in the Swedish population (~59%), and to be able to ascertain that HRDetect has independent prognostic properties capable of discerning patients who will respond to standard-of-care from those that do not seem to gain any benefit from current treatments [51]. Furthermore, the WGS approach allows us to “see” other abnormalities in these breast cancers, some of which may be targetable with treatments that are otherwise not offered to women with breast cancer.
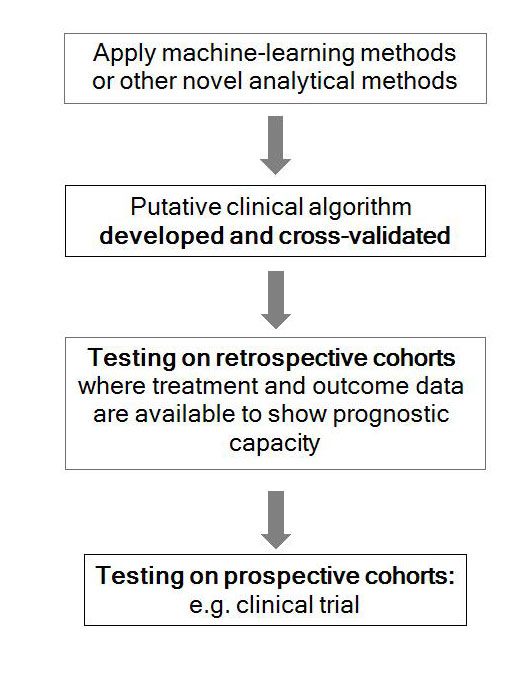
The example we provide above is one where we have reused large WGS datasets using machine-learning methods to develop a novel clinical algorithm, and then applied it to a clinical cohort in order to understand its true clinical value (fig. 2). Now rolled out into clinical trials, what we aim to do through the Josef Steiner Award is to build the infrastructure to allow us to perform these sorts of analyses more efficiently, to develop new algorithms as a result of having greater flexibility in exploring the data and to accelerate the translation of whole cancer genomics into the clinic.
Holistic genome interpretation
Today it is already possible to provide an individual WGS report in a matter of days, providing all the drivers, mutational signatures and even germline information for each patient. Novel algorithms can already provide clear read-outs of HR deficiency and MMR deficiency. In due course, more exhaustive holistic WGS reports should provide therapeutically relevant treatment strategies associated with the genomic results too. Presently, what are the hurdles that prevent using WGS reporting for cancer today?
One of the first concerns about WGS was the cost of sequencing. With the precipitous decline in sequencing costs in the last decade, this is now something of a myth. The cost of WGS is today less than that of a contrast-enhanced computed tomography (CT) scan of the chest, abdomen and pelvis. Yet this radiological test is performed routinely to gain the full anatomical picture of a patient’s disease status. Healthcare providers and medical insurance companies would not think twice about requesting or funding these investigations. Yet there is hesitation regarding WGS of solid tumours. There are admittedly additional costs associated with WGS, such as storage and backup of sequencing and intermediary files. Nevertheless, there is also added value. WGS data that are stored intelligently could serve as an ever-giving clinical research resource.
The second and more appropriate concern is the hurdle of analysis and interpretation. In this domain, however, there is substantial acceleration in how to perform WGS analysis and interpretation. Awards such as Steiner will certainly help our team to organise data and analyse it quickly to build more tools to assist in WGS interpretation.
Conclusions and future directions
It is quite possible, or even likely, that having a WGS for every (relevant) solid cancer will become a routine part of the diagnostic process for every patient within the next 10–20 years, if not sooner. There are already several large endeavours that have initiated WGS cancer sequencing research projects, including the 100,000 genomes project in the UK [52] and the Hartwig consortium in the Netherlands [53] among others. It is possible that these research projects will lead the way in transitioning into clinical practice. We should thus prepare for a future where WGS (or some form of genomics/transcriptomics) may become another assay like a set of bloods, an electrocardiogram, a staging CT scan or positron emission tomography scan, in the process of trying to comprehensively understand the patient’s cancer clinical picture.
To achieve this vision, first we must provide the infrastructure and support to train the next generation of molecular genomic interpreters, whether they are pathologists, geneticists or an altogether new breed of scientific/medical experts. Apart from computational support, we need to develop standard operating procedures for data handling and analysis, statistical and academic frameworks to operate from, and legal and/or ethical guidelines, to name a few areas of development. What they will learn to do is to read/interpret a whole cancer genome, like a radiologist would do for an X-ray or CT scan.
Second, clinical trials of chemotherapeutic agents that incorporate improved genomic profiling of tumours are required. This is not a trivial exercise and years of work are ahead of us before we will be in a position to match therapies to genomic status more effectively in the future.
Third, we must do the right thing by our future clinicians and scientists, which is to build the best infrastructure to support modern analytical methods. It is possible to structure data so that we maximise learning from every case going forward. The human genome is so vast and there is much that we do not yet understand. Genomic data are perfect for exploration using machine-learning methods, to develop artificial intelligence that will help the clinicians/scientists of the future with diagnostics. We must build the foundations to suit that future.
References
1
Bentley
DR
,
Balasubramanian
S
,
Swerdlow
HP
,
Smith
GP
,
Milton
J
,
Brown
CG
, et al.
Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–9. doi:.https://doi.org/10.1038/nature07517
2
Nik-Zainal
S
,
Alexandrov
LB
,
Wedge
DC
,
Van Loo
P
,
Greenman
CD
,
Raine
K
, et al.; Breast Cancer Working Group of the International Cancer Genome Consortium. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149(5):979–93. doi:.https://doi.org/10.1016/j.cell.2012.04.024
3
Nik-Zainal
S
,
Van Loo
P
,
Wedge
DC
,
Alexandrov
LB
,
Greenman
CD
,
Lau
KW
, et al.; Breast Cancer Working Group of the International Cancer Genome Consortium. The life history of 21 breast cancers. Cell. 2012;149(5):994–1007. doi:.https://doi.org/10.1016/j.cell.2012.04.023
4
Stephens
PJ
,
Tarpey
PS
,
Davies
H
,
Van Loo
P
,
Greenman
C
,
Wedge
DC
, et al.; Oslo Breast Cancer Consortium (OSBREAC). The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–4. doi:.https://doi.org/10.1038/nature11017
5
Zehir
A
,
Benayed
R
,
Shah
RH
,
Syed
A
,
Middha
S
,
Kim
HR
, et al.
Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13. doi:.https://doi.org/10.1038/nm.4333
6
Stratton
MR
,
Campbell
PJ
,
Futreal
PA
. The cancer genome. Nature. 2009;458(7239):719–24. doi:.https://doi.org/10.1038/nature07943
7
Wood
LD
,
Parsons
DW
,
Jones
S
,
Lin
J
,
Sjöblom
T
,
Leary
RJ
, et al.
The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–13. doi:.https://doi.org/10.1126/science.1145720
8
Hanahan
D
,
Weinberg
RA
. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi:.https://doi.org/10.1016/S0092-8674(00)81683-9
9
Nowell
PC
. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8. doi:.https://doi.org/10.1126/science.959840
10
Lawrence
MS
,
Stojanov
P
,
Mermel
CH
,
Robinson
JT
,
Garraway
LA
,
Golub
TR
, et al.
Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi:.https://doi.org/10.1038/nature12912
11
Kandoth
C
,
Schultz
N
,
Cherniack
AD
,
Akbani
R
,
Liu
Y
,
Shen
H
, et al., Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi:. Correction in: Nature. 2013;500:242
https://doi.org/10.1038/nature12113
12
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi:.https://doi.org/10.1038/nature11252
13
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi:.https://doi.org/10.1038/nature10166
14
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–25. doi:.https://doi.org/10.1038/nature11404
15
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22. doi:.https://doi.org/10.1038/nature12965
16
Tarpey
PS
,
Behjati
S
,
Cooke
SL
,
Van Loo
P
,
Wedge
DC
,
Pillay
N
, et al.
Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45(8):923–6. doi:.https://doi.org/10.1038/ng.2668
17
Papaemmanuil
E
,
Cazzola
M
,
Boultwood
J
,
Malcovati
L
,
Vyas
P
,
Bowen
D
, et al.; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–95. doi:.https://doi.org/10.1056/NEJMoa1103283
18
Alexandrov
LB
,
Nik-Zainal
S
,
Wedge
DC
,
Aparicio
SA
,
Behjati
S
,
Biankin
AV
, et al.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. doi:.https://doi.org/10.1038/nature12477
19
Helleday
T
,
Eshtad
S
,
Nik-Zainal
S
. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15(9):585–98. doi:.https://doi.org/10.1038/nrg3729
20
Kucab
JE
,
Zou
X
,
Morganella
S
,
Joel
M
,
Nanda
AS
,
Nagy
E
, et al.
A Compendium of Mutational Signatures of Environmental Agents. Cell. 2019;177(4):821–836.e16. doi:.https://doi.org/10.1016/j.cell.2019.03.001
21
Zou
X
,
Owusu
M
,
Harris
R
,
Jackson
SP
,
Loizou
JI
,
Nik-Zainal
S
. Validating the concept of mutational signatures with isogenic cell models. Nat Commun. 2018;9(1):1744. doi:.https://doi.org/10.1038/s41467-018-04052-8
22Alexandrov LB, Kim J, Haradhvala N, Huang NM, Ng AWT, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. Bioarkiv. 2018. Available at: https://doi.org/10.1101/322859.
23
Nik-Zainal
S
,
Davies
H
,
Staaf
J
,
Ramakrishna
M
,
Glodzik
D
,
Zou
X
, et al.
Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi:. Correction in: Nature. 2019;566:E1
https://doi.org/10.1038/nature17676
24
Macintyre
G
,
Goranova
TE
,
De Silva
D
,
Ennis
D
,
Piskorz
AM
,
Eldridge
M
, et al.
Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet. 2018;50(9):1262–70. doi:.https://doi.org/10.1038/s41588-018-0179-8
25
Tubio
JMC
,
Li
Y
,
Ju
YS
,
Martincorena
I
,
Cooke
SL
,
Tojo
M
, et al.; ICGC Breast Cancer Group; ICGC Bone Cancer Group; ICGC Prostate Cancer Group. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345(6196):1251343. doi:.https://doi.org/10.1126/science.1251343
26
Dowen
JM
,
Fan
ZP
,
Hnisz
D
,
Ren
G
,
Abraham
BJ
,
Zhang
LN
, et al.
Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374–87. doi:.https://doi.org/10.1016/j.cell.2014.09.030
27
Debruyne
DN
,
Dries
R
,
Sengupta
S
,
Seruggia
D
,
Gao
Y
,
Sharma
B
, et al.
BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature. 2019;572(7771):676–80. doi:.https://doi.org/10.1038/s41586-019-1472-0
28
Glodzik
D
,
Morganella
S
,
Davies
H
,
Simpson
PT
,
Li
Y
,
Zou
X
, et al.
A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat Genet. 2017;49(3):341–8. doi:. Correction in: Nat Genet. 2017;49:1661
https://doi.org/10.1038/ng.3771
29
Rheinbay
E
,
Parasuraman
P
,
Grimsby
J
,
Tiao
G
,
Engreitz
JM
,
Kim
J
, et al.
Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547(7661):55–60. doi:.https://doi.org/10.1038/nature22992
30
Glodzik
D
,
Purdie
C
,
Rye
IH
,
Simpson
PT
,
Staaf
J
,
Span
PN
, et al.
Mutational mechanisms of amplifications revealed by analysis of clustered rearrangements in breast cancers. Ann Oncol. 2018;29(11):2223–31. doi:.https://doi.org/10.1093/annonc/mdy404
31
Liu
S
,
Kwon
M
,
Mannino
M
,
Yang
N
,
Renda
F
,
Khodjakov
A
, et al.
Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature. 2018;561(7724):551–5. doi:.https://doi.org/10.1038/s41586-018-0534-z
32
Rosenthal
R
,
Cadieux
EL
,
Salgado
R
,
Bakir
MA
,
Moore
DA
,
Hiley
CT
, et al.; TRACERx consortium. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567(7749):479–85. doi:.https://doi.org/10.1038/s41586-019-1032-7
33
McGranahan
N
,
Rosenthal
R
,
Hiley
CT
,
Rowan
AJ
,
Watkins
TBK
,
Wilson
GA
, et al.; TRACERx Consortium. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171(6):1259–1271.e11. doi:.https://doi.org/10.1016/j.cell.2017.10.001
34
Foulkes
WD
,
Knoppers
BM
,
Turnbull
C
. Population genetic testing for cancer susceptibility: founder mutations to genomes. Nat Rev Clin Oncol. 2016;13(1):41–54. doi:.https://doi.org/10.1038/nrclinonc.2015.173
35
Nik-Zainal
S
,
Wedge
DC
,
Alexandrov
LB
,
Petljak
M
,
Butler
AP
,
Bolli
N
, et al.
Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet. 2014;46(5):487–91. doi:.https://doi.org/10.1038/ng.2955
36
Davies
H
,
Glodzik
D
,
Morganella
S
,
Yates
LR
,
Staaf
J
,
Zou
X
, et al.
HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–25. doi:.https://doi.org/10.1038/nm.4292
37
Lambert
SA
,
Abraham
G
,
Inouye
M
. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;ddz187. doi:.https://doi.org/10.1093/hmg/ddz187
38
Gerlinger
M
,
Rowan
AJ
,
Horswell
S
,
Larkin
J
,
Endesfelder
D
,
Gronroos
E
, et al.
Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. doi:.https://doi.org/10.1056/NEJMoa1113205
39
Caravagna
G
,
Giarratano
Y
,
Ramazzotti
D
,
Tomlinson
I
,
Graham
TA
,
Sanguinetti
G
, et al.
Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat Methods. 2018;15(9):707–14. doi:.https://doi.org/10.1038/s41592-018-0108-x
40
De Mattos-Arruda
L
,
Sammut
SJ
,
Ross
EM
,
Bashford-Rogers
R
,
Greenstein
E
,
Markus
H
, et al.
The Genomic and Immune Landscapes of Lethal Metastatic Breast Cancer. Cell Rep. 2019;27(9):2690–2708.e10. doi:.https://doi.org/10.1016/j.celrep.2019.04.098
41
Roth
A
,
Khattra
J
,
Yap
D
,
Wan
A
,
Laks
E
,
Biele
J
, et al.
PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11(4):396–8. doi:.https://doi.org/10.1038/nmeth.2883
42
Martincorena
I
,
Fowler
JC
,
Wabik
A
,
Lawson
ARJ
,
Abascal
F
,
Hall
MWJ
, et al.
Somatic mutant clones colonize the human esophagus with age. Science. 2018;362(6417):911–7. doi:.https://doi.org/10.1126/science.aau3879
43
Martincorena
I
,
Roshan
A
,
Gerstung
M
,
Ellis
P
,
Van Loo
P
,
McLaren
S
, et al.
Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–6. doi:.https://doi.org/10.1126/science.aaa6806
44
Tomasetti
C
,
Vogelstein
B
,
Parmigiani
G
. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA. 2013;110(6):1999–2004. doi:.https://doi.org/10.1073/pnas.1221068110
45
Yizhak
K
,
Aguet
F
,
Kim
J
,
Hess
JM
,
Kübler
K
,
Grimsby
J
, et al.
RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364(6444):eaaw0726. doi:.https://doi.org/10.1126/science.aaw0726
46
Domchek
SM
. Reversion Mutations with Clinical Use of PARP Inhibitors: Many Genes, Many Versions. Cancer Discov. 2017;7(9):937–9. doi:.https://doi.org/10.1158/2159-8290.CD-17-0734
47
Waddell
N
,
Pajic
M
,
Patch
AM
,
Chang
DK
,
Kassahn
KS
,
Bailey
P
, et al.; Australian Pancreatic Cancer Genome Initiative. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi:.https://doi.org/10.1038/nature14169
48
Campbell
PJ
,
Getz
G
,
Stuart
JM
,
Korbel
JO
,
Stein
LD
. Pan-cancer analysis of whole genomes. bioRxiv. 2017.
49
Davies
H
,
Morganella
S
,
Purdie
CA
,
Jang
SJ
,
Borgen
E
,
Russnes
H
, et al.
Whole-Genome Sequencing Reveals Breast Cancers with Mismatch Repair Deficiency. Cancer Res. 2017;77(18):4755–62. doi:.https://doi.org/10.1158/0008-5472.CAN-17-1083
50
Morganella
S
,
Alexandrov
LB
,
Glodzik
D
,
Zou
X
,
Davies
H
,
Staaf
J
, et al.
The topography of mutational processes in breast cancer genomes. Nat Commun. 2016;7(1):11383. doi:.https://doi.org/10.1038/ncomms11383
51
Staaf
J
,
Glodzik
D
,
Bosch
A
,
Vallon-Christersson
J
,
Reuterswärd
C
,
Häkkinen
J
, et al.
Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat Med. 2019;25(10):1526–33. doi:.https://doi.org/10.1038/s41591-019-0582-4
52
Siva
N
. UK gears up to decode 100,000 genomes from NHS patients. Lancet. 2015;385(9963):103–4. doi:.https://doi.org/10.1016/S0140-6736(14)62453-3
53Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole genome analyses of metastatic solid tumors. Bioarkiv. 2019. Available at: https://doi.org/10.1101/415133.