The role of autophagy in HER2-targeted therapy
DOI: https://doi.org/10.4414/smw.2019.20138
Félice A.
Janserab, Mario P.
Tschanab*, Rupert
Langera*
aInstitute of Pathology, University of Bern, Switzerland
bGraduate School for Cellular and Biomedical Sciences, University of Bern, Switzerland
Summary
Macroautophagy (hereafter referred to as autophagy) is a highly conserved, intracellular degradation process characterised by de novo formation of autophagosomes. These double membraned organelles engulf and deliver cargo, for example damaged organelles and protein aggregates, to lysosomes for degradation and recycling. Autophagy is primarily a stress response mechanism activated to survive unfavourable conditions such as starvation or hypoxia. In addition, autophagy functions in differentiation, immune responses against invading microorganisms and tissue remodelling in mammalian cells. Besides its cytoprotective nature, and depending on the context, autophagy can as well support cell death. Based on autophagy’s cytoprotective, cytotoxic and developmental influences, it does not come as a surprise that this mechanism is involved in tumourigenesis, tumour development and the response to anticancer therapies. HER2 is a receptor tyrosine kinase that activates downstream signalling pathways involved in cellular survival, growth and proliferation. Amplification of the gene and subsequent overexpression of the HER2 protein lead to increased activation of downstream signalling and are implicated in several cancer types. HER2-targeted therapies are valuable treatment options for HER2 amplified cancers. However, pre-existing and acquired resistance remain a clinical challenge. Autophagy has been discussed in several scenarios in HER2 amplified cancers. Generally, HER2+ tumours have been shown to exhibit low levels of proteins essential for autophagy. Moreover, a protein involved in autophagy activation, Beclin-1, was shown to interact directly with HER2 at the cellular membrane. The signalling cascade activated by HER2 also activates mTOR, a negative regulator of autophagy. In the context of resistance formation against HER2-targeting treatment, autophagy has often been reported to be upregulated, and resistance has been shown to be abrogated through autophagy inhibition. Since the autophagy inhibitors chloroquine and hydroxychloroquine are approved drugs for the treatment of malaria, autophagy inhibition is discussed as an option to enhance the effect of certain anticancer treatments or to overcome resistance against cancer therapies. In this review we focus on autophagy and its role in the response to HER2-targeted therapies for breast and gastrointestinal tumours.
Autophagy
Macroautophagy, often simply referred to as autophagy, is a multistep process involved in cellular homeostasis and adaptation to stress conditions. As a catabolic process, autophagy maintains the nutrient homeostasis of the cell and participates in the quality control of proteins and organelles [1]. Upon cellular stress, for instance by starvation, hypoxia, genotoxic or proteotoxic stress, autophagy is upregulated as an adaptive cell response [2, 3]. The process is characterised by the formation of a double membraned vesicle, the autophagosome, which engulfs cytoplasmic material. The autophagosome fuses with the lysosome, which ultimately leads to the degradation of its content. In the lysosome, proteins are degraded by cathepsins, which are a group of proteases activated at low pH values typical of lysosomes [4]. Thus, the content of the so-called autolysosome is recycled for biosynthesis and/or energy production. The macroautophagic pathway is divided into distinct steps (fig. 1): (a) nucleation of the isolation membrane, (b) expansion of the membrane, (c) closure and maturation of the autophagosome, (d) fusion of the autophagosome with the lysosome, and (e) degradation and recycling of the delivered cargo [5, 6]. The evolutionarily conserved degradation pathway involves at least 16–20 core autophagy (ATG) genes. The ATG proteins encoded by these genes are classified into functional groups that act at different stages of autophagy [7, 8].
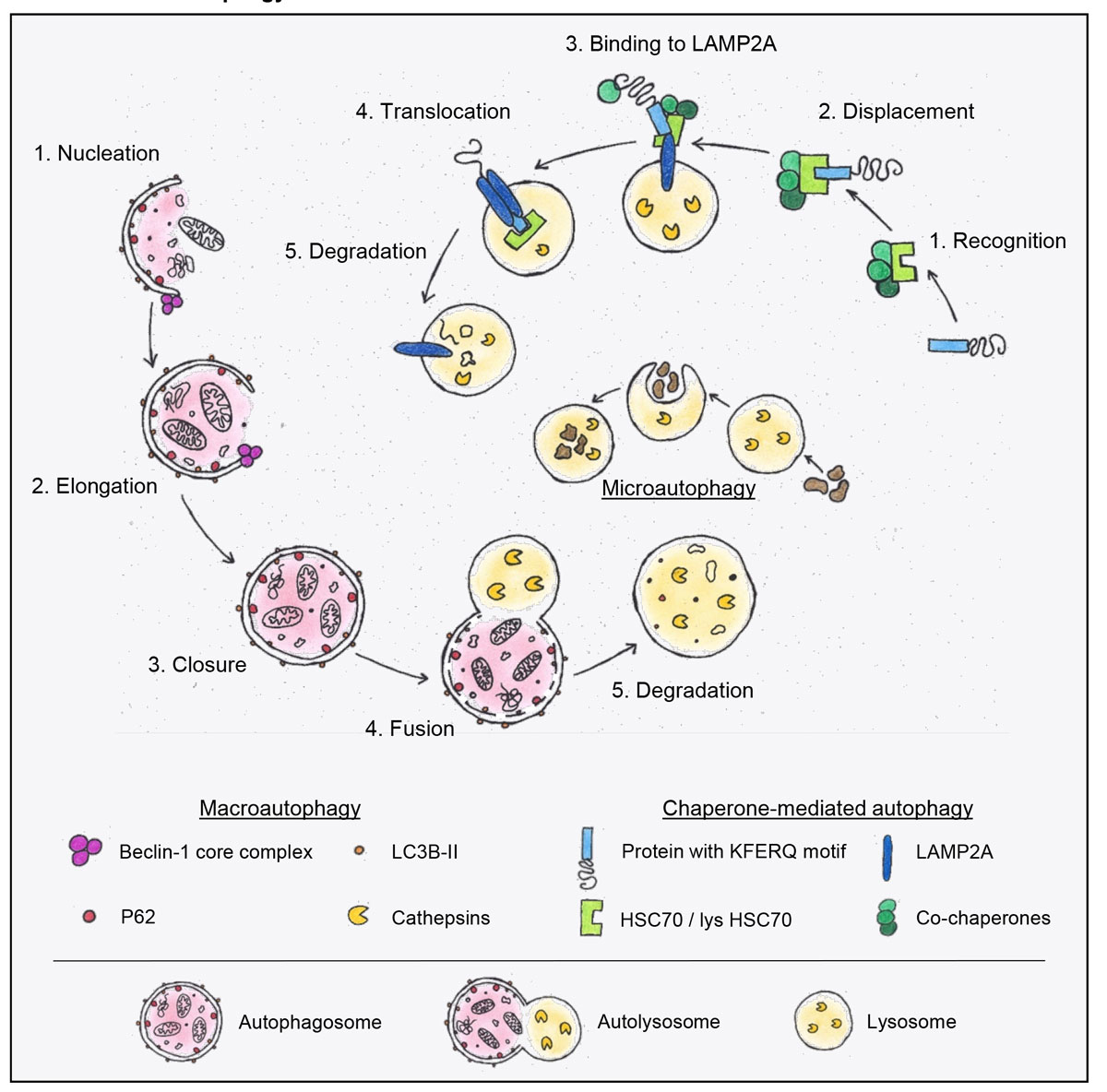
Figure 1
Schematic presentation of macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy. On the left hand side, an overview of macroautophagy is depicted in red. The isolation membrane forms during the nucleation process. An important player in nucleation and elongation is the Beclin-1 core complex. During the elongation of the double membrane LC3B is lipidated and allows the binding of adaptor proteins such as p62. After autophagosome closure, the autophagosome fuses with the lysosome, followed by degradation and recycling of its contents. The degradation is mediated by cathepsins. These proteases become activated in the low pH found in lysosomes. In the middle of the diagram, microautophagy, a process characterised by the direct uptake of cytosolic material into the lysosome, is depicted. On the right, the chaperone mediated autophagy (CMA) pathway is outlined. In this process, proteins containing a KFERQ amino acid sequence motif are recognised by a chaperone complex containing HSC70. The complex is then translocated to the lysosome and, with the help of the LAMP2A complex, incorporated into the lysosome and degraded.
p62 = sequestosome 1, SQSTM1; LC3B = microtubule-associated protein light chain 3B; HSC70 = heat shock 70 kDa protein 8; LAMP2A = lysosome-associated membrane protein 2A
One of the important players in this multistep process is unc-51-like autophagy activating kinase 1 (ULK1, the mammalian orthologue of yeast ATG1). It is the activating kinase in the autophagy initiating complex [9, 10]. The phosphorylation of downstream players by this complex leads to the elongation of the isolation membrane and allows the recruitment of another multiprotein complex containing Beclin-1 and the catalytic subunit VPS34 [11, 12]. Beclin-1 is monoallelically deleted or downregulated in various tumour types, such as breast and ovarian cancer, indicating its tumour suppressor function [13, 14]. The isolation membrane elongates through incorporation of phospholipids from different sources, such as the endoplasmic reticulum (ER). As autophagy is a dynamic process, it is difficult to capture the actual “autophagic flux” describing the rate of degradation by using only ATG gene expression data. Therefore, the microtubule-associated protein light chain 3B (LC3B) is frequently used as an autophagy marker. The LC3B protein is processed during active autophagy. As an ATG8 family member, it is lipidated to LC3B-II and then integrated into the growing autophagosomal membrane by its conjugation to phosphatidylethanolamine. LC3B-II enables the elongation of the isolation membrane as well as cargo recruitment [15, 16]. The non-lipidated LC3B-I and the lipidated LC3B-II can be differentiated by Western blotting. Thus, increasing autophagosome formation can be visualised via Western blotting and indicated by higher LC3B-II to LC3B-I ratios. Additional LC3B-based techniques include assessment of LC3B dot formation upon ectopic expression of GFP-LC3B or of endogenous LC3B, using immunofluorescent microscopy or immunohistochemistry [17].
Under certain conditions, such as starvation, autophagy is rather unspecific. In the last decade, however, specific degradation of cargo by macroautophagy, so-called selective autophagy, has been described. Different cargos, such as mitochondria, lipid droplets, ribosomes, protein aggregates or individual proteins can be degraded via selective autophagy with the help of cargo receptors. These cargo adaptor proteins directly link the cargo to the autophagosome. In most cases the cargo either contains a so-called LC3-interacting region (LIR) that can bind to LC3B or the cargo is labelled with a ubiquitin tag. In the latter case, the degradation is mediated by an adapter protein that has a ubiquitin binding site as well as an LIR [18]. Sequestosome 1 (SQSTM1, also known as p62) represents just such an autophagy adaptor that targets cargo to the growing autophagosomal membrane. P62 is degraded together with the cargo, which allows using degradation of p62 as a marker for autophagic flux [19, 20].
Besides macroautophagy there are at least two additional forms of autophagy, namely chaperone mediated autophagy (CMA) and microautophagy. These three pathways differ in the way the cargo is delivered to the lysosome (fig. 1). During CMA, cytosolic proteins are recognised by heat shock 70 kDa protein 8 (HSC70) through a specific amino acid sequence, the so-called KFERQ motif. Subsequently, the protein is shuttled to the lysosome and is finally translocated via another protein complex including lysosome-associated membrane protein 2A (LAMP2A) into the lysosome (fig. 1) [21, 22]. Microautophagy is characterised by the direct lysosomal uptake of cytoplasmic entities [23].
In addition to its role in homeostasis and stress adaptation, autophagy also plays a role in cellular differentiation, immune response against invading microorganisms or tissue remodelling [24, 25]. Aberrant autophagy has been reported in connection with various diseases, such as inflammation, cancer formation or neurodegeneration [2, 26]. In this context it is important to mention that autophagy and apoptosis are closely linked. The anti-apoptotic protein Bcl-2, which is often upregulated in cancer, directly interacts with the key autophagy gene Beclin-1. By binding and sequestering Beclin-1 and thus inhibiting it from initiating autophagy, Bcl-2 acts as a negative regulator of autophagy [27]. Moreover, additional ATG proteins, such as ATG5 or ATG12, interact with Bcl-2 family members, indicating a complex crosstalk between the two pathways [28, 29].
To summarise, the term autophagy describes several lysosomal degradation pathways that differ in their cargo delivery to the lysosome. The best-studied form of autophagy is macroautophagy. The conserved multistep process is responsible for cellular homeostasis at basal conditions and can be upregulated upon different cellular stresses. In recent years, macroautophagy has been shown not only to perform bulk degradation but also to exert high cargo specificity. The cellular stress response mechanism autophagy is interconnected with apoptosis, and aberrant autophagy has been linked to various diseases.
Autophagy and cancer
Autophagy plays a dual role in cancer development and progression. Autophagy guards cellular homeostasis and therefore contributes to the prevention of malignant transformation. On the other hand, it seems to play a rather tumour-promoting role in established tumours (fig. 2) [30]. Evidence to support a tumour suppressor function of autophagy stems from murine models defective in essential autophagy genes. For instance, Beclin-1+/-
animals spontaneously develop malignancies such as lymphomas or lung carcinomas [31, 32]. Moreover, mice with liver-specific knockout of Atg7 or a systemic mosaic deletion of Atg5 develop benign hepatic neoplasms [33]. Additionally, autophagy suppresses the accumulation of genetic and genomic defects caused by reactive oxygen species (ROS) through removal of dysfunctional mitochondria and redox-active aggregates of ubiquitinated proteins [34, 35]. Moreover, by eliminating dysfunctional mitochondria autophagy ensures optimal energy supply, which counteracts the metabolic rewiring often observed during malignant transformation [35]. Further, autophagy is involved in the maintenance of normal stem cells. The above-mentioned Beclin-1+/-
mice, for example, show an expansion of progenitor-like mammary epithelial cells [36, 37]. Autophagy is also involved in the degradation of aggregate-prone oncogenes, such as forms of mutated TP53, p62, PML-RARA or BCR-ABL1 [34, 38–40]. It is required in several aspects of anticancer immunosurveillance, and thus in the elimination of potentially tumourigenic cells by the immune system [41]. Additionally, autophagy plays a key role in first line defence against bacterial or viral infection. Multiple potentially carcinogenic pathogens, such as Salmonella enterica, Helicobacter pylori or Chlamydia pneumoniae, can activate autophagy upon infection [42–45].
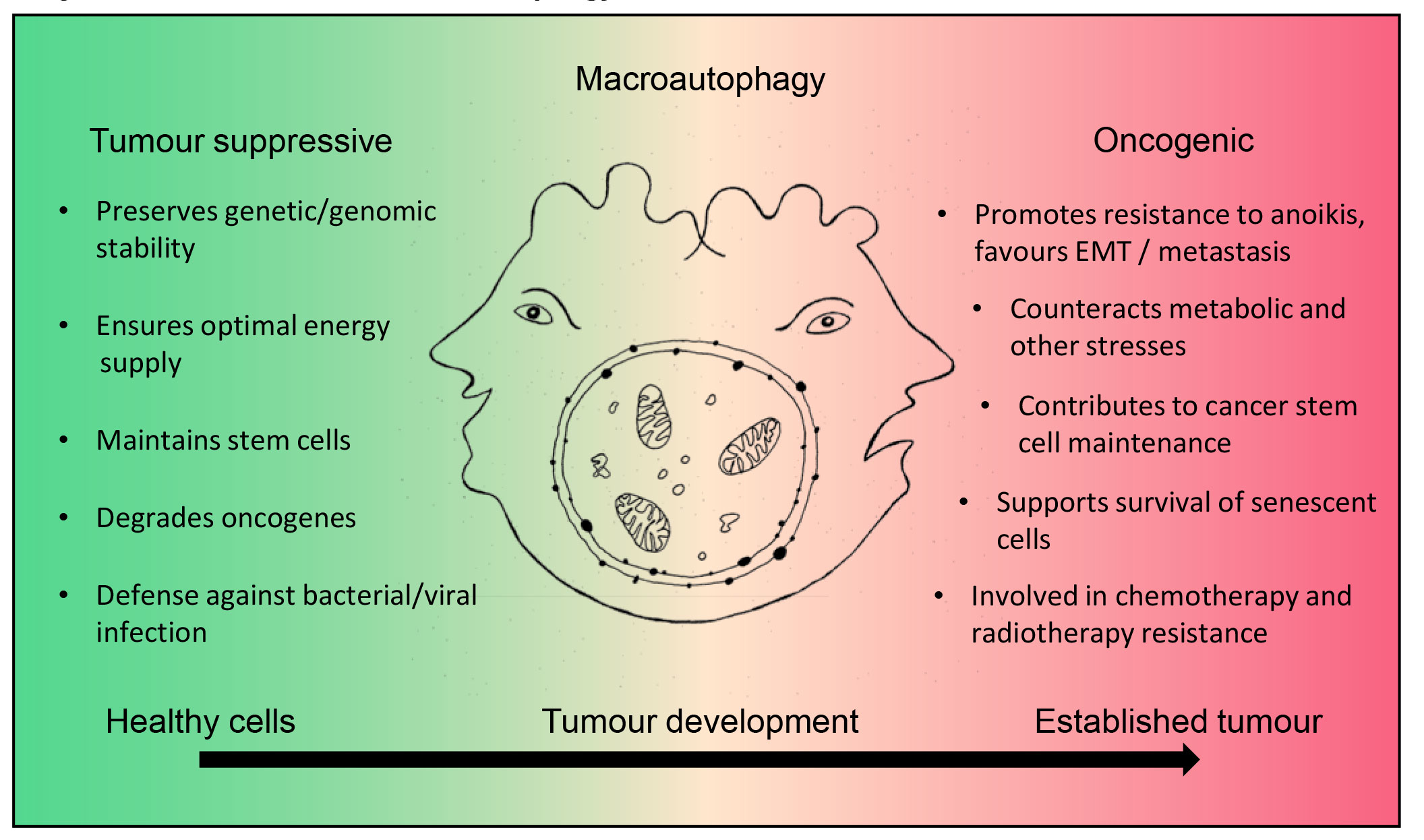
Figure 2
The Janus-faced role of autophagy in cancer. In healthy cells macroautophagy has a tumour suppressor function. As a catabolic survival mechanism, it ensures optimal energy supply, preserves genetic and genomic stability, maintains normal stem cells, is involved in the degradation of oncogenes, and is the first line defence against bacterial or viral infection. During tumour formation downregulation of autophagy is frequently observed, possibly supporting tumor development. In established tumours, however, autophagy functions can be restored and instead support tumour development. This occurs, for example, via (a) supporting EMT and metastasis, (b) rendering cells resistant to anoikis (programmed cell death upon detachment), (c) counteracting metabolic and oxidative stress, (d) maintaining cancer stem cells and supporting the senescent cell state, and (e) promoting chemo- as well as radiotherapy resistance.
There are indications that the activation of oncoproteins, and, similarly, the inactivation of tumour suppressor proteins can attenuate autophagy. This reduced autophagic activity supports early phases of oncogenesis [46]. Anti-apoptotic Bcl-2 family members, such as Bcl-2 or Bcl-XL, that are upregulated in various cancer types, also inhibit autophagy through sequestration of Beclin-1 [27]. MDM2 represents another proto-oncogene that negatively affects autophagy. High MDM2 levels inactivate the TP53 tumour suppressor. Inactivated TP53 then fails to activate transcription of its target ATG genes [47]. Furthermore, several receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR) or v-erb-b2 avian erythroblastic leukaemia viral oncogene homologue 2 (ERBB2, also known as HER2), or downstream signal transducers that are often overexpressed in solid tumours inhibit autophagy by activating its negative regulator mTORC1 [48]. The tumour suppressor phosphatase and tensin homologue (PTEN) is often inactivated in cancers. This phosphatase promotes autophagy by antagonising PI3K signalling that negatively regulates autophagy [49]. The transcription factor forkhead box O1 (FOXO1) represents another tumour suppressor essential for stress-induced autophagy that is mutated in diffuse large B-cell lymphomas [50].
In neoplastic cells, however, restored autophagy response allows cancer cells to cope with intracellular and environmental stress (fig. 2, right panel) [51, 52]. Thus, in advanced human tumours high autophagic flux correlates with an invasive, metastatic phenotype and poor survival rates [53]. In mouse experiments, highly metastatic hepatocellular carcinoma cell lines with inhibited Beclin-1 or Atg5 expression are unable to survive in the metastatic niche, in contrast to their autophagy-competent counterparts [54]. In KRAS-driven pancreatic adenocarcinoma cells, autophagy is upregulated upon oncogene ablation to counterbalance the metabolic stress occurring upon shutdown of oncogenic KRAS signalling [55]. Breast cancer stem cells from mammosphere cultures are also characterised by elevated autophagic flux. Importantly, their ability to form tumours in vivo seems to depend on proficient autophagy, as they are not able to form tumours upon genetic inhibition of Beclin-1 or ATG4A [56, 57]. Autophagy-deficient tumours are generally more sensitive to chemotherapeutic agents and to radiotherapy compared with their autophagy-proficient counterparts [58, 59]. Senescent cancer cells, which do not proliferate but can still support relapse by influencing the tumour microenvironment, depend on autophagy for survival [60].
In summary, autophagy in healthy cells prevents tumour development, and its downregulation may contribute to early oncogenesis. In late tumour development cancer cells hijack autophagy to play instead an oncogenic role (fig. 2). In this context, autophagy can protect cancer cells from anticancer treatment and may thus contribute to therapy resistance.
Several compounds that inhibit autophagy at different stages are known. The only clinically approved autophagy inhibitor is the antimalarial drug chloroquine and its derivative hydroxychloroquine. These drugs interfere with lysosomal acidification and thus prevent the degradation of autophagosomes [61, 62]. Hydroxychloroquine is preferred over chloroquine in clinical trials owing to its lower toxicity [63, 64]. Several preclinical in vitro and in vivo studies have shown an antineoplastic effect of hydroxychloroquine in combination with various clinically approved drugs [65]. However, the use of chloroquine and hydroxychloroquine as autophagy inhibitors is controversial, since both reagents exert autophagy inhibition-independent effects as well. Thus, chloroquine can sensitise cancer cells to chemotherapeutic drugs independent of autophagy inhibition [66, 67]. This was illustrated in a 2012 study, where chloroquine was used to sensitise mouse breast cancer cells to several established anticancer treatments. The observed sensitisation was independent of autophagy inhibition as it could not be mimicked with ATG12 or Beclin-1 knockdown or treatment with Bafilomycin A1 (BafA), another autophagy inhibitor [66]. Furthermore, a preclinical study evaluating the pharmacodynamics of hydroxychloroquine in pet dogs with lymphoma showed that the plasma concentration of hydroxychloroquine does not correlate with drug concentration in the tumours. This discrepancy consequently also applies to the extent of autophagy inhibition in the tumour tissue [68]. Another open question concerning the use of chloroquine and hydroxychloroquine as autophagy inhibitors in the clinic is whether it is better to block the process at early or at late autophagy stages. Early autophagosomal structures can serve as scaffolds for inducing apoptosis and necroptosis. Thus, the accumulation of autophagosomes could promote these pathways in some cases [69]. Since chloroquine and hydroxychloroquine inhibit autophagy at a late stage, and their mechanism of action is not well understood, new specific inhibitors with improved pharmacodynamics that also target early autophagy kinases, such as VPS34 or ULK1, are being developed and evaluated in preclinical studies [70, 71].
Human epidermal growth factor receptor 2 (HER2) and its role in human cancers
HER2 (also known as ERBB2) is a transmembrane receptor tyrosine kinase of the EGFR (epidermal growth factor receptor) family, consisting of EGFR (ERBB1), HER2 (ERBB2), HER3 (ERBB3) and HER4 (ERBB4) [72, 73]. These receptor tyrosine kinases act in the epithelium as signal transducers between mesenchymal and epithelial cells. The EGFR receptors form homo- and heterodimers which transphosphorylate each other upon activation. This further stimulates intracellular downstream pathways, such as mitogen-activated protein kinase (MAPK) signalling cascades RAS/MEK/ERK, PI3K/AKT/TOR or STAT transcription factors, that are involved in proliferation, survival and differentiation [74, 75]. Examples of ligands shed by the mesenchyme are Neuregulins that bind to HER3 and HER4. Although HER2 is the most potent oncogene of the family, to date no high affinity ligand for HER2 has been found. However, its conformation resembles a ligand-activated state, which favours dimerisation [76, 77]. This makes HER2 the preferred dimerisation partner for the other three family members [78]. As the signalling system may be fine-tuned by the partners of the heterodimer, the abundance of HER2 is crucial. Importantly, the HER2-HER3 heterodimer is the most transforming and mitogenic heterodimer of the EGFR family [79, 80]. The potent proliferation signalling generated by the EGFR network is often corrupted in cancer cells. Overexpression or constitutive action of the individual receptors is observed in a variety of tumours [81]. HER2 is overexpressed in several types of tumours such as breast, gastric, oesophageal, lung, bladder or endometrial cancer [82]. However, in breast cancer the association of ERBB2 amplification and subsequent overexpression of HER2 is the best studied. About 20% of breast tumours show ERBB2 gene amplification [83]. The amplification correlates with high risk of recurrence and disease-related death and thus a poor prognosis [84, 85]. HER2+ tumours form a separate subclass of breast cancer that is eligible for HER2-targeted treatment. These tumours are typically oestrogen receptor negative. Moreover, HER2 expression is the most important predictive factor for response to HER2-targeted therapies [86]. Similarly, HER2 overexpression correlates with pathological features such as lymphatic invasion, high grade or large tumour size in gastric cancer [87]. Besides the ERBB2 amplification, somatic mutations are observed in several tumour types, such as breast, lung, gastric and bladder cancer. Most of these mutations are missense mutations in the tyrosine kinase or extracellular domain, rendering the receptor constantly active [88].
HER2-targeted therapies
HER2-targeting drugs to treat HER2+ tumours can be divided into two classes: (a) HER2-directed antibodies, such as trastuzumab, and (b) small-molecule inhibitors targeting the kinase activity of the receptor, such as lapatinib [89]. Trastuzumab is a humanised antibody that binds an extracellular epitope of HER2. Binding of the antibody uncouples HER2-containing dimers, which leads to partial inhibition of downstream signalling. In addition, trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) [90–92]. As early as 1998, trastuzumab was approved for metastatic breast cancer, and in 2006 approval for adjuvant therapy of early breast cancer followed. Trastuzumab has been successfully integrated into standard therapy for HER2+ breast cancer, either in a perioperative or metastatic setting [93–96]. In 2010, trastuzumab was approved for advanced gastric and gastro-oesophageal cancer. Previously, the ToGa (trastuzumab for gastric cancer) study showed a significant overall survival benefit for patients with metastatic HER2+ gastric cancer when the drug was added to standard chemotherapy [97]. A new generation of HER2 antibody, pertuzumab, recognises a different epitope of HER2. Its binding leads to the blockage of ligand-induced HER2-HER3 dimerisation and thus inhibits partially downstream signalling [98]. As trastuzumab and pertuzumab target different epitopes, the combination of the two antibodies showed a synergistic effect in preclinical studies and clinical trials [99–101]. Since 2012, pertuzumab has been approved for the treatment of HER2+ metastatic breast cancer [102]. Another derivative of trastuzumab is trastuzumab emtansine (T-DM1). This is an antibody-drug conjugate whereby trastuzumab is bound to maytansinoid, a drug inhibiting microtubule polymerisation [103]. This new antibody-drug conjugate binds to the epitope with an affinity similar to trastuzumab, and, in addition to blocking signal transduction and induction of ADCC, the drug mediates the inhibition of microtubules [104]. In 2013, T-DM1 was approved for advanced HER2+ breast cancer [105].
In contrast to trastuzumab, lapatinib is a small-molecule kinase inhibitor that binds reversibly to the ATP-binding side of EGFR and HER2. Lapatinib disables HER2 downstream signalling, being effective even in HER2+ cancers that have progressed after trastuzumab treatment. Lapatinib was approved for the treatment of advanced breast cancer in 2006 [106]. Further developed derivatives are afatinib and neratinib. Both small-molecule inhibitors bind irreversibly to the ATP-binding site of RTKs. Whereas neratinib binds only to HER2, afatinib binds to both HER2 and EGFR [107, 108]. Neratinib was approved for the adjuvant treatment of HER2+ breast cancer after trastuzumab progression in 2017 [109]. Afatinib, as a dual EGFR-HER2 inhibitor, is approved for the treatment of non-small-cell lung carcinoma [110]. Besides the considerable success of HER2-targeting drugs and their improvements, resistance formation remains a clinical challenge.
Resistance mechanisms against HER2 inhibition
Although HER2-targeting treatment provides considerable benefit for patients with HER2+ tumours, most tumours ultimately progress to treatment resistance [111]. Resistance may be pre-existing or drug-induced (acquired). Generally, pre-existing resistance tends to occur through alterations of the receptor itself or modifications of downstream signalling pathways [112, 113]. On the other hand, acquired resistance is more diverse. Here, resistance instead occurs through bypass mechanisms as increased expression of other family members (EGFR, HER3) or different receptor tyrosine kinases, such as hepatocyte growth factor receptor (MET) [114–116]. It is important to mention that all the resistance mechanisms described above can be the cause of pre-existing as well as of acquired resistance [117–119].
Expression of a truncated form of HER2, so-called p95-HER2, which lacks the trastuzumab binding epitope, represents an alteration of the HER2 receptor mediating treatment resistance [113, 120]. P95-HER2 is associated with low response rates to trastuzumab. However, since the kinase activity of the receptor remains intact, these tumours are still sensitive to kinase inhibitors such as lapatinib [121]. Another HER2 alteration that mediates resistance is an ERBB2 gene splice variant lacking exon 16. These receptors retain the epitopes recognised by trastuzumab, but HER2 homodimers containing this isoform are more stable than their wild-type counterparts. Therefore, trastuzumab-mediated disruption of the homodimers is circumvented and the homodimers remain activated to stimulate downstream targets such as Src kinase [122, 123].
Another family of resistance mechanisms consists of so-called bypass track pathways. Here, downstream signalling pathways are kept activated by mechanisms “bypassing” the inhibited receptor tyrosine kinase [124]. Examples are the amplification of MET or the enhanced stimulation of MET by its ligand, hepatocyte growth factor (HGF) [125]. Similarly, upregulation of EGFR and/or HER3 renders cancer cells resistant to HER2-targeting treatment. In this scenario, more active EGFR/HER3 receptors are available that activate downstream signalling pathways [126]. Moreover, aberrantly activated intracellular kinases of the HER2 signalling pathway can bypass HER2 inhibition. Prominent examples are mutations in the phosphatidylinositol 3-kinase (PI3K) pathway. Such alterations are observed in about 30% of HER2+ breast cancer patients [127]. Patients bearing PI3K catalytic subunit alpha (PI3KCA) gene mutations do not benefit from HER2-targeting therapies. As was shown in the randomised phase III EMILIA and Neo-ALLTO trials, where this patient group did not benefit from lapatinib or trastuzumab, respectively [128, 129].
Resistance can also emerge from aberration of the apoptosis pathways. Inhibition of driver oncogenes such as HER2 ultimately leads to apoptosis. Accordingly, expression levels of the pro-apoptotic BH3-only protein Bcl-2-like protein 11 (BIM) are predictive for the response of HER2-overexpressing breast cancer cells to HER2-targeting treatment. Thus, in a small patient cohort where lapatinib was used as a single agent treatment for metastatic HER2+ breast cancer, low BIM expression levels before treatment correlated with inefficient treatment due to inadequate apoptotic response [130, 131]. Moreover, resistance against HER2-targeted treatment in HER2+ breast cancers involves anti-apoptotic Bcl-2 as well as pro-apoptotic BH3-only family members. Accordingly, Bcl-2 is up- and Bax downregulated in trastuzumab-resistant breast cancer cells [132]. As already mentioned, several links between apoptosis and autophagy exist, indicating crosstalk between the two pathways [133]. The aberrant expression of proteins is thus involved in apoptosis influence as well autophagy activity. The anti-apoptotic protein Bcl-2 is often upregulated in tumours. Through binding and subsequent sequestration of the autophagy protein Beclin-1, Bcl-2 can negatively regulate autophagy initiation [27]. High Bcl-2 levels can allow co-existence of low apoptosis as well as low autophagy. The pro-apoptotic Bcl-2 family member Bax can negatively regulate autophagy initiation through caspase-mediated cleavage of Beclin-1 as well [134]. This connection would indicate that activation of apoptosis results in autophagy suppression. Additionally, Bcl-2 family members regulate a non-canonical form of autophagy leading to necroptosis, an apoptosis-independent form of programmed cell death [135]. Here the autophagy machinery serves as a scaffold for the formation of the cell death-inducing signalling complex, the necrosome [136].
Resistance mechanisms against HER2-targeting treatment are multifaceted. Some of them include an aberration of the receptor itself, whereas others bypass HER2 signalling. Examples are the overexpression of other tyrosine kinase receptors or constitutive activation of downstream targets [111]. Resistance may arise from aberrations in the apoptosis pathway as well. As apoptosis and autophagy are interconnected pathways, expression levels of apoptosis proteins may also influence autophagic flux. However, the connection between the pathways is complex and not yet fully understood [69].
HER2 and autophagy – possible candidates for combination therapy?
HER2-targeted therapies are best studied in breast cancer. However, they are also approved for the treatment of HER2+ gastric cancer and tumours of the gastro-oesophageal junction and can be applied off-label for HER2-positive oesophageal adenocarcinoma [137]. Decreased autophagy supports the development of HER2+ breast cancer. Firstly, an association between the loss of the tumour suppressor and autophagy protein Beclin-1 with HER2 gene amplification was found [138]. Secondly, decreased Beclin-1 mRNA expression in mammary tumours is not only associated with worse disease-free survival but is also more common in HER2+ breast cancer [139]. Supporting these notions, a study investigating human and mouse breast cancer cells found that low Beclin-1 mRNA levels correlate with HER2 overexpression, and that HER2-amplified tumours exhibit a low autophagy gene expression signature, independent of Beclin-1 mRNA expression. Results from xenograft experiments in this study suggest that in HER2+ tumours autophagy is downregulated even in a Beclin-1 wild type background [140]. Moreover, it was found that HER2 directly interacts with Beclin-1 in breast cancer cells, and that this interaction inhibits autophagy. Disruption of this interaction with Tat-Beclin-1, an autophagy-inducing peptide, caused a cessation of tumour growth in xenograft models [141].
During breast cancer therapy, autophagy has been shown to support resistance to chemotherapeutic agents [142–144]. Similarly, in the context of HER2-targeting treatment, autophagy is mainly discussed as a resistance mechanism. An analysis of a large collection of breast cancer cell lines showed that the transcript levels of ATG12 were upregulated in trastuzumab-non-responsive HER2-overexpressing cells as compared with treatment-sensitive cell lines [145]. Furthermore, trastuzumab-resistant breast cancer spheroids are characterised by increased autophagic activity and show increased sensitivity to autophagy inhibition [146, 147]. Similarly, breast cancer cells rendered resistant to lapatinib exhibited an activation of autophagy and could be re-sensitised to the drug by autophagy inhibition [148, 149]. In our group, we observed a similar phenomenon for HER2-amplified oesophageal adenocarcinoma (EAC) cells. In a lapatinib-resistant EAC cell line (OE19LapR) we observed a general upregulation of basal autophagy levels compared to the parental cell line (OE19P). Upon autophagy inhibition, OE19LapR could be re-sensitised to lapatinib treatment to the level of parental cells [150]. We were able to corroborate these findings by growing both cell lines in a chick chorioallantoic membrane (CAM) xenograft assay. The CAM assay is a 3D in ovo cell culture model, where tumour cells are grown in a scaffold on an extraembryonic membrane of the chick embryo [151]. Generally, OE19LapR cells formed microtumours that contained more cells, were more vascularised and less necrotic. LC3B and p62 dot formation compared to a homogeneous staining is considered to be an indication for active autophagy in IHC [152]. In the microtumours formed by our two EAC cell lines, OE19LapR and OE19P, higher autophagy levels were indicated by dot-like staining in the OE19LapR tumours compared to the OE19P tumours, where the staining was more homogenous (fig. 3). Conversely, a recent study in gastric cancer cells reported inhibition of autophagic flux upon trastuzumab treatment. A pool of resistant cancer cells showed lower basal levels of autophagy than the parental cells, and autophagy inhibition induced more cell death in the parental cell line than in the trastuzumab refractory cells [153]. Moreover, the activation of autophagy in the context of HER2 inhibitor resistance is discussed as part of a metabolic shift in resistant cells. Trastuzumab-resistant breast cancer cells with upregulated autophagy were shown to have a significantly higher expression of the catalytic subunit of AMPK, AMPKα1 [154, 155]. Cells treated with receptor tyrosine kinase inhibitors activate AMPK as a response to growth factor deprivation, which leads, among other effects, to the inhibition of protein synthesis. This is lethal to HER2-amplified breast cancer cells depending on glycolysis [156]. AMPK not only coordinates the adaptive response to ATP depletion but also modulates the activity of autophagy regulators such as ULK1 and mTOR [157]. Thus, increased activity of AMPK could result in a shift to catabolism and prepare cells resistant to HER2 inhibition to activate protective autophagy and overcome the acute bioenergetic crisis resulting from HER2 inhibition [158]. Current data on the role of autophagy in HER2+ tumour formation and possible resistance against HER2 inhibition are not fully conclusive and the role of autophagy in the formation and progression of HER2+ carcinogenesis warrants further studies [159, 160].
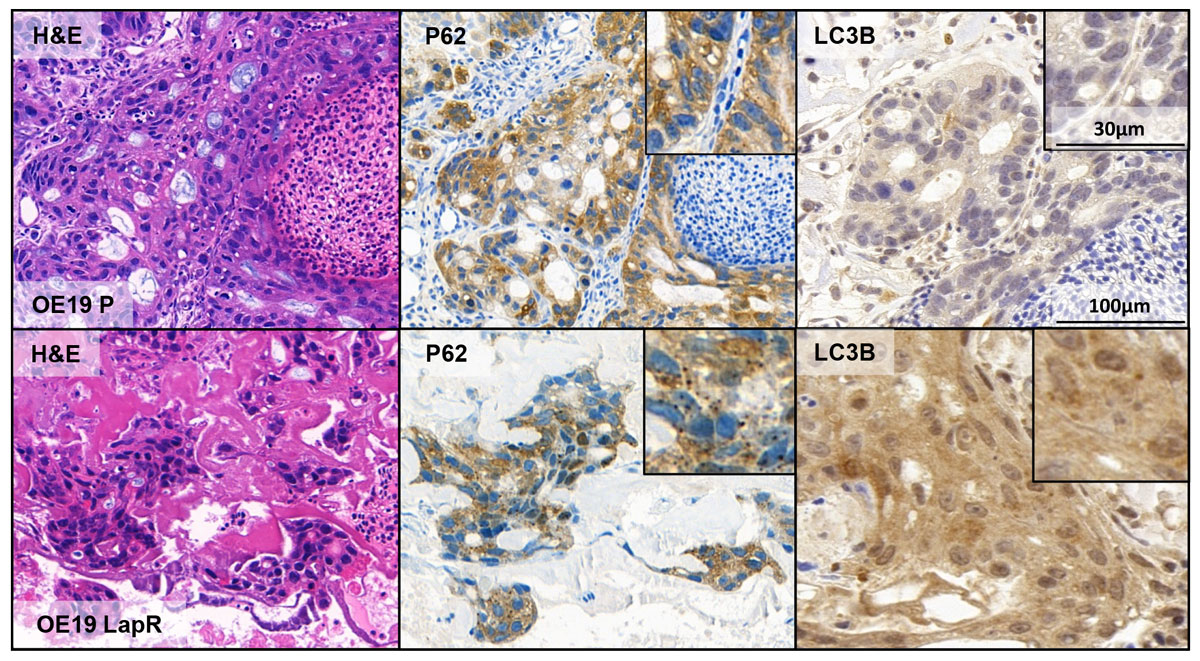
Figure 3
Increased autophagy in lapatinib-resistant OE19 oesophageal cancer cells. Microtumours developed from OE19 parental (OE19P) and OE19 lapatinib-resistant (OE19LapR) cells are shown in the upper and lower panels, respectively. Depicted from left to right are: H&E, p62 and LC3B staining. Lapatinib-resistant OE19 cells display p62 and LC3B dot formation indicative of increased autophagic activity compared to the parental cell line. The staining intensity of p62 corresponds to a score of 3+ and of LC3B to a score of 2+, according to Schläfli et al. [152].
H&E = haematoxylin and eosin; p62 = sequestosome 1, SQSTM1; LC3B = microtubule-associated protein light chain 3B
On the basis of several preclinical studies demonstrating the antineoplastic effect of autophagy inhibition in combination with a variety of anticancer treatments, first phase I clinical trials studying the safety and antineoplastic effect of the autophagy inhibitors chloroquine and hydroxychloroquine were conducted. In 2014, it was shown that hydroxychloroquine could be safely combined with cytotoxic chemotherapeutics [68, 161, 162]. First clinical results of studies including chloroquine or hydroxychloroquine for anticancer treatment in combination with standard treatment are promising [163]. At the moment there are no clinical trials involving hydroxychloroquine in HER2+ cancers. However, there are several clinical trials investigating the effect of hydroxychloroquine on breast cancer. One of them is the CLEVER pilot trial, which is a phase II trial of hydroxychloroquine in combination with everolimus, an mTOR inhibitor, for the prevention of recurrent breast cancer (NCT03032406). The GLACIER trial is testing the efficacy of gedatolisib (a PI3K/mTOR inhibitor) and hydroxychloroquine on early recurrent breast cancer (NCT03400254). Another study is investigating the efficacy of hydroxychloroquine in metastatico estrogen-receptor-positive breast cancer that has progressed after hormonal therapy (NCT02414776).
Various pieces of evidence suggest a connection between autophagy and HER2. The autophagy-initiating protein Beclin-1 was shown to be low-expressed in HER2+ breast cancer. Moreover, independent of Beclin-1 expression, HER2+ breast cancer cells were shown to exhibit low expression of ATGs. Additionally, the direct interaction of Beclin-1 and HER2 was shown to influence autophagy activity [141]. In the context of resistance to HER2-targeted treatment, autophagy is often reported as a resistance mechanism upregulated in resistant cells. Some of these studies showed that autophagy inhibition led to a re-sensitisation to HER2-targeting drugs [148]. The results from previous clinical studies with hydroxychloroquine, indicate that the inhibition of autophagy might be a valuable new avenue for breast cancer treatment. Clearly, there is a need for more specific and potent autophagy inhibitors. Clinical trials investigating the inhibition of autophagy in HER2+ breast cancer are still lacking, but results from ongoing trials in other breast cancer subtypes and the above-mentioned preclinical data might drive research in the direction of combining autophagy inhibition with HER2-targeting treatment in HER2-amplified cancers.
Concluding remarks
Generally, the role of macroautophagy in tumour development, progression and resistance formation against cancer therapy is ambivalent [46]. The HER2 oncogene is overexpressed in different tumour types, and drugs specifically targeting this receptor tyrosine kinase have been successfully applied in cancer therapy [98]. However, progression and resistance formation against these drugs ultimately occurs. Resistance mechanisms are multifaceted, and ways to prevent or overcome resistance are urgently needed [159]. Beclin-1, an autophagy initiating protein was reported to directly interact with HER2. Moreover, this interaction was shown to influence autophagy activity [141]. Additionally, Beclin-1 expression was reported to be lower in HER2+ tumours. In the context of resistance to HER2-targeting treatments, autophagy was reported to be upregulated. Even though contradictory data exists, most in vitro studies report an upregulation of autophagy in resistant cells, which can be abrogated to some extent by inhibiting autophagy [145, 147, 148]. Still, the understanding of autophagy’s role in the development and progression of HER2+ cancers is in its infancy. Future studies deciphering the networks connecting HER2 and autophagy are therefore needed. Two autophagy-inhibiting agents, chloroquine and hydroxychloroquine, are available for clinical application. They have been evaluated for cancer treatment alone and in different combinations in a number of clinical trials. However, it is not yet known whether the beneficial effects observed during hydroxychloroquine and chloroquine treatment are based on their autophagy inhibition function or on their effects on other pathways [64]. Moreover, the inhibitory effect of hydroxychloroquine on the lysosome could lead to defective lysosomal function and thus cause lysosomal storage disease [164, 165]. The development of specific autophagy inhibitors is still in an early phase, but using conditional ATG knockout animals it has been shown that systemic inhibition of essential autophagy genes is possible during a certain therapeutic window [166]. In addition, a better understanding of the crosstalk between autophagy and apoptosis could help to shape the development of new, more specific autophagy targeting agents for antineoplastic treatment. With improved autophagy inhibitors and a better understanding of the processes involved, new quests will undoubtedly arise, such as the search for better and more reliable biomarkers as indicators of macroautophagic flux in human tissue. The combination of LC3B and p62 has been proposed as an approximate approach to characterising different autophagy status in tissue samples [152, 167]. However, other marker proteins such as Beclin-1, ULK1 or ATG5 have also been used to detect autophagy in tissue via IHC (excellently reviewed in [168]). One issue of these attempts to assess autophagy via IHC is that the expression of some autophagy-related proteins does not change upon autophagy induction. Moreover, the expression levels of these proteins are cell-type and tissue-specific, and since most autophagy related genes are also involved in other cellular pathways, an autophagy specific marker or marker combination has not yet been identified. For future studies on autophagy-targeting therapies, however, such biomarkers would be of great value in deciding which tumours should be treated with autophagy-inhibiting or possibly even autophagy-inducing agents.
Acknowledgements
The authors acknowledge the Translational Research Unit (TRU) of the Institute of Pathology, University of Bern, for excellent technical support in this project. The support of the TRANSAUTOPHAGY COST Action CA15138 and Life Sciences Switzerland, Section Autophagy is highly appreciated.
References
1
Guo
JY
,
Teng
X
,
Laddha
SV
,
Ma
S
,
Van Nostrand
SC
,
Yang
Y
, et al.
Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016;30(15):1704–17. doi:.https://doi.org/10.1101/gad.283416.116
2
Yin
Z
,
Pascual
C
,
Klionsky
DJ
. Autophagy: machinery and regulation. Microb Cell. 2016;3(12):588–96. doi:.https://doi.org/10.15698/mic2016.12.546
3
Abada
A
,
Elazar
Z
. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014;15(8):839–52. doi:.https://doi.org/10.15252/embr.201439076
4
Patel
S
,
Homaei
A
,
El-Seedi
HR
,
Akhtar
N
. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed Pharmacother. 2018;105:526–32. doi:.https://doi.org/10.1016/j.biopha.2018.05.148
5
Mizushima
N
,
Komatsu
M
. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. doi:.https://doi.org/10.1016/j.cell.2011.10.026
6
Chen
Y
,
Yu
L
. Recent progress in autophagic lysosome reformation. Traffic. 2017;18(6):358–61. doi:.https://doi.org/10.1111/tra.12484
7
Dikic
I
,
Elazar
Z
. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–64. doi:.https://doi.org/10.1038/s41580-018-0003-4
8
Yu
L
,
Chen
Y
,
Tooze
SA
. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018;14(2):207–15. doi:.https://doi.org/10.1080/15548627.2017.1378838
9
Papinski
D
,
Schuschnig
M
,
Reiter
W
,
Wilhelm
L
,
Barnes
CA
,
Maiolica
A
, et al.
Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53(3):471–83. doi:.https://doi.org/10.1016/j.molcel.2013.12.011
10
Karanasios
E
,
Walker
SA
,
Okkenhaug
H
,
Manifava
M
,
Hummel
E
,
Zimmermann
H
, et al.
Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. 2016;7(1):12420. doi:.https://doi.org/10.1038/ncomms12420
11
Lamb
CA
,
Yoshimori
T
,
Tooze
SA
. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–74. doi:.https://doi.org/10.1038/nrm3696
12
Kihara
A
,
Kabeya
Y
,
Ohsumi
Y
,
Yoshimori
T
. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2(4):330–5. doi:.https://doi.org/10.1093/embo-reports/kve061
13
Liang
XH
,
Jackson
S
,
Seaman
M
,
Brown
K
,
Kempkes
B
,
Hibshoosh
H
, et al.
Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–6. doi:.https://doi.org/10.1038/45257
14
Miracco
C
,
Cosci
E
,
Oliveri
G
,
Luzi
P
,
Pacenti
L
,
Monciatti
I
, et al.
Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30(2):429–36.
15
Tanida
I
,
Ueno
T
,
Kominami
E
. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi:.https://doi.org/10.1007/978-1-59745-157-4_4
16
Cherra
SJ, 3rd
,
Kulich
SM
,
Uechi
G
,
Balasubramani
M
,
Mountzouris
J
,
Day
BW
, et al.
Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190(4):533–9. doi:.https://doi.org/10.1083/jcb.201002108
17
Klionsky
DJ
,
Abdelmohsen
K
,
Abe
A
,
Abedin
MJ
,
Abeliovich
H
,
Acevedo Arozena
A
, et al.
Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. doi:.https://doi.org/10.1080/15548627.2015.1100356
18
Zaffagnini
G
,
Martens
S
. Mechanisms of Selective Autophagy. J Mol Biol. 2016;428(9 Pt A):1714–24. doi:.https://doi.org/10.1016/j.jmb.2016.02.004
19
Bjørkøy
G
,
Lamark
T
,
Pankiv
S
,
Øvervatn
A
,
Brech
A
,
Johansen
T
. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi:.https://doi.org/10.1016/S0076-6879(08)03612-4
20
Birgisdottir
ÅB
,
Lamark
T
,
Johansen
T
. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126(Pt 15):3237–47.
21
Kaushik
S
,
Cuervo
AM
. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–17. doi:.https://doi.org/10.1016/j.tcb.2012.05.006
22
Saha
T
. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy. 2012;8(11):1643–56. doi:.https://doi.org/10.4161/auto.21654
23
Uttenweiler
A
,
Mayer
A
. Microautophagy in the yeast Saccharomyces cerevisiae. Methods Mol Biol. 2008;445:245–59. doi:.https://doi.org/10.1007/978-1-59745-157-4_16
24
Gomes
LC
,
Dikic
I
. Autophagy in antimicrobial immunity. Mol Cell. 2014;54(2):224–33. doi:.https://doi.org/10.1016/j.molcel.2014.03.009
25
Niida
M
,
Tanaka
M
,
Kamitani
T
. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol. 2010;47(14):2378–87. doi:.https://doi.org/10.1016/j.molimm.2010.05.004
26
Rubinsztein
DC
,
Codogno
P
,
Levine
B
. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–30. doi:.https://doi.org/10.1038/nrd3802
27
Pattingre
S
,
Tassa
A
,
Qu
X
,
Garuti
R
,
Liang
XH
,
Mizushima
N
, et al.
Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39. doi:.https://doi.org/10.1016/j.cell.2005.07.002
28
Rubinstein
AD
,
Eisenstein
M
,
Ber
Y
,
Bialik
S
,
Kimchi
A
. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44(5):698–709. doi:.https://doi.org/10.1016/j.molcel.2011.10.014
29
Pyo
J-O
,
Jang
MH
,
Kwon
YK
,
Lee
HJ
,
Jun
JI
,
Woo
HN
, et al.
Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280(21):20722–9. doi:.https://doi.org/10.1074/jbc.M413934200
30
Mizushima
N
,
Levine
B
,
Cuervo
AM
,
Klionsky
DJ
. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75. doi:.https://doi.org/10.1038/nature06639
31
Yue
Z
,
Jin
S
,
Yang
C
,
Levine
AJ
,
Heintz
N
. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100(25):15077–82. doi:.https://doi.org/10.1073/pnas.2436255100
32
Qu
X
,
Yu
J
,
Bhagat
G
,
Furuya
N
,
Hibshoosh
H
,
Troxel
A
, et al.
Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20. doi:.https://doi.org/10.1172/JCI20039
33
Takamura
A
,
Komatsu
M
,
Hara
T
,
Sakamoto
A
,
Kishi
C
,
Waguri
S
, et al.
Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi:.https://doi.org/10.1101/gad.2016211
34
Mathew
R
,
Karp
CM
,
Beaudoin
B
,
Vuong
N
,
Chen
G
,
Chen
HY
, et al.
Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75. doi:.https://doi.org/10.1016/j.cell.2009.03.048
35
Green
DR
,
Galluzzi
L
,
Kroemer
G
. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–12. doi:.https://doi.org/10.1126/science.1201940
36
Mortensen
M
,
Soilleux
EJ
,
Djordjevic
G
,
Tripp
R
,
Lutteropp
M
,
Sadighi-Akha
E
, et al.
The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455–67. doi:.https://doi.org/10.1084/jem.20101145
37
Cicchini
M
,
Chakrabarti
R
,
Kongara
S
,
Price
S
,
Nahar
R
,
Lozy
F
, et al.
Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–52. doi:.https://doi.org/10.4161/auto.34398
38
Rodriguez
OC
,
Choudhury
S
,
Kolukula
V
,
Vietsch
EE
,
Catania
J
,
Preet
A
, et al.
Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11(23):4436–46. doi:.https://doi.org/10.4161/cc.22778
39
Isakson
P
,
Bjørås
M
,
Bøe
SO
,
Simonsen
A
. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116(13):2324–31. doi:.https://doi.org/10.1182/blood-2010-01-261040
40
Goussetis
DJ
,
Gounaris
E
,
Wu
EJ
,
Vakana
E
,
Sharma
B
,
Bogyo
M
, et al.
Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120(17):3555–62. doi:.https://doi.org/10.1182/blood-2012-01-402578
41
Ma
Y
,
Galluzzi
L
,
Zitvogel
L
,
Kroemer
G
. Autophagy and cellular immune responses. Immunity. 2013;39(2):211–27. doi:.https://doi.org/10.1016/j.immuni.2013.07.017
42
Deretic
V
,
Saitoh
T
,
Akira
S
. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–37. doi:.https://doi.org/10.1038/nri3532
43
Yasir
M
,
Pachikara
ND
,
Bao
X
,
Pan
Z
,
Fan
H
. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun. 2011;79(10):4019–28. doi:.https://doi.org/10.1128/IAI.05308-11
44
Zhang
L
,
Sung
JJ
,
Yu
J
,
Ng
SC
,
Wong
SH
,
Cho
CH
, et al.
Xenophagy in Helicobacter pylori- and Epstein-Barr virus-induced gastric cancer. J Pathol. 2014;233(2):103–12. doi:.https://doi.org/10.1002/path.4351
45
Conway
KL
,
Kuballa
P
,
Song
JH
,
Patel
KK
,
Castoreno
AB
,
Yilmaz
OH
, et al.
Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology. 2013;145(6):1347–57. doi:.https://doi.org/10.1053/j.gastro.2013.08.035
46
Maiuri
MC
,
Tasdemir
E
,
Criollo
A
,
Morselli
E
,
Vicencio
JM
,
Carnuccio
R
, et al.
Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16(1):87–93. doi:.https://doi.org/10.1038/cdd.2008.131
47
Vousden
KH
,
Lane
DP
. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–83. doi:.https://doi.org/10.1038/nrm2147
48
Laplante
M
,
Sabatini
DM
. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi:.https://doi.org/10.1016/j.cell.2012.03.017
49
Arico
S
,
Petiot
A
,
Bauvy
C
,
Dubbelhuis
PF
,
Meijer
AJ
,
Codogno
P
, et al.
The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276(38):35243–6. doi:.https://doi.org/10.1074/jbc.C100319200
50
Zhao
Y
,
Yang
J
,
Liao
W
,
Liu
X
,
Zhang
H
,
Wang
S
, et al.
Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665–75. doi:.https://doi.org/10.1038/ncb2069
51
Cai
Q
,
Yan
L
,
Xu
Y
. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2015;34(25):3315–24. doi:.https://doi.org/10.1038/onc.2014.264
52
Kroemer
G
,
Mariño
G
,
Levine
B
. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93. doi:.https://doi.org/10.1016/j.molcel.2010.09.023
53
Lazova
R
,
Camp
RL
,
Klump
V
,
Siddiqui
SF
,
Amaravadi
RK
,
Pawelek
JM
. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18(2):370–9. doi:.https://doi.org/10.1158/1078-0432.CCR-11-1282
54
Peng
Y-F
,
Shi
YH
,
Ding
ZB
,
Ke
AW
,
Gu
CY
,
Hui
B
, et al.
Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9(12):2056–68. doi:.https://doi.org/10.4161/auto.26398
55
Viale
A
,
Pettazzoni
P
,
Lyssiotis
CA
,
Ying
H
,
Sánchez
N
,
Marchesini
M
, et al.
Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32. doi:.https://doi.org/10.1038/nature13611
56
Wolf
J
,
Dewi
DL
,
Fredebohm
J
,
Müller-Decker
K
,
Flechtenmacher
C
,
Hoheisel
JD
, et al.
A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013;15(6):R109. doi:.https://doi.org/10.1186/bcr3576
57
Gong
C
,
Bauvy
C
,
Tonelli
G
,
Yue
W
,
Deloménie
C
,
Nicolas
V
, et al.
Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32(18):2261–72, 1–11. doi:.https://doi.org/10.1038/onc.2012.252
58
Levy
JMM
,
Thompson
JC
,
Griesinger
AM
,
Amani
V
,
Donson
AM
,
Birks
DK
, et al.
Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–80. doi:.https://doi.org/10.1158/2159-8290.CD-14-0049
59
Ko
A
,
Kanehisa
A
,
Martins
I
,
Senovilla
L
,
Chargari
C
,
Dugue
D
, et al.
Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014;21(1):92–9. doi:.https://doi.org/10.1038/cdd.2013.124
60
Young
ARJ
,
Narita
M
,
Ferreira
M
,
Kirschner
K
,
Sadaie
M
,
Darot
JF
, et al.
Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi:.https://doi.org/10.1101/gad.519709
61
Mauthe
M
,
Orhon
I
,
Rocchi
C
,
Zhou
X
,
Luhr
M
,
Hijlkema
KJ
, et al.
Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–55. doi:.https://doi.org/10.1080/15548627.2018.1474314
62
Wang
Y
,
Peng
RQ
,
Li
DD
,
Ding
Y
,
Wu
XQ
,
Zeng
YX
, et al.
Chloroquine enhances the cytotoxicity of topotecan by inhibiting autophagy in lung cancer cells. Chin J Cancer. 2011;30(10):690–700. doi:.https://doi.org/10.5732/cjc.011.10056
63
Finbloom
DS
,
Silver
K
,
Newsome
DA
,
Gunkel
R
. Comparison of hydroxychloroquine and chloroquine use and the development of retinal toxicity. J Rheumatol. 1985;12(4):692–4.
64
Manic
G
,
Obrist
F
,
Kroemer
G
,
Vitale
I
,
Galluzzi
L
. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol. 2014;1(1):e29911. doi:.https://doi.org/10.4161/mco.29911
65
Goldberg
SB
,
Supko
JG
,
Neal
JW
,
Muzikansky
A
,
Digumarthy
S
,
Fidias
P
, et al.
A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol. 2012;7(10):1602–8. doi:.https://doi.org/10.1097/JTO.0b013e318262de4a
66
Maycotte
P
,
Aryal
S
,
Cummings
CT
,
Thorburn
J
,
Morgan
MJ
,
Thorburn
A
. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–12. doi:.https://doi.org/10.4161/auto.8.2.18554
67
Eng
CH
,
Wang
Z
,
Tkach
D
,
Toral-Barza
L
,
Ugwonali
S
,
Liu
S
, et al.
Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA. 2016;113(1):182–7. doi:.https://doi.org/10.1073/pnas.1515617113
68
Barnard
RA
,
Wittenburg
LA
,
Amaravadi
RK
,
Gustafson
DL
,
Thorburn
A
,
Thamm
DH
. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10(8):1415–25. doi:.https://doi.org/10.4161/auto.29165
69
Goodall
ML
,
Fitzwalter
BE
,
Zahedi
S
,
Wu
M
,
Rodriguez
D
,
Mulcahy-Levy
JM
, et al.
The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev Cell. 2016;37(4):337–49. doi:.https://doi.org/10.1016/j.devcel.2016.04.018
70
Ronan
B
,
Flamand
O
,
Vescovi
L
,
Dureuil
C
,
Durand
L
,
Fassy
F
, et al.
A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10(12):1013–9. doi:.https://doi.org/10.1038/nchembio.1681
71
Martin
KR
,
Celano
SL
,
Solitro
AR
,
Gunaydin
H
,
Scott
M
,
O’Hagan
RC
, et al.
A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience. 2018;8:74–84. doi:.https://doi.org/10.1016/j.isci.2018.09.012
72
Yarden
Y
,
Sliwkowski
MX
. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37. doi:.https://doi.org/10.1038/35052073
73
Arteaga
CL
,
Engelman
JA
. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303. doi:.https://doi.org/10.1016/j.ccr.2014.02.025
74
Borg
JP
,
Marchetto
S
,
Le Bivic
A
,
Ollendorff
V
,
Jaulin-Bastard
F
,
Saito
H
, et al.
ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2(7):407–14. doi:.https://doi.org/10.1038/35017038
75
Yarden
Y
,
Pines
G
. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–63. doi:.https://doi.org/10.1038/nrc3309
76
Cho
H-S
,
Mason
K
,
Ramyar
KX
,
Stanley
AM
,
Gabelli
SB
,
Denney
DW, Jr
, et al.
Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924):756–60. doi:.https://doi.org/10.1038/nature01392
77
Garrett
TPJ
,
McKern
NM
,
Lou
M
,
Elleman
TC
,
Adams
TE
,
Lovrecz
GO
, et al.
The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11(2):495–505. doi:.https://doi.org/10.1016/S1097-2765(03)00048-0
78
Olayioye
MA
,
Neve
RM
,
Lane
HA
,
Hynes
NE
. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19(13):3159–67. doi:.https://doi.org/10.1093/emboj/19.13.3159
79
Alimandi
M
,
Romano
A
,
Curia
MC
,
Muraro
R
,
Fedi
P
,
Aaronson
SA
, et al.
Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10(9):1813–21.
80
Pinkas-Kramarski
R
,
Soussan
L
,
Waterman
H
,
Levkowitz
G
,
Alroy
I
,
Klapper
L
, et al.
Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996;15(10):2452–67. doi:.https://doi.org/10.1002/j.1460-2075.1996.tb00603.x
81
Dong
J
,
Opresko
LK
,
Dempsey
PJ
,
Lauffenburger
DA
,
Coffey
RJ
,
Wiley
HS
. Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc Natl Acad Sci USA. 1999;96(11):6235–40. doi:.https://doi.org/10.1073/pnas.96.11.6235
82
Yan
M
,
Schwaederle
M
,
Arguello
D
,
Millis
SZ
,
Gatalica
Z
,
Kurzrock
R
. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34(1):157–64. doi:.https://doi.org/10.1007/s10555-015-9552-6
83
Slamon
DJ
,
Clark
GM
,
Wong
SG
,
Levin
WJ
,
Ullrich
A
,
McGuire
WL
. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi:.https://doi.org/10.1126/science.3798106
84
Ross
JS
,
Fletcher
JA
. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells. 1998;16(6):413–28. doi:.https://doi.org/10.1002/stem.160413
85
Press
MF
,
Bernstein
L
,
Thomas
PA
,
Meisner
LF
,
Zhou
JY
,
Ma
Y
, et al.
HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J Clin Oncol. 1997;15(8):2894–904. doi:.https://doi.org/10.1200/JCO.1997.15.8.2894
86
Wolff
AC
,
Hammond
ME
,
Hicks
DG
,
Dowsett
M
,
McShane
LM
,
Allison
KH
, et al.; American Society of Clinical Oncology; College of American Pathologists. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. doi:.https://doi.org/10.1200/JCO.2013.50.9984
87
Begnami
MD
,
Fukuda
E
,
Fregnani
JH
,
Nonogaki
S
,
Montagnini
AL
,
da Costa
WL, Jr
, et al.
Prognostic implications of altered human epidermal growth factor receptors (HERs) in gastric carcinomas: HER2 and HER3 are predictors of poor outcome. J Clin Oncol. 2011;29(22):3030–6. doi:.https://doi.org/10.1200/JCO.2010.33.6313
88
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:.https://doi.org/10.1038/nature11412
89
Carey
LA
. Breast cancer: HER2--a good addiction. Nat Rev Clin Oncol. 2012;9(4):196–7. doi:.https://doi.org/10.1038/nrclinonc.2012.36
90
Clynes
RA
,
Towers
TL
,
Presta
LG
,
Ravetch
JV
. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–6. doi:.https://doi.org/10.1038/74704
91
Molina
MA
,
Codony-Servat
J
,
Albanell
J
,
Rojo
F
,
Arribas
J
,
Baselga
J
. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61(12):4744–9.
92
Yakes
FM
,
Chinratanalab
W
,
Ritter
CA
,
King
W
,
Seelig
S
,
Arteaga
CL
. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62(14):4132–41.
93
Slamon
D
,
Eiermann
W
,
Robert
N
,
Pienkowski
T
,
Martin
M
,
Press
M
, et al.; Breast Cancer International Research Group. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–83. doi:.https://doi.org/10.1056/NEJMoa0910383
94
Piccart-Gebhart
MJ
,
Procter
M
,
Leyland-Jones
B
,
Goldhirsch
A
,
Untch
M
,
Smith
I
, et al.; Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–72. doi:.https://doi.org/10.1056/NEJMoa052306
95
Romond
EH
,
Perez
EA
,
Bryant
J
,
Suman
VJ
,
Geyer
CE, Jr
,
Davidson
NE
, et al.
Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–84. doi:.https://doi.org/10.1056/NEJMoa052122
96
Slamon
DJ
,
Leyland-Jones
B
,
Shak
S
,
Fuchs
H
,
Paton
V
,
Bajamonde
A
, et al.
Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92. doi:.https://doi.org/10.1056/NEJM200103153441101
97
Bang
Y-J
,
Van Cutsem
E
,
Feyereislova
A
,
Chung
HC
,
Shen
L
,
Sawaki
A
, et al.; ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–97. doi:.https://doi.org/10.1016/S0140-6736(10)61121-X
98
Agus
DB
,
Akita
RW
,
Fox
WD
,
Lewis
GD
,
Higgins
B
,
Pisacane
PI
, et al.
Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–37. doi:.https://doi.org/10.1016/S1535-6108(02)00097-1
99
Baselga
J
,
Cortés
J
,
Kim
SB
,
Im
SA
,
Hegg
R
,
Im
YH
, et al.; CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–19. doi:.https://doi.org/10.1056/NEJMoa1113216
100
Gianni
L
,
Pienkowski
T
,
Im
YH
,
Roman
L
,
Tseng
LM
,
Liu
MC
, et al.
Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. doi:.https://doi.org/10.1016/S1470-2045(11)70336-9
101
Richard
S
,
Selle
F
,
Lotz
JP
,
Khalil
A
,
Gligorov
J
,
Soares
DG
. Pertuzumab and trastuzumab: the rationale way to synergy. An Acad Bras Cienc. 2016;88(Suppl 1):565–77. doi:.https://doi.org/10.1590/0001-3765201620150178
102
Howie
LJ
,
Scher
NS
,
Amiri-Kordestani
L
,
Zhang
L
,
King-Kallimanis
BL
,
Choudhry
Y
, et al.
FDA Approval Summary: Pertuzumab for Adjuvant Treatment of HER2-Positive Early Breast Cancer. Clin Cancer Res. 2019;25(10):2949–55. doi:.https://doi.org/10.1158/1078-0432.CCR-18-3003
103
Lewis Phillips
GD
,
Li
G
,
Dugger
DL
,
Crocker
LM
,
Parsons
KL
,
Mai
E
, et al.
Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–90. doi:.https://doi.org/10.1158/0008-5472.CAN-08-1776
104
Junttila
TT
,
Li
G
,
Parsons
K
,
Phillips
GL
,
Sliwkowski
MX
. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–56. doi:.https://doi.org/10.1007/s10549-010-1090-x
105
Verma
S
,
Miles
D
,
Gianni
L
,
Krop
IE
,
Welslau
M
,
Baselga
J
, et al.; EMILIA Study Group. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91. doi:.https://doi.org/10.1056/NEJMoa1209124
106
Geyer
CE
,
Forster
J
,
Lindquist
D
,
Chan
S
,
Romieu
CG
,
Pienkowski
T
, et al.
Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43. doi:.https://doi.org/10.1056/NEJMoa064320
107
Minkovsky
N
,
Berezov
A
. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs. 2008;9(12):1336–46.
108
Burstein
HJ
,
Sun
Y
,
Dirix
LY
,
Jiang
Z
,
Paridaens
R
,
Tan
AR
, et al.
Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–7. doi:.https://doi.org/10.1200/JCO.2009.25.8707
109FDA approves neratinib for extended adjuvant treatment of early stage HER2-positive breast cancer. US Food and Drug Administration. 2018. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neratinib-extended-adjuvant-treatment-early-stage-her2-positive-breast-cancer
110
Sequist
LV
,
Yang
JC
,
Yamamoto
N
,
O’Byrne
K
,
Hirsh
V
,
Mok
T
, et al.
Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–34. doi:.https://doi.org/10.1200/JCO.2012.44.2806
111
Garrett
JT
,
Arteaga
CL
. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther. 2011;11(9):793–800. doi:.https://doi.org/10.4161/cbt.11.9.15045
112
Asić
K
. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit Rev Oncol Hematol. 2016;97:178–96. doi:.https://doi.org/10.1016/j.critrevonc.2015.08.004
113
Scaltriti
M
,
Rojo
F
,
Ocaña
A
,
Anido
J
,
Guzman
M
,
Cortes
J
, et al.
Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–38. doi:.https://doi.org/10.1093/jnci/djk134
114
Nahta
R
. Deciphering the role of insulin-like growth factor-I receptor in trastuzumab resistance. Chemother Res Pract. 2012;2012:648965. doi:.https://doi.org/10.1155/2012/648965
115
Huang
X
,
Gao
L
,
Wang
S
,
McManaman
JL
,
Thor
AD
,
Yang
X
, et al.
Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70(3):1204–14. doi:.https://doi.org/10.1158/0008-5472.CAN-09-3321
116
Shattuck
DL
,
Miller
JK
,
Carraway
KL, 3rd
,
Sweeney
C
. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–7. doi:.https://doi.org/10.1158/0008-5472.CAN-07-5962
117
Yang
Z
,
Guo
L
,
Liu
D
,
Sun
L
,
Chen
H
,
Deng
Q
, et al.
Acquisition of resistance to trastuzumab in gastric cancer cells is associated with activation of IL-6/STAT3/Jagged-1/Notch positive feedback loop. Oncotarget. 2015;6(7):5072–87. doi:.https://doi.org/10.18632/oncotarget.3241
118
Yano
S
,
Yamada
T
,
Takeuchi
S
,
Tachibana
K
,
Minami
Y
,
Yatabe
Y
, et al.
Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6(12):2011–7. doi:.https://doi.org/10.1097/JTO.0b013e31823ab0dd
119
Sequist
LV
,
Waltman
BA
,
Dias-Santagata
D
,
Digumarthy
S
,
Turke
AB
,
Fidias
P
, et al.
Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:.https://doi.org/10.1126/scitranslmed.3002003
120
Anido
J
,
Scaltriti
M
,
Bech Serra
JJ
,
Santiago Josefat
B
,
Todo
FR
,
Baselga
J
, et al.
Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006;25(13):3234–44. doi:.https://doi.org/10.1038/sj.emboj.7601191
121
Scaltriti
M
,
Chandarlapaty
S
,
Prudkin
L
,
Aura
C
,
Jimenez
J
,
Angelini
PD
, et al.
Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010;16(9):2688–95. doi:.https://doi.org/10.1158/1078-0432.CCR-09-3407
122
Mitra
D
,
Brumlik
MJ
,
Okamgba
SU
,
Zhu
Y
,
Duplessis
TT
,
Parvani
JG
, et al.
An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–62. doi:.https://doi.org/10.1158/1535-7163.MCT-09-0295
123
Kwong
KY
,
Hung
MC
. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23(2):62–8. doi:.https://doi.org/10.1002/(SICI)1098-2744(199810)23:2<62::AID-MC2>3.0.CO;2-O
124
Niederst
MJ
,
Engelman
JA
. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6(294):re6. doi:.https://doi.org/10.1126/scisignal.2004652
125
Minuti
G
,
Cappuzzo
F
,
Duchnowska
R
,
Jassem
J
,
Fabi
A
,
O’Brien
T
, et al.
Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107(5):793–9. doi:.https://doi.org/10.1038/bjc.2012.335
126
Ritter
CA
,
Perez-Torres
M
,
Rinehart
C
,
Guix
M
,
Dugger
T
,
Engelman
JA
, et al.
Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13(16):4909–19. doi:.https://doi.org/10.1158/1078-0432.CCR-07-0701
127
Engelman
JA
. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi:.https://doi.org/10.1038/nrc2664
128
Majewski
IJ
,
Nuciforo
P
,
Mittempergher
L
,
Bosma
AJ
,
Eidtmann
H
,
Holmes
E
, et al.
PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol. 2015;33(12):1334–9. doi:.https://doi.org/10.1200/JCO.2014.55.2158
129
Baselga
J
,
Lewis Phillips
GD
,
Verma
S
,
Ro
J
,
Huober
J
,
Guardino
AE
, et al.
Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin Cancer Res. 2016;22(15):3755–63. doi:.https://doi.org/10.1158/1078-0432.CCR-15-2499
130
Faber
AC
,
Corcoran
RB
,
Ebi
H
,
Sequist
LV
,
Waltman
BA
,
Chung
E
, et al.
BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1(4):352–65. doi:.https://doi.org/10.1158/2159-8290.CD-11-0106
131
Tanizaki
J
,
Okamoto
I
,
Fumita
S
,
Okamoto
W
,
Nishio
K
,
Nakagawa
K
. Roles of BIM induction and survivin downregulation in lapatinib-induced apoptosis in breast cancer cells with HER2 amplification. Oncogene. 2011;30(39):4097–106. doi:.https://doi.org/10.1038/onc.2011.111
132
Crawford
A
,
Nahta
R
. Targeting Bcl-2 in Herceptin-Resistant Breast Cancer Cell Lines. Curr Pharmacogenomics Person Med. 2011;9(3):184–90. doi:.https://doi.org/10.2174/187569211796957584
133
Marquez
RT
,
Xu
L
. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2(2):214–21.
134
Luo
S
,
Rubinsztein
DC
. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17(2):268–77. doi:.https://doi.org/10.1038/cdd.2009.121
135
Shimizu
S
,
Kanaseki
T
,
Mizushima
N
,
Mizuta
T
,
Arakawa-Kobayashi
S
,
Thompson
CB
, et al.
Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–8. doi:.https://doi.org/10.1038/ncb1192
136
Goodall
ML
,
Cramer
SD
,
Thorburn
A
. Autophagy complexes cell death by necroptosis. Oncotarget. 2016;7(32):50818–9. doi:.https://doi.org/10.18632/oncotarget.10640
137
Lordick
F
,
Mariette
C
,
Haustermans
K
,
Obermannová
R
,
Arnold
D
; ESMO Guidelines Committee. Oesophageal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(suppl 5):v50–7. doi:.https://doi.org/10.1093/annonc/mdw329
138
Negri
T
,
Tarantino
E
,
Orsenigo
M
,
Reid
JF
,
Gariboldi
M
,
Zambetti
M
, et al.
Chromosome band 17q21 in breast cancer: significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosomes Cancer. 2010;49(10):901–9. doi:.https://doi.org/10.1002/gcc.20798
139
Tang
H
,
Sebti
S
,
Titone
R
,
Zhou
Y
,
Isidoro
C
,
Ross
TS
, et al.
Decreased BECN1 mRNA Expression in Human Breast Cancer is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis. EBioMedicine. 2015;2(3):255–63. doi:.https://doi.org/10.1016/j.ebiom.2015.01.008
140
Lozy
F
,
Cai-McRae
X
,
Teplova
I
,
Price
S
,
Reddy
A
,
Bhanot
G
, et al.
ERBB2 overexpression suppresses stress-induced autophagy and renders ERBB2-induced mammary tumorigenesis independent of monoallelic Becn1 loss. Autophagy. 2014;10(4):662–76. doi:.https://doi.org/10.4161/auto.27867
141
Vega-Rubín-de-Celis
S
,
Zou
Z
,
Fernández
ÁF
,
Ci
B
,
Kim
M
,
Xiao
G
, et al.
Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc Natl Acad Sci USA. 2018;115(16):4176–81. doi:.https://doi.org/10.1073/pnas.1717800115
142
Janku
F
,
McConkey
DJ
,
Hong
DS
,
Kurzrock
R
. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8(9):528–39. doi:.https://doi.org/10.1038/nrclinonc.2011.71
143
Zhou
S
,
Zhao
L
,
Kuang
M
,
Zhang
B
,
Liang
Z
,
Yi
T
, et al.
Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde?
Cancer Lett. 2012;323(2):115–27. doi:.https://doi.org/10.1016/j.canlet.2012.02.017
144
Eng
CH
,
Abraham
RT
. The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene. 2011;30(47):4687–96. doi:.https://doi.org/10.1038/onc.2011.220
145
Cufí
S
,
Vazquez-Martin
A
,
Oliveras-Ferraros
C
,
Corominas-Faja
B
,
Urruticoechea
A
,
Martin-Castillo
B
, et al.
Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget. 2012;3(12):1600–14. doi:.https://doi.org/10.18632/oncotarget.742
146
Rodríguez
CE
,
Reidel
SI
,
Bal de Kier Joffé
ED
,
Jasnis
MA
,
Fiszman
GL
. Autophagy Protects from Trastuzumab-Induced Cytotoxicity in HER2 Overexpressing Breast Tumor Spheroids. PLoS One. 2015;10(9):e0137920. doi:.https://doi.org/10.1371/journal.pone.0137920
147
Vazquez-Martin
A
,
Oliveras-Ferraros
C
,
Menendez
JA
. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One. 2009;4(7):e6251. doi:.https://doi.org/10.1371/journal.pone.0006251
148
Chen
S
,
Zhu
X
,
Qiao
H
,
Ye
M
,
Lai
X
,
Yu
S
, et al.
Protective autophagy promotes the resistance of HER2-positive breast cancer cells to lapatinib. Tumour Biol. 2016;37(2):2321–31. doi:.https://doi.org/10.1007/s13277-015-3800-9
149
Chen
S
,
Li
X
,
Feng
J
,
Chang
Y
,
Wang
Z
,
Wen
A
. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med Hypotheses. 2011;77(2):206–8. doi:.https://doi.org/10.1016/j.mehy.2011.04.013
150
Janser
FA
,
Adams
O
,
Bütler
V
,
Schläfli
AM
,
Dislich
B
,
Seiler
CA
, et al.
Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int J Mol Sci. 2018;19(10):3069. doi:.https://doi.org/10.3390/ijms19103069
151
Ribatti
D
. The chick embryo chorioallantoic membrane as a model for tumor biology. Exp Cell Res. 2014;328(2):314–24. doi:.https://doi.org/10.1016/j.yexcr.2014.06.010
152
Schläfli
AM
,
Berezowska
S
,
Adams
O
,
Langer
R
,
Tschan
MP
. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur J Histochem. 2015;59(2):2481. doi:.https://doi.org/10.4081/ejh.2015.2481
153
Ye
H
,
Chai
X
,
Wang
X
,
Zheng
Q
,
Zheng
D
,
Wu
F
, et al.
Autophagy flux inhibition augments gastric cancer resistance to the anti-human epidermal growth factor receptor 2 antibody trastuzumab. Oncol Lett. 2018;15(4):4143–50. doi:.https://doi.org/10.3892/ol.2018.7891
154
Hardie
DG
,
Ross
FA
,
Hawley
SA
. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. doi:.https://doi.org/10.1038/nrm3311
155
Jones
RG
,
Thompson
CB
. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–48. doi:.https://doi.org/10.1101/gad.1756509
156
Spector
NL
,
Yarden
Y
,
Smith
B
,
Lyass
L
,
Trusk
P
,
Pry
K
, et al.
Activation of AMP-activated protein kinase by human EGF receptor 2/EGF receptor tyrosine kinase inhibitor protects cardiac cells. Proc Natl Acad Sci USA. 2007;104(25):10607–12. doi:.https://doi.org/10.1073/pnas.0701286104
157
Paquette
M
,
El-Houjeiri
L
,
Pause
A
. mTOR Pathways in Cancer and Autophagy. Cancers (Basel). 2018;10(1):18. doi:.https://doi.org/10.3390/cancers10010018
158
Jeon
S-M
,
Hay
N
. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015;38(3):346–57. doi:.https://doi.org/10.1007/s12272-015-0549-z
159
Shi
H
,
Zhang
W
,
Zhi
Q
,
Jiang
M
. Lapatinib resistance in HER2+ cancers: latest findings and new concepts on molecular mechanisms. Tumour Biol. 2016;37(12):15411–31. doi:.https://doi.org/10.1007/s13277-016-5467-2
160
Cui
J
,
Hu
YF
,
Feng
XM
,
Tian
T
,
Guo
YH
,
Ma
JW
, et al.
EGFR inhibitors and autophagy in cancer treatment. Tumour Biol. 2014;35(12):11701–9. doi:.https://doi.org/10.1007/s13277-014-2660-z
161
Rangwala
R
,
Leone
R
,
Chang
YC
,
Fecher
LA
,
Schuchter
LM
,
Kramer
A
, et al.
Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1369–79. doi:.https://doi.org/10.4161/auto.29118
162
Wolpin
BM
,
Rubinson
DA
,
Wang
X
,
Chan
JA
,
Cleary
JM
,
Enzinger
PC
, et al.
Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19(6):637–8. doi:.https://doi.org/10.1634/theoncologist.2014-0086
163
Verbaanderd
C
,
Maes
H
,
Schaaf
MB
,
Sukhatme
VP
,
Pantziarka
P
,
Sukhatme
V
, et al.
Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience. 2017;11:781. doi:.https://doi.org/10.3332/ecancer.2017.781
164
Pellegrini
P
,
Strambi
A
,
Zipoli
C
,
Hägg-Olofsson
M
,
Buoncervello
M
,
Linder
S
, et al.
Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10(4):562–71. doi:.https://doi.org/10.4161/auto.27901
165
Onyenwoke
RU
,
Brenman
JE
. Lysosomal Storage Diseases-Regulating Neurodegeneration. J Exp Neurosci. 2016;9(Suppl 2):81–91.
166
Amaravadi
RK
,
Lippincott-Schwartz
J
,
Yin
XM
,
Weiss
WA
,
Takebe
N
,
Timmer
W
, et al.
Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–66. doi:.https://doi.org/10.1158/1078-0432.CCR-10-2634
167
Liu
J-L
,
Chen
FF
,
Lung
J
,
Lo
CH
,
Lee
FH
,
Lu
YC
, et al.
Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br J Cancer. 2014;111(5):944–54. doi:.https://doi.org/10.1038/bjc.2014.355
168
Martinet
W
,
Roth
L
,
De Meyer
GRY
. Standard Immunohistochemical Assays to Assess Autophagy in Mammalian Tissue. Cells. 2017;6(3):17. doi:.https://doi.org/10.3390/cells6030017