A simple guide to the interpretation of the significance of the association of a disease with a particular HLA allele
DOI: https://doi.org/10.4414/smw.2019.20128
Division of Rheumatology, Columbia University, New York, NY, USA
Summary
This review is a simple guide to the deeper meaning of the association of a disease with a particular HLA allele. We will first review some principles of the function of the adaptive immune system, and some basic notions of autoimmune disease. In this, we will focus on the tripartite unity of the human leukocyte antigen (HLA) molecule, the peptide, and the T cell receptor that recognizes the complex of peptide and HLA molecule, placing emphasis on the function of the T cell receptor. We will also touch on the evolutionary forces shaping our adaptive immune system. Then we will apply this background to some current HLA data concerning the heterogeneity of psoriatic arthritis and the extent to which it is relayed to psoriasis, to illustrate how these principles are used to advance our understanding of the disease.
Introduction
Finding that a disease is associated with a human leukocyte antigen (HLA) allele tells us, firstly, with a few exceptions, that underlying the disease is a T cell driven adaptive immune response to specific peptides. This adaptive immune response is directed either against an infectious pathogen, i.e. non-self, or is autoimmune in nature, and its more specific nature may well have therapeutic implications. Secondly, at the clinical level, because many complex diseases are diagnosed or classified by syndromic clinical criteria, the presence of a strong HLA association provides an important additional diagnostic criterion, and may also reveal unexpected insight into disease heterogeneity. This review is a simple guide to the deeper meaning of the association of a disease with a particular HLA allele. In it we will use the term “HLA” in this guide rather than the equivalent, but more general term “major histocompatibility complex” (MHC).
Each individual’s immune system is nearly unique because the T cells are selected on self-peptides contained in self-HLA molecules
Selection of the individual’s T cell repertoire on self-peptides presented by self-HLA molecules
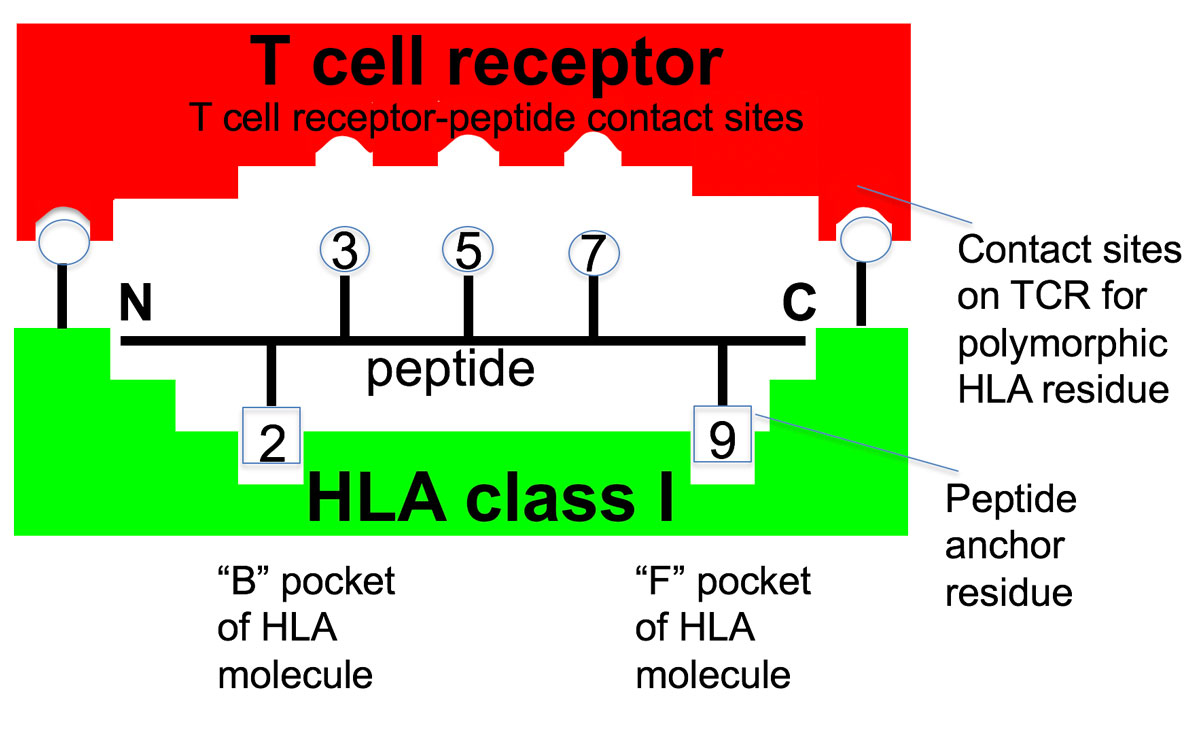
T cell clones comprising the adaptive immune system function by specifically recognizing virtually every pathogen protein by one or more of its constituent peptides. The challenge of the immense diversity of pathogen peptides resulted in a series of biologic processes that distinguish the adaptive immune system from all other systems in the body. The first of these biologic processes is that to generate the trillions of different T cell clones that are specific for different peptides requires random somatic generation through DNA recombinatorial mechanisms of the T cell peptide clonal receptors (TCR) that distinguish each clone. This, because there is insufficient room in the germ line genome to encode each of the billions of different TCRs as individual genes. The second is that since the peptides recognized are generally nine amino acids in length they require a protein structure to bind these different short peptide ligands in a specific manner to ensure their appropriate recognition by the TCR. This is the role of the HLA molecule. Amongst the universe of peptides to which T cells in general could respond, the particular peptides that the T cells of any individual can perceive are specified by the particular binding properties of that person’s HLA molecules. Typically, two amino acid side chains of the peptide fit into pockets in the HLA molecule, tightly anchoring to form a peptide-HLA complex (fig. 1). The peptide-HLA complex is specifically recognized by the T cell TCR through specific contact sites on the peptide and on the HLA molecule. The third, and perhaps the most critical for understanding autoimmune disease, is that since all the T cell clones comprising the repertoire are developed prior to encountering the pathogen, self-peptides are used as the surrogate for the pathogen peptide. These self-peptides bound to self-HLA molecules serve as examples or prototypes of all the possible pathogen peptides that the immune system will encounter and select the individual’s T cell clones that will comprise their T cell repertoire to be used in future immune responses against some of these pathogens. The fourth is that the process of random TCR generation necessitates a selection process that positively selects potentially useful clones for that individual with TCR that effectively recognize self-peptides bound to self-HLA molecules. Additionally, this process includes several mechanisms that eliminate or inactivate clones with overtly strong reactivity to self-peptides presented by self-HLA molecules.
Accordingly, the T cell repertoires of the adaptive immune system that are selected on self-peptides bound to self-HLA molecules are, at the level of the recognition event of the peptide-HLA molecule, incapable of distinguishing between an HLA molecule bearing a self-peptide or a pathogen peptide. The T cell, distinguished by a TCR that is likely incapable of distinguishing between self- or pathogen-peptide, is then guided by a variety of accessory and co-stimulatory molecules to specifically react to pathogen peptides, and in turn to the pathogen. This is accomplished by the microenvironment established in lymphoid tissues in large part determined by dendritic and other antigen-presenting cells that initially encountered the pathogen and its constituent peptides in a peripheral site. These T cells that initiate the immune response were initially selected, perhaps years earlier, in the thymus by self-peptide analogues of the pathogen peptide.
Since all T cell clones are selected on self-peptides, the development of autoimmunity and an autoimmune disease is increasingly being viewed as an untoward consequence of a pro-inflammatory microenvironment that leads to the inappropriate triggering of these previously quiescent T cell clones that were selected on self-peptides. This is in contrast to the earlier concept of a dichotomous state of “autotoxicus” characterized by the intrinsic development of “abnormal” overtly self-reacting clones that mediate autoimmunity, in contrast to the “normal” T cell clones populating healthy individuals that are incapable of recognizing self.
However, in the instance of an autoimmune disease, the specificity of a particular activated T cell clone for self-peptides derived from a given protein defines that protein as the fundamental target tissue of the autoimmune response. The character of the autoimmune response is also dependent on whether the T cell is of CD8 or CD4 lineage, and how it is induced to differentiate down particular pathways towards its effector phenotype, for example TH1. TH17 or in the provision of help for B cells to produce autoantibodies that result in the distinctive clinical phenotypes of immune injury underlying the various autoimmune diseases.
Pathogen drive has diversified the HLA system to render the T cell repertoire of each individual nearly unique in terms of its ability to recognize different peptides
Whereas the selection of the individual’s T cell repertoire on self-peptides contained in HLA molecules is the first element contributing to the nature of each person’s adaptive immune system, the critical second element is the number of different HLA loci and the immense diversity of alternative alleles for each of these loci across the species comprising the HLA gene complex. In principle, each of the HLA molecules, encoded by the different HLA alleles, preferentially binds a different repertoire of peptides and this property of preferential binding underlies the association of a given autoimmune disease with a particular HLA allele.
Different varieties of HLA molecules within the individual, their function, and the genes encoding them
The four million base pair long HLA gene complex is located on the short arm of the 6th chromosome, and is a remarkable gene-rich region of the genome. It contains some 220 genes, many of which function in aspects of the immune response. Figure 2 illustrates the overall genetic organization of the HLA gene complex. The principal illustrated HLA molecules are divided into two main functionally different classes, class I and class II. These are distinguished by whether the HLA molecule presents peptides obtained from the cell cytoplasm to CD8 T cells (Class I HLA molecules) or present peptides obtained from proteins that were engulfed from the environment of the cell and presented to CD4 T cells (Class II HLA molecules). The two different classes of HLA molecules reflect the two fundamentally different immunosurveillance challenges faced by the immune system: Identification of a cell harboring an intracellular pathogen, such as a virus that commandeered the replicative machinery of the cell, versus the presence of an extracellular pathogen such as bacteria that were engulfed by a macrophage.
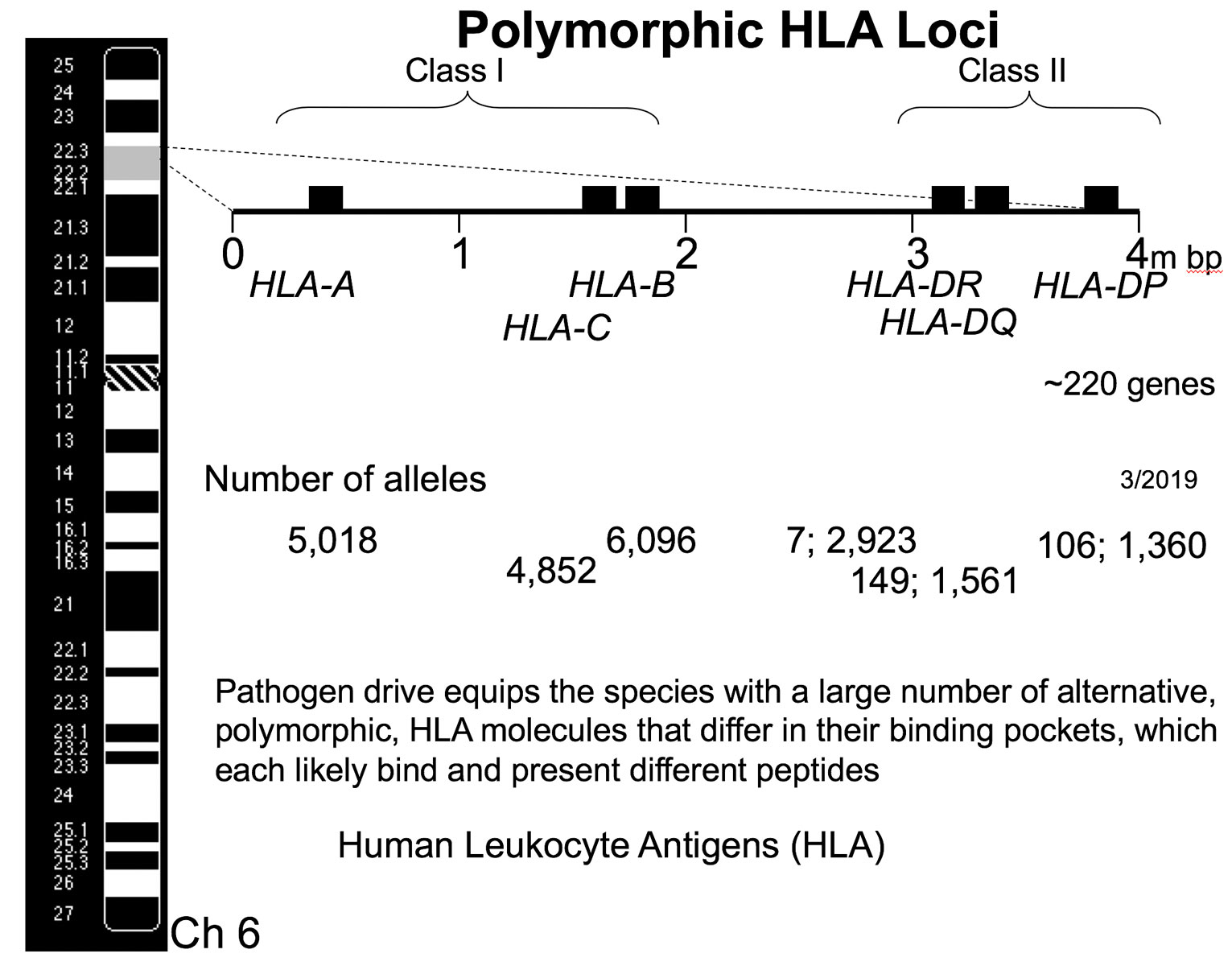
Figure 2 The overall genetic organization of the HLA gene complex, located on the short arm of chromosome 6. This gene complex is approximately 4 million base pairs in length and the principal illustrated HLA loci are divided into two main functionally different classes, class I and class II. These classes reflect the two fundamentally different immunosurveillance challenges faced by the immune system: identification of an infected cell and an extracellular pathogen. There are three class I loci: HLA-A, HLA-B and HLA-C. The molecules encoded by each of these three loci selects their own repertoire of T cells that recognize different peptides in the context of the respective HLA class I molecule. There are three types of class II HLA molecules designated HLA-DR, HLA-DQ, and HLA-DP, however they consist of two chains, alpha (A) and beta (B), and are synthesized by separate loci. Only the expressed polymorphic HLA loci are illustrated in this simplified figure. The large number of alternative gene forms, or alleles, found at each of these loci is illustrated.
Since each different type of HLA molecule selects its own repertoire of T cells according to the binding of a different set of peptides, the resulting diversity in T cell recognition properties constitutes an evolutionary advantage in dealing with a pathogen. To diversify the peptide-presenting structures in the individual evolutionary processes led to the three types of class I HLA molecules designated HLA-A, HLA-B, and HLA-C. These are encoded, respectively, at the three HLA-A, HLA-B and HLA-C loci. This single alpha chain encodes the entire peptide-binding domain of the molecule. Each of these three subtypes of HLA molecules selects their own repertoire of T cells. These T cells recognize different peptides in the context of the respective HLA class I molecule. Because the genes encoding these molecules are inherited from both parents and are co-dominantly expressed, we consequently have six sets of different peptide-presenting class I HLA molecules that have each selected their own unique repertoire of T cells.
Analogously, there are three types of class II HLA molecules designated HLA-DR, HLA-DQ, and HLA-DP, that similarly select distinct T cell repertoires. Although the overall conformation of the class II HLA molecule resembles that of the class I molecule, they consist of two chains, alpha (A) and beta (B), that are synthesized by separate loci in the class II region, and together form the peptide binding domain of the molecule. These chains are encoded by HLA-DRA, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and HLA-DPB1 loci. Only the expressed polymorphic HLA loci are illustrated in the simplified figure 2. Also, not illustrated for reasons of space, are those essentially monomorphic HLA genes that have important immune functions, some additional polymorphic loci such as DRB3, etc., as well as a number of HLA pseudogenes that continue to play a dynamic genomic role. Some of these genetic terms used in figure 2 are defined in table 1. The reader is referred to references such as [1] for additional details about the HLA system.
Table 1 Nomenclature used to describe the genetic polymorphisms of HLA genes.
|
Alleles: the alternative forms of a gene found in different individuals (the gene name is usually italicized) |
|
Allotypes or allomorphs: the different or alternative protein forms encoded by alleles |
Genotype: the collection of genes in an individual, often referring to the two
alleles of a single locus present on the respective maternal and paternal chromosomes |
|
Haplotype: the genes (alleles) on a chromosome or region of the chromosome contributed by one parent. It is usually referred to by naming the principal alleles in the region. It can span across both class I and class II regions. |
|
Gene loci exhibit linkage, a measure of their genetic distance |
|
Linkage disequilibrium: certain alleles of different genes comprising a haplotype are found together significantly more (or less) frequently than expected by chance in certain populations and geographic regions. These stable haplotypes are referred to as conserved haplotypes. |
Different varieties of HLA molecules within the species
Because a given variety of HLA molecule can only bind and present a certain repertoire of peptides limited by the specificity of its pockets to bond certain peptide amino acid side chains, the evolutionary response of the species to the diversity of pathogen proteins and peptides has driven the development of a very large number of alternative gene forms at each locus, termed “alleles”, which encode alternative molecules termed “allomorphs”. These allomorphs are distinguished by major differences in their preferences to bind different varieties of peptides, based on the nature of the amino acids forming the pockets in the HLA molecules that govern their preference for particular anchor amino acids, and consequently for different peptides. The number of alternative alleles for each of the loci illustrated in figure 2 that have been identified to date across all humankind is orders of magnitude greater than the number of alleles at any other site in the genome. This immense genetic diversity reflects the power of the evolutionary drive to make the recognition properties of the adaptive immune system of each individual different from those of another person. This strategy of diversifying the antigen-presenting characteristics of HLA molecules in each individual offers the species a powerful mechanism for surviving a virulent pandemic.
A large proportion of the HLA alleles are present in the population at a frequency significantly greater than could be accounted for by random mutation; that is, they are considered to be genetically polymorphic. The two most common mechanisms affecting the frequency of the various HLA alleles in different populations are:
- Frequency-dependent selection:
- Heterozygote advantage:
Accordingly, there is no single HLA allomorph that is universally and intrinsically superior to any other in all circumstances. It is simply that in the context of a pathogen in a specific environment, one allele is more effective at binding peptides of that pathogen. This confers a survival advantage to that person for that pathogen and specific environment. However, even in this environment there is a contravening principle. Genetic pluralism in a population (the presence of a variety of different HLA alleles), rather than uniformity is evolutionarily advantageous because a new pathogen could mutate to accommodate to the recognition properties encoded by a too-preponderant allomorph. Accordingly, the HLA-B*27 allele, though associated with ankylosing spondylitis, has clearly been positively selected in some geographic regions, likely because of the advantage it confers in the response to certain infectious agents.
This genetic diversity of the various HLA alleles across humankind directly leads to the concept of “immunologic self”, the nearly unique set of self-peptides and self-HLA molecules resulting from the massive genetic diversity of HLA alleles that generates, and in turn is recognized by, the individual’s unique adaptive immune system T cell repertoires. The major selective advantage to the species of this near immunological uniqueness is in dealing with infection, since there is essentially no set of stereotyped recognition structures shared by different individuals in the species. The consequence for the physician treating a given autoimmune disease associated with a particular HLA allele is that the overall autoimmune response is influenced by the T cell repertoire and peptide binding specificities of the other HLA alleles in the individual, rendering the autoimmune response different from that of other patients that share the same disease susceptibility allele.
How different HLA allomorphs confer different functionality on the adaptive immune system
Different HLA alleles confer different functional properties on the adaptive immune system by specifying molecular allomorphs that have different peptide binding abilities. With some exceptions, the molecules encoded by “each” HLA allele generally differ in their amino acid sequence around the anchoring peptide binding pockets, and each HLA allomorphic molecule binds a different set of peptides. This is primarily influenced by the amino acids of the class I HLA molecule that form the major B and F pockets, which confer the main specificity for binding peptides through complementary interactions with the second amino acid of the peptide (P2) and the ninth position (P9) (see fig. 1). In contrast, the amino acid side chains at positions 3, 5, and 7 of the peptide are illustrated as interacting with the TCR. Importantly, about two thirds of the sites of specific interaction between the TCR and the peptide-HLA complex are with the amino acids in the HLA molecule that surround the peptide binding groove. This accounts for the dual specificity of a T cell clone for both peptide specifically bound to the HLA molecule, and the surrounding regions of the polymorphic HLA molecule.
Conserved haplotypes
The last topic in HLA genetics to become familiar with is the concept of conserved “ancestral” haplotypes. A haplotype refers to the unit of haploid chromosomal inheritance containing the genes provided by the mother or the father. One might anticipate that any HLA-A allele would be found on a haplotype with any HLA-B allele, and this is generally true across all humankind, but because crossing over between loci in the HLA region is quite infrequent, different peoples originating from different geographic areas are strongly characterized by particular combinations of certain HLA-A and HLA-B alleles, a phenomenon called linkage disequilibrium. This largely reflects different pathogen selection mechanisms acting in different geographic regions, founder effects, as well as subsequent effects due to migration, etc. For example, among northern Europeans, the HLA-B*08:01 allele is nearly always found on the same 6th chromosome with the HLA-C*07:01 allele, constituting a conserved haplotype. Thus, in populations of northern European ancestry, an association of a disease phenotype with HLA-B*08:01 will also be manifest as an association with HLA-C*07:01. Linkage disequilibrium and the attendant formation of conserved haplotypes complicate the interpretation of HLA associations, but correspondingly are at the heart of the genetic architecture responsible for a given person’s adaptive immune system.
Autoimmune disease
Since the entire T cell system is selected on self-peptides and self-HLA, it is inherently autoreactive and if triggered, these T cells will be responsible for autoimmunity. In effect, autoimmune diseases are just another adaptive immune response, but with two major differences: They are directed to a self-peptide, not a pathogen peptide, and they evolve slowly, and often inexorably, over a number of years. The key to their understanding and therapy is to think of them as immune responses reflecting the individuality of each person’s immune system, and not as conceptually fixed disease entities.
One classification of autoimmune diseases is based on the class of HLA molecule implicated in susceptibility. For example, HLA Class I associated diseases include ankylosing spondylitis, psoriasis and psoriatic arthritis. Each of these diseases are characterized by the presence of clonally expanded CD8 T cells in lesional sites and the absence of autoantibodies, suggesting that the critical event underlying these diseases is presentation of a self-peptide by class I molecules to a CD8 T cell. In contrast, there is a large group of diseases whose occurrence is associated with the inheritance of a particular class II HLA allele, including systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis and Type I diabetes mellitus. These class II HLA allele-associated diseases are characterized by CD4 T cells in lesional sites and usually characterized by autoantibodies, reflecting their differentiation to a stage where they are capable of providing specific help to a B cell.
This close association of susceptibility to different autoimmune diseases with different HLA alleles implies both that the autoimmune response is determined by the person’s HLA allotype reflecting the ability to bind a specific self-peptide, and that the protein/organ containing the peptide becomes the target of the autoimmune response. We emphasize the major role of the HLA alleles in disease susceptibility, but it is critical to understand that it is the distinctive recognition features of the T cell repertoires selected by the individual’s HLA allomorphs, and the specific identification of target antigens by the T cells comprising this repertoire that actually are responsible for the particular disease.
The natural history of each autoimmune disease or this adaptive immune response to self-molecules can be divided into three phases. The first is that of genetic predisposition in which the various genes, including those of the HLA complex, determine the intrinsic potential for developing a particular autoimmune disease by selecting a particular repertoire of T cells. The second phase, asymptomatic autoimmunity results from the activation of T cell clones to respond to self-peptides, primarily through signals provided by co-stimulatory molecules. This phase is characterized by an enhanced level of recognition and T cell response to self-antigens. In the instance where the peptide driving the autoimmune response is presented by class II HLA molecules and correspondingly is recognized by T cells of the CD4 lineage, the inflammatory milieu directs some of the CD4 T cells to differentiate into follicular helper T cells that provide help to B cells. This may result in the production of autoantibodies to antigens that are clinically tested, accounting for the sometimes clinically perplexing problem of, for example an asymptomatic individual positive for antinuclear antibodies. Accordingly, the asymptomatic person with autoantibodies to nuclear antigens, citrullinated peptides, or thyroglobulin, etc. would be considered instances of an individual in the phase of asymptomatic autoimmunity. In a subset of people with asymptomatic autoimmunity this phase progresses in intensity with further T cell clonal expansion, spreading, and in some instances further B cell help, until the autoimmune response achieves a state where it begins to mediate tissue injury, denoting entrance into the third phase of a recognizable autoimmune disease.
The psoriasis phenotype: psoriasis and psoriatic arthritis
Psoriasis is a prototypic autoimmune disease that is among the most common of the autoimmune diseases. Figure 3 summarizes the main conclusion of extensive studies on the genetic basis of psoriasis, showing disease susceptibility maps into the HLA-C locus, and specifically with the HLA-C*06:02 allele [2–4]. Three common psoriasis susceptibility ancestral haplotypes are illustrated in which the C*06:02 allele is in linkage disequilibrium with either of three HLA-B locus alleles: B*13:01, B*37:01 or B*57:01. Several additional neighboring gene loci in the gene rich HLA region are also depicted that were earlier candidates for psoriasis susceptibility.

Figure 3 Psoriasis HLA genetics, showing a more detailed map of the class I region around the HLA-B and HLA-C loci, in the region of Psors 1, the major candidate region for psoriasis susceptibility identified by earlier linkage studies. More recent studies primarily map susceptibility into the HLA-C locus, eliminating some of the depicted candidate loci. More specifically, susceptibility has been shown to be associated with the HLA-C*06:02 allele, which is present at a frequency ~60% in most series ascertained on psoriasis. The frequency increases to ≥70% if late onset disease is eliminated. Three main conserved PsC susceptibility haplotypes containing C*06:02 are indicated.
Based on prospective studies largely carried out in psoriasis cohorts, many textbooks describe psoriatic arthritis as developing in 15–20% of psoriasis patients after the passage of 10–15 years. With this as background, we set out to ask and more specifically, is the genotype of psoriatic arthritis identical to that of psoriasis? Does the phenotypic frequency of the HLA- C*06:02 allele in psoriatic arthritis parallel that found in psoriasis? We used DNA-based allele typing. The reader is referred to this [5] and other cited primary references for greater methodologic details. Briefly, the approach was to compare the HLA genotypes of two cohorts in the homogeneous Irish population with psoriasis that were meticulously characterized by Oliver FitzGerald and David Kane; University College Dublin using CASPAR criteria for inclusion. The psoriasis cohort (n = 214) was without features of arthritis, spondylitis or enthesitis and presented to a dermatologic clinic, whereas the psoriatic arthritis cohort (n = 359) presented to a rheumatology clinic. Of the psoriasis cohort, 57.5% had the HLA-C*06:02 allele compared with 19.3% in a control population (odds ratio [OR] 6.61, p <0.0001), a value consistent with that reported in the dermatologic literature for the northern European population. This is depicted in figure 4. The frequency of HLA-C*06:02 in the psoriatic arthritis cohort was only 27.9%, significantly lower than that in the psoriasis cohort (OR 0.30, p <0.0001), but still significantly increased over that of the control population (OR 1.9, p <0.0001). This confirms that the HLA-C*06:02 allele is associated with psoriatic arthritis. There were two main implications of this study: The psoriasis phenotype is not genetically homogeneous (p = 9.94 × 10-1) [2], and psoriatic arthritis itself is heterogeneous, with only a smaller subset genetically closely related to those with cutaneous psoriasis [5].
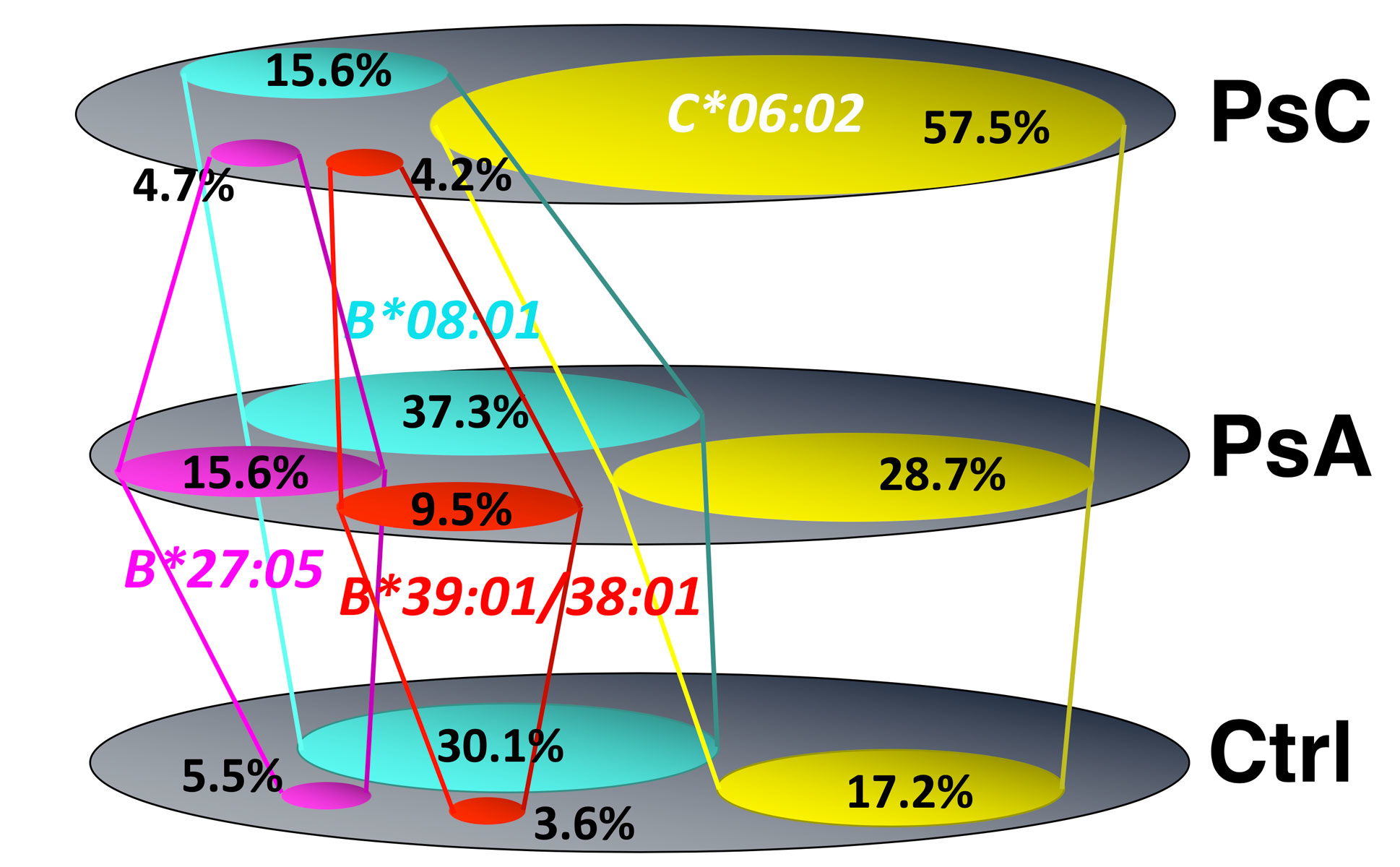
Figure 4 A comparison of the frequencies of the principal HLA-B and HLA-C loci alleles positively associated with susceptibility to psoriatic arthritis depicted as Venn diagrams, emphasizing the difference between allele frequencies in healthy controls and psoriasis. It shows the psoriasis phenotype is not genetically homogeneous and that psoriatic arthritis (PsA) itself is genetically heterogeneous, with only a smaller subset genetically closely related to those with cutaneous psoriasis (PsC).
The next question was, which HLA alleles are present in the balance of psoriatic arthritis patients who have not inherited C*06:02? These results are sketched in figure 4 and showed HLA-B*27:05:02, and HLA-B*39:01:01 and B*38:01:01 are indeed susceptibility alleles for psoriatic arthritis, as has been previously described, but are not high-risk alleles for psoriasis in the absence of musculoskeletal involvement. Moreover, HLA-B*08:01 contributes to psoriatic arthritis susceptibility, but interestingly, unlike HLA-B*27:05:02, B*39:01:01 and B*38:01:01 or C*06:02, the B*08:01 allele appears relatively “protective” for the development of psoriasis in the absence of arthritis. This result provides strong support for the conclusion that psoriatic arthritis is genetically heterogeneous and analogous results have been obtained by other groups [6].
This finding that psoriasis is genetically heterogeneous with the musculoskeletal-cutaneous phenotype of psoriatic arthritis defined by several HLA-B locus alleles and C*06:02 suggests that both HLA-B locus alleles and HLA-C locus alleles play a role in driving the psoriatic arthritis association, but with some differences in the resulting clinical phenotype, as will be discussed in a little more detail subsequently. The phenotype conferred by HLA-C*06:02 was identified as one of highly penetrant severe skin disease and lower penetrance musculoskeletal disease, whereas HLA-B*27:05:02, HLA-B*39:01:01 and HLA-B*08:01 confer equivalent penetrance for milder skin and more severe musculoskeletal disease. Several HLA-B alleles including B*44:02, B*44:03 and B*40:01 were found at significantly decreased frequency in the psoriatic arthritis cohort, suggesting they appear “protective” for the development of psoriatic arthritis [5]. It is important to emphasize that this study is based on a northern European population and that given the marked variation of these alleles in different geographic regions across the globe, different associations will likely be present in these various populations.
A potential molecular basis for susceptibility
A potential molecular basis to psoriasis and psoriatic arthritis susceptibility is suggested by comparing the architecture of the peptide-binding pockets and their peptide-binding preference for the various HLA allomorphs encoded by the alleles positively and negatively associated with psoriatic arthritis susceptibility. Figure 5 shows the single amino acid code for a portion of the amino acid residues of the B pocket of the HLA molecules and the principal patterns of peptide binding by HLA-B and HLA-C molecules associated with psoriatic arthritis susceptibility. The preferential peptide-binding specificity of the allomorphs for binding particular amino acid side chains at the second position of the peptide, P2, is shown. At positions 45, 67 and 74 in the HLA-B alpha chain forming critical parts of the B pocket, HLA-B*27:05:02, HLA-B*39:01:01 and B*38:01:01 are characterized by the same amino acid residues, containing negatively charged glutamic acid (at position 45) and aspartic acid (at position 74), and these allomorphs have been defined to preferentially bind peptides containing positively charged arginine or histidine at P2 [7, 8]. Specifically, the negatively charged amino acids in the HLA molecule pocket create a charge environment that non-covalently interacts with the opposite, positively charged amino acids at P2 of peptides via the formation of salt bridges, conferring high affinity binding to these allomorphs of peptides that have an arginine at this position. Conversely, the B*44 allomorphs associated with significantly decreased susceptibility to psoriatic arthritis are characterized with positively charged lysine at position 45, and this confers the preference to bind peptides with negatively charged amino acids at P2 of the peptide, glutamic and aspartic acids [5]. This pattern suggests that the inheritance of alleles encoding allomorphs with the ability to bind a self-peptide with arginine at P2 is a critical step in defining the predisposition to develop the molecular basis of the susceptibility to develop psoriatic arthritis, and that T cell repertoires developing in individuals on HLA-B allomorphs that preferentially bind peptides with negatively charged amino acid residues at P2 significantly decrease the likelihood of developing psoriatic arthritis.
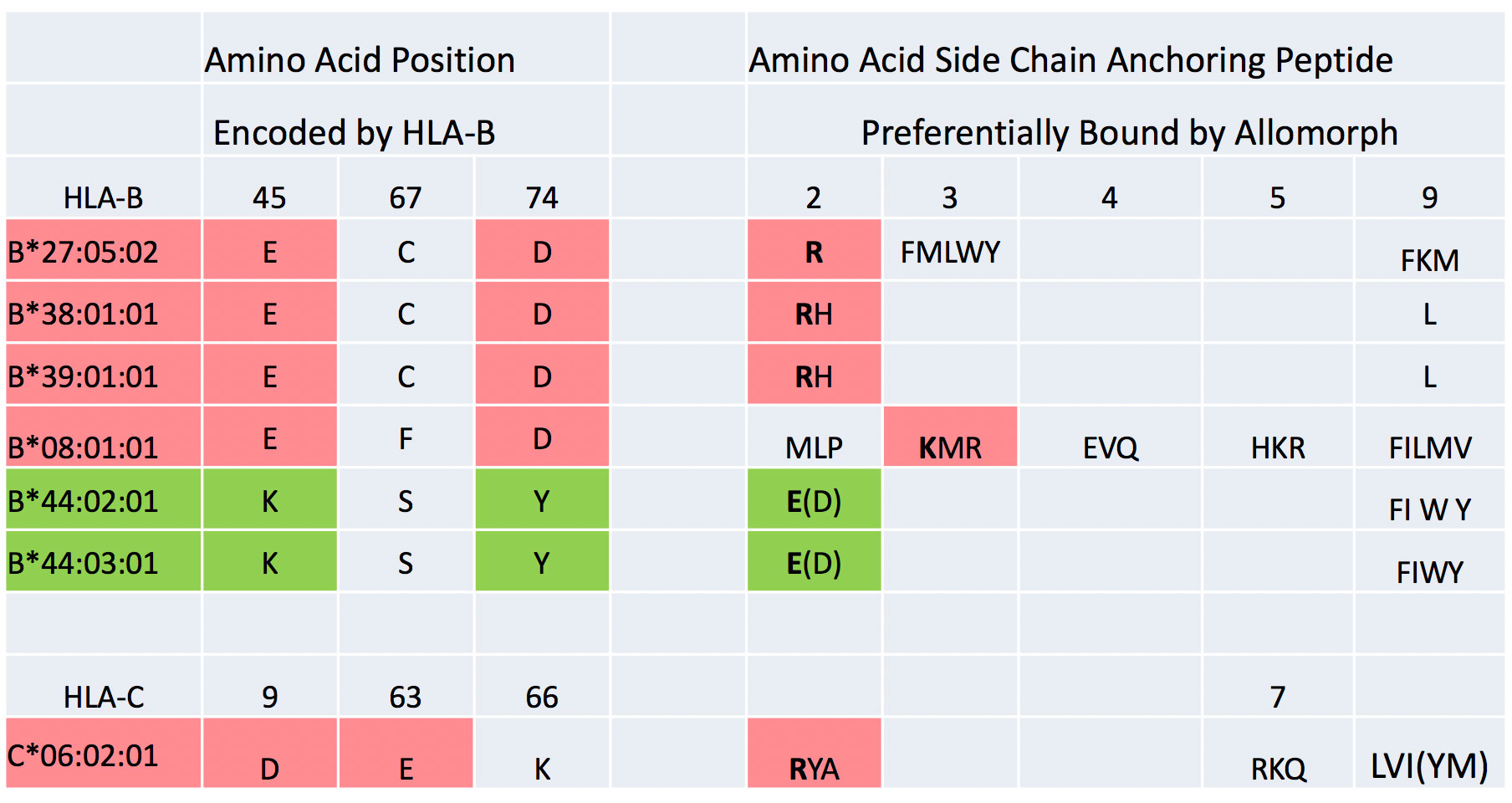
Figure 5 Illustrates the single amino acid code for a portion of the amino acid residues of the B pocket of the HLA molecules and the principal patterns of peptide binding by the HLA-B and HLA-C allomorphic molecules associated with significantly increased and decreased psoriatic arthritis susceptibility. The preferential peptide binding specificity of the allomorphs for binding particular amino acid side chains at the second position of the peptide P2 is shown. At positions 45, 67 and 74 in the HLA-B alpha chain that forming critical parts of the B pocket, HLA-B*27:05:02, HLA-B*39:01:01 and B*38:01:01 have the same negatively charged amino acid residues, and these allomorphs have been defined to preferentially bind peptides containing positively charged arginine or histidine at P2. Conversely, the B*44 allomorphs associated with significantly decreased susceptibility to psoriatic arthritis are characterized with positively charged lysine at position 45, conferring the preference to bind peptides with negatively charged amino acids at P2. The somewhat related properties of B*08:01:01 and C*06:02:01 are shown.
The HLA-C*06:02 molecule differs slightly in features from the HLA-B molecule. It is also characterized by a negatively charged “B” pocket, in this case by an aspartic acid at position 9 in the alpha chain and by crystallography this residue forms a salt bridge with positively charged arginine at P2, the preferred anchoring side chain for binding peptides [9]. In this respect, there are intriguing analogies with the P2 binding properties of HLA-B*27:05:02, HLA-B*39:01:01 and B*38:01:01. However, HLA-C*06:02 differs from the previously described allomorphs in that it also anchors peptides by arginine, lysine or glutamine at P7. Future work will have to determine whether these intriguing analogies reflect binding of the similar peptides critical to the development of the autoimmune response underlying psoriatic arthritis. Similarly, HLA-B*08:01:01 molecules somewhat resemble HLA-B*27:05:02, HLA-B*39:01:01 and B*38:01:01 in having negatively charged residues at positions 45 and 74 and this is characterized by a preference for peptides containing lysine, methionine or arginine, but the preferential anchor site is P3. Here also, current knowledge is not sufficient to interpret precisely whether these clues suggest preferential binding of a particular specific peptide. From the dominant nature of the association of each HLA allele with susceptibility, we infer that the binding property of the self-peptides by the allomorphs during thymic development of the repertoire results in a dominant recognition property of these particular repertoires, specified by each susceptibility allomorph of particular of self-peptides.
The relation of psoriatic arthritis susceptibility genotypes to clinical subphenotypes.
Different genetic susceptibility genes imply different disease mechanisms, and perhaps different clinical courses and therapeutic responses to particular biologics. Accordingly, we asked, is the phenotypic heterogeneity among patients with psoriatic arthritis related to their inheritance of different HLA alleles?? The first clue of a genotype-phenotype relationship in psoriatic arthritis was the finding that the onset of arthritis developed closer to the appearance of psoriasis in the HLA-B*27:05:02 or B*39:01:01 subset than in the HLA-HLA-C*06:02 subset of psoriatic arthritis cases [5]. The mean number of years between the onset of psoriasis and diagnosis of psoriatic arthritis was 11.3 years among HLA-C*06:02 individuals and 3.8 years in those who were HLA-B*27:05:02 or B*39:01:01. This suggested the time interval between psoriasis and diagnosis of psoriatic arthritis is a genetically determined quantitative trait. The phenotype conferred by HLA-C*06:02 can accordingly be further defined as one of highly penetrant severe skin disease and lower penetrance and considerably delayed onset of musculoskeletal disease, while that conferred by HLA-B*27:05:02 or B*39:01:01 can defined to indicate the more synchronous appearance of skin and musculoskeletal disease.
Since both the clinical phenotype and the HLA genotype were heterogeneous, one could ask whether a particular phenotype was associated with a specific HLA allele. Table 2 summarizes the reported univariate associations between certain clinical subphenotypes and psoriatic arthritis susceptibility alleles, and shows the differing genetic architecture underlying the development of clinical subphenotypes of psoriatic arthritis [10, 11]. One of the clinical features of psoriatic arthritis is the development of sacroiliitis, which more commonly is asymmetric, in contrast to that of ankylosing spondylitis, which is almost invariably bilateral as determined by conventional X-ray studies. In psoriatic arthritis, as in the instance of ankylosing spondylitis, symmetric sacroiliitis was significantly associated only with the presence of HLA-B*27:05:02 (OR 4.16, p <0.001), whereas the presence of C*06:02 was borderline negatively associated with this form of sacroiliitis. In striking contrast, asymmetric sacroiliitis was significantly increased in frequency only among those who inherited HLA-B*08:01, with no positive association with HLA-B*27:05:02 or with C*06:02.
Table 2 Differing genetic architecture underlying development of clinical subtypes of psoriatic arthritis.
| . |
Asymmetric sacroiliitis vs no sacroiliitis
|
Symmetric sacroiliitis vs no sacroiliitis
|
Enthesitis
|
Dactylitis
|
Nail disease
|
|
OR
|
p-value
|
OR
|
p-value
|
OR
|
p-value
|
OR
|
p-value
|
OR
|
p-value
|
|
HLA-B*08:01:0
|
1.97
|
0.008
|
0.49 |
0.07 |
0.81 |
33 |
1.57
|
0.019
|
2 0
|
003
|
|
HLA-B*27:05:02
|
1.24 |
0.57 |
4.16
|
<0.001
|
3.65
|
<0.001
|
1.97
|
0.014
|
0.95 |
0.86 |
|
HLA-B*38:01:01
|
2.87 |
0.065 |
1.92 |
0.42 |
1.25 |
0.56 |
1.08 |
0.87 |
0.75 |
0.6 |
|
HLA-B*39:01:0
|
0.4 |
0.22 |
1.06 |
0.93 |
0.97 |
0.95 |
0.87 |
0.72 |
0.64 |
0.27 |
|
HLA-C*06:02
|
0.54
|
0.36
|
0.47
|
0.053
|
1.09 |
0.68 |
0.67
|
0.04
|
0.52
|
0.002
|
Enthesitis, determined by physical examination, was significantly associated with HLA-B*27:05:02, whereas dactylitis was significantly positively associated with either HLA-B*27:05:02 or B*08:01, and nail disease was only positively associated with, HLA-B*08:01. Again, the presence of C*06:02 was negatively associated with nail disease and dactylitis [12].
The implication of this relation between genotype and subphenotype is that a given subphenotype, such as enthesitis, reflects a specific autoimmune attack on the molecules comprising the enthesis by T cells that were selected on HLA-B*27:05:02 molecules in the thymus, and among this repertoire of T cells might well have been some that were positively selected on peptides from self-molecules identical to those found in the enthesis. In this sense, the intriguing difference between symmetric and asymmetric sacroiliitis in terms of a completely different HLA association accounts for the overall difference in the pattern of axial involvement between psoriatic arthritis and ankylosing spondylitis, and suggests that although at a given specific site the involvement appears radiographically equivalent, that it is the result of a different autoimmune response, and differs in the molecules targeted by each immune response. A further implication of this concept would be that the spine involvement in an HLA-B*27:05:02 psoriatic arthritis patient would be more analogous to that of ankylosing spondylitis in terms of natural history and therapeutic response than that of a HLA-B*08:01 psoriatic arthritis patient. Since the CASPAR classification criteria to ascertain the diagnosis of psoriatic arthritis relies on the presence of clinical subphenotypes such as sacroiliitis, enthesitis, dactylitis and nail disease that are dependent on the individual’s HLA type (see table 2), the determination of the development of psoriatic arthritis among those with psoriasis, who are predominantly HLA-C*06:02, might by falsely underestimated because this population is less likely to exhibit these clinical subphenotypes [11].
It is likely that the HLA genetics will determine the clinical response of psoriatic arthritis to biologic agents and other newer therapeutic agents exhibiting heightened specificity for a particular inflammatory pathway. This has clearly been found to be the case in psoriasis where HLA-C*06:02 -positive patients had a significantly superior response to the interleukin-12–interleukin-23 blocking biologic ustekinumab, whereas HLA-C*06:02 -negative patients were significantly more likely to respond to the tumor necrosis factor inhibitor adalimumab, than to ustekinumab [13, 14].
Genotypic interactions among susceptibility haplotypes determine psoriatic arthritis severity
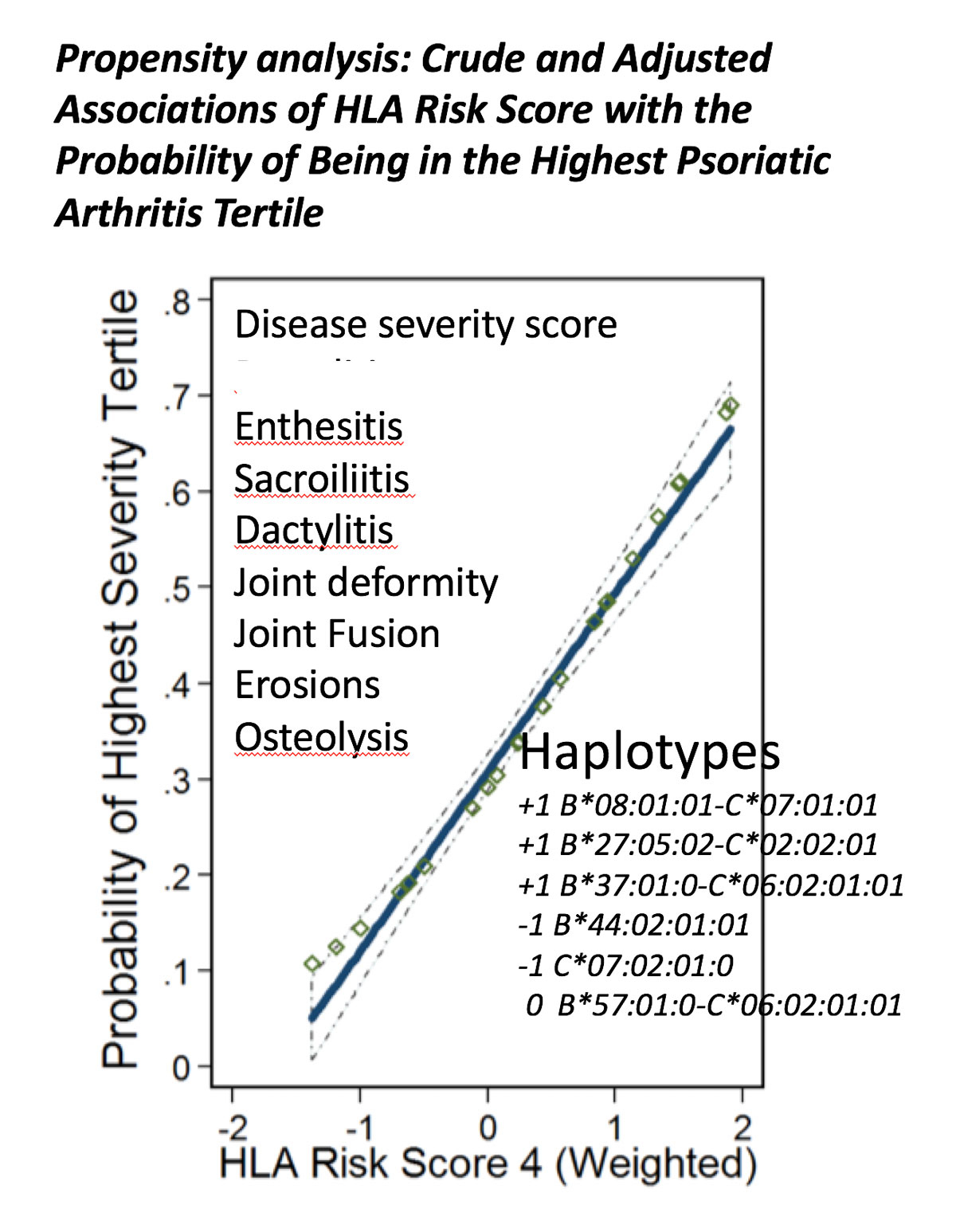
Since we inherit one haploid chromosome from each of our parents, the question arises of how to determine the contribution of both maternal and paternal alleles on the resultant phenotype and severity of clinical disease. One initial simplified approach to answer this question was to use propensity analysis to assess the positive and negative contribution of risk alleles on both chromosomes to the resulting severity of the psoriatic arthritis phenotype. A disease severity score was developed based on the presence of enthesitis, sacroiliitis, dactylitis, joint deformity, joint fusion, erosions or osteolysis. The patients were then ranked into severity tertiles, enabling address of the question: what combination of HLA haplotypes or alleles resulted in the greatest probability of being in the highest severity tertile? An HLA weighted risk score was developed that gave 1 point for each instance of the presence of the HLA-B*08:01-C*07:01 ancestral haplotype, the HLA-B*27:05:02-C*02:02 ancestral haplotype and the HLA-B*37:01-C*06:02 ancestral haplotype, which were separately associated with one or more psoriatic arthritis subphenotypes, 0 for the presence of the HLA-B*57:01-C*06:02 ancestral haplotype, which was neither positively or negatively associated with a phenotype, and −1 for the presence of B*44:02:01 or C*07:02 alleles, which were negatively associated with the features in the disease severity score. Thus, an individual could have an HLA allele risk score ranging from −2 to +2 and a person who was homozygous for B*27:05:02-C*02:02 receives a score of 2. The result for all the patients in the cohort is illustrated in figure 6 and shows the contribution of six positive or negative risk alleles/haplotypes on both chromosomes to the probability of having the greatest psoriatic arthritis severity, which is best explained by this additive model in which the positive and negative risk alleles independently contribute to the composite phenotype of more severe disease [10]. Of course, more detailed analyses using larger cohorts with additional alleles are required to better delineate the relationship between genotype and phenotype.
This suggests that in an individual with psoriatic arthritis, each T cell repertoire resulting from selection on a particular HLA molecule and associated with a psoriatic arthritis subphenotype functions in parallel in the autoimmune response to different self-molecules. However, the repertoires do not function independently of one another. HLA alleles such as B*44:02:01 that are associated with a decrease risk of developing psoriatic arthritis if present in an individual with a risk allele such as B*27:05:02, diminish the effect of the risk allele on conferring a severe disease phenotype. Conversely, other alleles such as B*08:01, enhance the effect of a risk allele. Accordingly, we conceptualize psoriatic arthritis as a complex autoimmune-mediated disease with a variety of subphenotypes that are under the control of different HLA alleles, reflecting the presence immune responses directed to different target molecules, but acting in concert to result in a spectrum of severity.
Conclusion
Pondering the answer to the question, “how did my psoriatic arthritis come to be?” takes us on an exploration of the deeper meaning of the association of a disease with a particular HLA allele. The apparent seamless function of the adaptive immune response belies its astonishing genetic intricacy, which itself is an evolutionary response to pathogen challenge. The allelic complexity of the HLA system results in the near uniqueness of each individual’s immune system because the T cells are selected on self-peptides contained in self-HLA molecules. However, the impressive survival advantage of the adaptive immune system comes at the cost of autoimmunity and autoimmune disease because of the self-selected character of the adaptive immune system’s T cell repertoires. In some circumstances this results in the persistent and expanding self-directed immune response that underlies the particular clinical phenotype of the patient’s disease, and represents a therapeutic challenge to down-modulate. One can anticipate that in the next decade the knowledge hinted at in this simple guide will become much more clearly etched and therapeutically relevant.
References
1Monos DS, Winchester R. The Major Histocompatibility Complex. In: Rich RR, ed. Clinical Immunology: Principles and Practice. 5 ed. New York, NY: Elsevier; 2018.
2
Nair
RP
,
Stuart
PE
,
Nistor
I
,
Hiremagalore
R
,
Chia
NVC
,
Jenisch
S
, et al.
Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78(5):827–51. doi:.https://doi.org/10.1086/503821
3
Okada
Y
,
Han
B
,
Tsoi
LC
,
Stuart
PE
,
Ellinghaus
E
,
Tejasvi
T
, et al.
Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes. Am J Hum Genet. 2014;95(2):162–72. doi:.https://doi.org/10.1016/j.ajhg.2014.07.002
4
Tsoi
LC
,
Stuart
PE
,
Tian
C
,
Gudjonsson
JE
,
Das
S
,
Zawistowski
M
, et al.
Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat Commun. 2017;8(1):15382. doi:.https://doi.org/10.1038/ncomms15382
5
Winchester
R
,
Minevich
G
,
Steshenko
V
,
Kirby
B
,
Kane
D
,
Greenberg
DA
, et al.
HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum. 2012;64(4):1134–44. doi:.https://doi.org/10.1002/art.33415
6
Eder
L
,
Chandran
V
,
Pellet
F
,
Shanmugarajah
S
,
Rosen
CF
,
Bull
SB
, et al.
Human leucocyte antigen risk alleles for psoriatic arthritis among patients with psoriasis. Ann Rheum Dis. 2012;71(1):50–5. doi:.https://doi.org/10.1136/ard.2011.155044
7
Vita
R
,
Overton
JA
,
Greenbaum
JA
,
Ponomarenko
J
,
Clark
JD
,
Cantrell
JR
, et al.
The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43(Database issue):D405–12. doi:.https://doi.org/10.1093/nar/gku938
8
Schuler
MM
,
Nastke
MD
,
Stevanović
S
. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol. 2007;409:75–93. doi:.https://doi.org/10.1007/978-1-60327-118-9_5
9
Mobbs
JI
,
Illing
PT
,
Dudek
NL
,
Brooks
AG
,
Baker
DG
,
Purcell
AW
, et al.
The molecular basis for peptide repertoire selection in the human leucocyte antigen (HLA) C*06:02 molecule. J Biol Chem. 2017;292(42):17203–15. doi:.https://doi.org/10.1074/jbc.M117.806976
10
Haroon
M
,
Winchester
R
,
Giles
JT
,
Heffernan
E
,
FitzGerald
O
. Certain class I HLA alleles and haplotypes implicated in susceptibility play a role in determining specific features of the psoriatic arthritis phenotype. Ann Rheum Dis. 2016;75(1):155–62. doi:.https://doi.org/10.1136/annrheumdis-2014-205461
11
FitzGerald
O
,
Haroon
M
,
Giles
JT
,
Winchester
R
. Concepts of pathogenesis in psoriatic arthritis: genotype determines clinical phenotype. Arthritis Res Ther. 2015;17(1):115–25. doi:.https://doi.org/10.1186/s13075-015-0640-3
12
Winchester
R
,
Giles
J
,
Jadon
D
,
Haroon
M
,
McHugh
N
,
FitzGerald
O
. Implications of the diversity of class I HLA associations in psoriatic arthritis. Clin Immunol. 2016;172:29–33. doi:.https://doi.org/10.1016/j.clim.2016.07.019
13
Talamonti
M
,
Botti
E
,
Galluzzo
M
,
Teoli
M
,
Spallone
G
,
Bavetta
M
, et al.
Pharmacogenetics of psoriasis: HLA-Cw6 but not LCE3B/3C deletion nor TNFAIP3 polymorphism predisposes to clinical response to interleukin 12/23 blocker ustekinumab. Br J Dermatol. 2013;169(2):458–63. doi:.https://doi.org/10.1111/bjd.12331
14
Dand
N
,
Duckworth
M
,
Baudry
D
, et al.
HLA-C*06:02 genotype is a predictive biomarker of biologic treatment response in psoriasis. J Allergy Clin Immunol. 2019;143(6):2120–30.