Prevalence of genetic susceptibility for breast and ovarian cancer in a non-cancer related study population: secondary germline findings from a Swiss single centre cohort
DOI: https://doi.org/10.4414/smw.2019.20092
Dennis
Kraemera, Silvia
Azzarello-Burria, Katharina
Steindla, Paranchai
Boonsawata, Markus
Zweiera, Konstantin J.
Dedesb, Pascal
Joseta, Daniel
Finkb, Anita
Raucha
aInstitute of Medical Genetics (IMG), University of Zurich, Schlieren-Zurich, Switzerland
bDepartment of Gynaecology, University Hospital Zurich, Switzerland
Summary
BACKGROUND
Since the advent of high-throughput sequencing technologies, organised germline screening, independent of the personal and family cancer history, has been frequently proposed. Since ethnic and geographic populations significantly differ in their mutation spectra and prevalence, one critical prerequisite would be the knowledge of the expected carrier frequencies.
OBJECTIVE
For the first time, in a retrospective non-cancer related cohort from a single Swiss genetic centre, we systematically assessed the prevalence of secondary findings in 19 genes (BRCA1/2 plus 17 non-BRCA genes) previously designated by the US National Comprehensive Cancer Network (NCCN) for hereditary breast and ovarian cancer (HBOC) germline testing.
DESIGN
A total of 400 individuals without a cancer diagnosis undergoing whole-exome sequencing (WES) analysis for neurodevelopmental disorders (NDDs) from 2015 to 2017 at IMG Zurich were included after quality assessment. Among these, 180 were unaffected parental couples, 27 unaffected parental singles and 13 NDD index patients (mean age 43 years). The majority of the cohort was of Caucasian ethnicity (n = 336, 84.0%) and of Northwest European ancestry (n = 202, 50.5%), for 70 of whom (42.5%) an autochthonous Swiss descent was assumed. For WES filtering of rare, potentially actionable secondary variants in HBOC genes, an overall minor allele frequency (MAF) below 0.65% was used as cut-off. Each rare variant was manually evaluated according to the recommended ACGM-AMP standards, with some adaptations including “hypomorphic” as an additional distinct pathogenicity class.
RESULTS
Overall, 526 rare secondary variants (339 different variants) were encountered, with the BRCA1/2 genes accounting for 27.2% of the total variant yield. If stratified for variant pathogenicity, for BRCA1/2, three pathogenic variants were found in three females of Italian ancestry (carrier frequency of 0.8%). In the non-BRCA genes, five carriers of (likely) pathogenic variants (1.3%) were identified, with two Swiss individuals harbouring the same CHEK2 Arg160Gly variant known to be recurrent among Caucasians. Hence, the overall carrier rate added up to 2.0%. Additionally, seven various hypomorphic HBOC predisposing alleles were detected in 22 individuals (5.5%).
CONCLUSION
We provide the first evidence of a high prevalence of HBOC-related cancer susceptibility in the heterogeneous Swiss general population and relevant subpopulations, particularly in individuals of Italian descent. These pioneering data may substantiate population-based HBOC screening in Switzerland.
Introduction
According to the Swiss Cancer Report 2015 [1], an average of more than 6200 females are diagnosed with breast or ovarian cancer in Switzerland per year, with breast cancer being the leading cancer in women and accounting for a third of all new cancer cases. It is assumed that approximately 5–10% of breast cancers and 10–15% of epithelial ovarian cancers arise in women with an underlying monogenic hereditary predisposition, frequently caused by deleterious germline variants in the cancer susceptibility genes BRCA1 and BRCA2 (designated as hereditary breast and ovarian cancer [HBOC] syndrome) [2]. Assuming an average prevalence of 5%, ~2000–4000 HBOC index cases are estimated to be currently living in Switzerland, according to the CASCADE study proposal for cascade cancer predisposition screening in Switzerland [3].
However, about one in five Swiss breast cancer patients are diagnosed when they are younger than 50 years [4, 5], which may indicate genetic susceptibility and, hence, may reflect a significant underdiagnosis of underlying germline alterations. Likewise, only an estimated 20–25% of familial breast cancer cases are explained by BRCA1/2 pathogenic variants and >60% of the hereditary predisposition remains unexplained [6, 7]. Nevertheless, in current practice, genetic testing is usually limited to the evaluation of patients meeting strict criteria for an individual or family cancer history, as specified by various (inter-)national guidelines [8–11]. These restrictions of the testing approach, which are validated for the minimum mutation detection rate of ≥10% as a consensus [11], have been more and more questioned by recent studies using gene-panel based diagnostic strategies [12–14]. To this end, were the US National Comprehensive Cancer Network (NCCN) HBOC susceptibility screening guideline endorsed 17 non-BRCA susceptibility genes, which were found to confer a high to moderate breast and/or ovarian cancer risk, partially within the context of clinically broader hereditary cancer syndromes (HCSs), such as Li-Fraumeni, Lynch and PTEN hamartoma tumour syndromes [8].
Given that HBOC patients are at high risk of cancers at a younger age and of high grade, routine screening programmes lack sufficient sensitivity for the detection of HBOC-associated lesions at early stages. Magnetic resonance imaging screening from an early age and risk-reducing measures such as chemo-prevention and prophylactic surgery, however, have been shown to lead to a substantial decrease of HBOC-associated morbidity and mortality [15, 16]. These preventive strategies, as well as options for tailored therapies such as poly-ADP-ribose polymerase inhibitors (PARPi) pharmacotherapy for BRCA1/2-associated malignancies [17], mean that diagnostic assessment for HCSs plays a crucial role in oncological care.
Therefore, since the advent of the high-throughput sequencing technologies, organised population-based screening for BRCA1/2 for populations harbouring a high pathogenic variant burden has been proposed several times. However, knowledge of the expected carrier frequency and possible recurrent (founder) mutations would be a critical prerequisite to giving precise estimations about the predictive values and determining if they are at diagnostically and economically reasonable levels [18–20].
Systematic investigations about specific carrier frequencies in the general – particularly autochthonous – Swiss population are nevertheless lacking. This paucity of reports even extends to the mutation frequency and spectrum in the setting of HBOC-affected cohorts originating from Switzerland and its different language areas, although these have been well studied in other, also non-Caucasian, populations.
This scarcity of studies prompted us to retrospectively assess secondary findings in the 19 NCCN-designated HBOC-related genes in a Swiss monocentric cohort of 400 individuals, almost half of whom were of assumed autochthonous Swiss descent, previously undergoing non-cancer related whole-exome sequencing (WES) analysis. Since these findings were not related to the initial reason for referral, but were identified via proactive searching, the term “secondary findings” was used throughout this study [21, 22]. Hence, for the first time, we provide data on the prevalence of HBOC-related genetic alterations in the general population in Switzerland, which may provide a first data basis for specifying carrier rates for genetic counselling or future cancer screening programmes. The present study indicated an unexpectedly high carrier frequency for genetic variants predisposing to HBOC, especially among the Swiss subpopulation of Italian descent.
Material and methods
Study population
WES data with consent for secondary analyses from 404 subjects enrolled between 2015 and 2017 in our local monocentric study on genetic causes of neurodevelopmental disorders (NDDs) were available for this study. As described below, the WES datasets of four subjects were excluded because of insufficient data quality. The study population therefore consisted of 400 independent individuals (198 males and 202 females; median age 43 years), of whom 180 (90.0%) were parental couples not affected by NDDs, 27 (6.8%) parental singles, and 13 (3.3%) NDD index patients (table 1). For five of these index patients, genetic NDD diagnosis had been established; in eight index cases, the definitive aetiology remained unclear. For the latter, hitherto known NDD-associated tumour syndromes including Fanconi anaemia could be virtually excluded by initial phenotype-driven and panel-agnostic WES, and chromosomal microarray analyses (CMA) (Affymetrix CytoScan® HD or 2.7M arrays) [23, 24]. In none of the investigated individuals was a personal cancer history documented; in 28 individuals (7.0%), however, single or multiple relatives of different relationship degrees were reported to be affected by various cancer entities (supplementary table S1, appendix 1). The study cohort mostly included Caucasians (n = 336, 84.0%) of North West European ancestry (n = 202, 50.5%), among whom 170 (42.5%) were assumed to be of autochthonous Swiss descent. Non-Caucasians (n = 64, 16.0%) predominantly originated from the Middle East / North Africa (n = 35; 8.8%). Consanguinity was noted for 10.0% of individuals with a degree of relationship ranging from the sixth to the third degree. Genetic testing was part of a research project approved by the local ethics committee. Written informed consent for all kinds of scientific research questions, as well as for publication of clinical information, was obtained from participants.
Table 1 Detailed demographic and recruitment data for all enrolled subjects.
| Total no. of investigated individuals |
400 |
| Median age |
43 years |
| |
n
|
%
|
| Sex |
Male |
198 |
49.5 |
| Female |
202 |
50.5 |
| Analysed by WES |
Parental couples |
180/220 (360/400) |
81.8 (90.0) |
| Parental singles |
27/220 (27/400) |
12.3 (6.8) |
| NDD index singles*
|
13/220 (13/400) |
5.9 (3.3) |
| Consanguinity of parental couples |
20/180 (40/400) |
11.1 (10.0) |
| Ancestry†
|
Caucasian |
All |
336 |
84.0 |
| North West European |
All |
202 |
50.5 |
| Autochthonous Swiss‡
|
170 |
42.5 |
| South European |
117 |
29.3 |
| East European |
17 |
4.3 |
| Non-Caucasian |
All |
64 |
16.0 |
| Middle Eastern / North African |
35 |
8.8 |
| East Asian |
1 |
0.3 |
| South Asian |
20 |
5.0 |
| Sub-Sahara African |
8 |
2.0 |
| American |
7 |
1.8 |
Whole-exome sequencing data analysis
WES analyses was performed on DNA extracted from peripheral blood lymphocytes using Agilent (Santa Clara, CA, USA) SureSelect XT Clinical Research Kit (v5 or v6) for capturing, followed by bi-directional paired-end sequencing of 125 base pairs (bps) on a HiSeq2500 platform (Illumina, San Diego, California, USA) and alignment to the hg19 reference genome using NextGENe software (Softgenetics, State College, Pennsylvania, USA). Cut-off values for alternate read fraction and minimum coverage were 16% of supporting reads and 20× read depth, respectively. WES data with less than 85% of the exome covered at ≥20× read depth were considered to be of insufficient quality. Accordingly, WES datasets of four subjects were excluded from this study in the course of quality control. In the remaining 400 WES datasets, on average 95.9% of the WES targeted bases were covered with ≥20× reads (87.1–98.9%) yielding an average read depth of 232× (72–635×) (supplementary figure S1, appendix 4). For the present investigation, the coding regions including 12 flanking intronic base pairs (CDS ±12 bps) of 19 genes related to HBOC syndrome, namely BRCA1/2 plus 17 non-BRCA genes specified by the NCCN guideline "Genetic/Familial High-Risk Assessment: Breast and Ovarian" [8] (v.2.2017; see fig. 1B), were evaluated using the NextGENe software. In our exploratory preliminary analysis of variants with a minor allele frequency (MAF) ≤2%, the low-risk BRCA2 nonsense allele NM_000059.3:c.9976A>T was the sequence alteration showing the highest overall MAF of 0.65% and that still had evidence of a potential clinical effect. An overall MAF threshold of ≤0.65% (gnomAD database, v2.0 and later) was thus set to define rare variants potentially predisposing to HBOC. In order to ascertain sequence quality and confidence, each variant was visually validated and those considered true positives all had a NextGENe overall mutation score above the default quality threshold (≥12). The annotation of the filtered sequence variants was performed using NextGENE and Alamut Visual software (v.2.10; Interactive Biosoftware, Rouen, France) or by manual curation (table S2, appendices 2A and 2B).
For variant annotation and pathogenicity assessment, the web resources as detailed at the end of the article were used.
Variant classification and Sanger sequencing validation
The clinical relevance of the identified sequence variants was manually classified according to the current standards recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) [25], with some adaptations. The variants were evaluated using a five-tier scheme and categorised as “pathogenic” (P), “likely pathogenic” (LP), “hypomorphic” (HM), “variant of uncertain significance” (VUS) and “(likely) benign” (B/LB) with the ACMG-AMP pathogenicity categories “benign” (B) and “likely benign” (LB) being subsumed to “(likely) benign” for simplicity, and a novel category “hypomorphic” being introduced. Given the empirical pathogenic variant burden in the Exome Aggregation Consortium (ExAC) database [26], the frequency levels “within pathogenic range” (≤8 total alleles in the ExAC database; corresponding to the moderate ACMG-AMP “PM2” criterion), “somewhat high” (>8 total alleles), “high” (overall MAF >0.1%), and “very high” (overall MAF >0.5%) were introduced as proposed by Nykamp et al. [27] for genes with autosomal-dominant inheritance. Also according to Nykamp et al., the MAF levels “high” and ”very high” (overall MAF >0.1%) were considered sufficient criteria for B/LB classification if there existed no additional corroborating evidence for variant pathogenicity. Variants were classified as hypomorphic if they had been recurrently observed and shown a low to moderate association with cancer in large case-control studies, with evidence for functional damaging effects but controversial assessment in the literature. Of note, the classification criteria proposed by the ENIGMA consortium for BRCA1/2 variants are solely intended to differentiate germline high-risk BRCA1/2 variants from those with low or no risk. These guidelines are therefore explicitly not intended to evaluate and classify non-high-risk variants associated with an intermediate or moderate risk (ENIGMA BRCA1/2 Gene Variant Classification Criteria, v2.5.1., 29 June 2017). In the absence of contradictory evidence, variant classifications reviewed by international expert consortia [28–32] were used as a stand-alone criterion. Variant interpretations provided in locus-specific databases (LSDBs) were taken into account for pathogenicity assessment only if they were reviewed by multiple submitters without interpretation conflicts (”two-star” review status in the NCBI ClinVar database; corresponding to the supporting ACMG-AMP “BP6” and “PP5” criteria). For the evaluation of computational evidence data, a prediction consensus was built considering seven functional and conservation prediction algorithms (see web resources). To this end, CADD Phred and GERP_RS scores of ≥20 and >2 were set as cut-off values for deleteriousness, as suggested elsewhere [33]. The prediction consensus was assigned as “damaging” (corresponding to the supporting ACMG-AMP “PP3” criterion) if at least six out of seven prediction algorithms indicated a deleterious variant effect. Variants for which 1/7 or none of the predictions were damaging were assumed to be neutral and considered to meet the supporting ACMG-AMP criterion “BP4”; any other cases were categorised as “undetermined” (for further details, see legend to table S2 in the appendix). Likewise, potential splice alterations at the proximal canonical splice site and at in silico predicted cryptic splice donor/acceptor sites (CSDS/CSAS) were monitored for all filtered variants by using a prediction consensus of four different splicing algorithms (see web resources). A variant was predicted to induce a potential splicing defect (corresponding to the supporting ACMG-AMP “PP3” criterion) if at least three algorithms scored above the relative cut-off values established for the score differences between wild-type and variant. For these difference scores, algorithm-specific thresholds were used as proposed by Tang et al. [34] and Baert et al. [35] and detailed in table S3 (appendix 3).
All variants finally classified as (likely) pathogenic were confirmed by bi-directional Sanger sequencing on an ABI3730 capillary sequencer (Applied Biosystems) using the SequencePilot v.4.4.0 software (JSI medical systems, Ettenheim, Germany) for evaluation.
Results
Overall, WES data evaluation for the 19 HBOC-related genes investigated identified 339 different rare variants (MAF ≤0.65%) with an absolute yield of 526 rare sequence alterations. With regard to the latter, the BRCA1 and BRCA2 genes accounted for 9.3% and 17.9% (combined fraction of 27.2%) of all detected rare variants, respectively (fig. 1). Among a total of 383 rare non-BRCA variants, including 246 different variants, most sequence alterations affected the ATM (12.2%) and NF1 genes (8.9%), followed by the CDH1 gene (7.8%). The majority of variants were missense (59.5%) followed by synonymous (26.8%), noncoding (9.9%), nonsense (2.5%), and frameshift sequence variations (1.0%) (table S2, appendix 2).
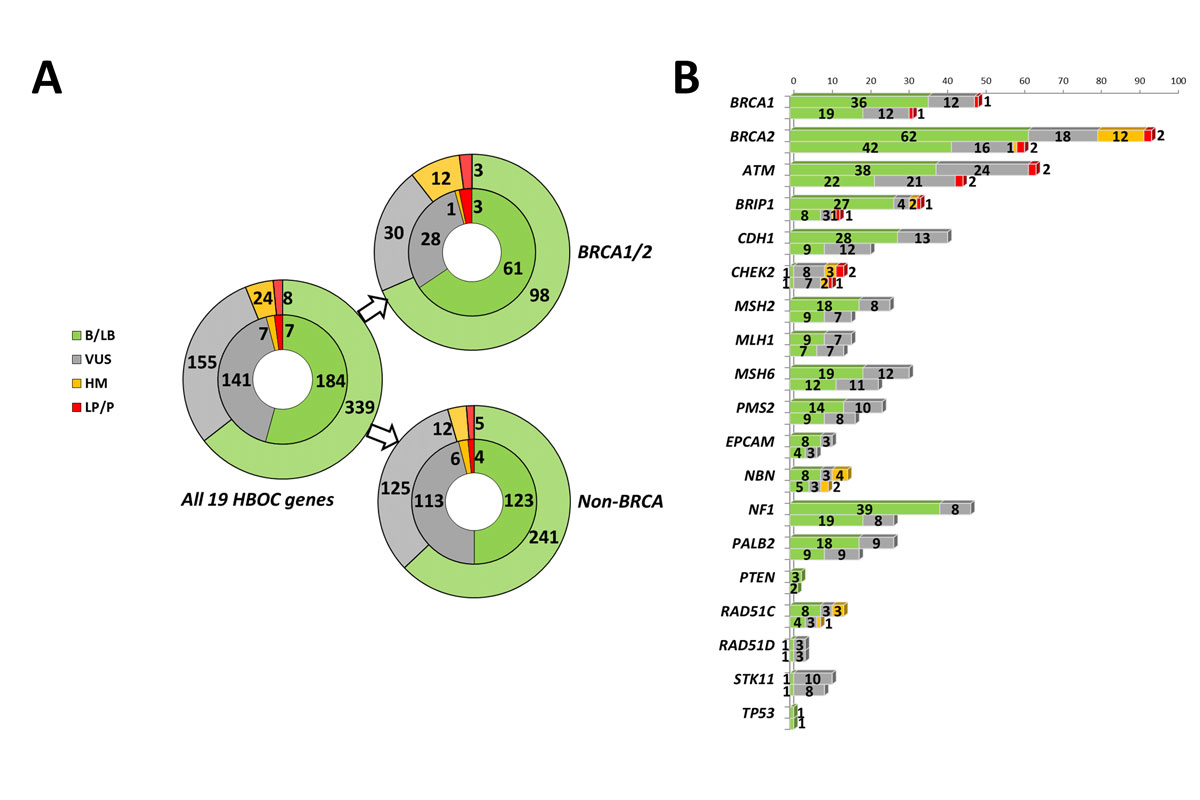
Figure 1 Distribution and classification of rare variants in HBOC-related genes. (A) Distribution of the secondary variants detected in the 19 NCCN-designated genes related to HBOC with stratification into the BRCA1/2 and non-BRCA genes and the different pathogenicity classes. In the outer circles, the absolute variant number is given; in the inner circles, the number of the different variants is shown. (B) Detailed pathogenicity class distribution for each evaluated gene with the absolute variant counts (upper bars) and the number of different variants (lower bars). For clarity purposes, no difference was made for the pathogenicity classes benign/likely benign and likely pathogenic/pathogenic. B = benign; HBOC = hereditary breast and ovarian cancer; HM = hypomorphic; LB = likely benign; LP = likely pathogenic; P = pathogenic; VUS = variant of unclear significance
According to our variant classification, a total of eight secondary LP/P findings were found (1.5% of all detected variants), among which three (0.6%) affected BRCA1/2 and five (1.0%) the non-BRCA genes, with the CHEK2 Arg117Gly variant observed in two unrelated individuals. Additionally, we detected seven different hypomorphic variants with the recurrent low-risk BRCA2 c.9976A>T nonsense allele accounting for one half of all hypomorphic findings (12/24 HM variant carriers). Furthermore, about one third of all rare variants were VUSs (155/526, 29.5%), of which 80.6% (125/155) were detected in the non-BRCA genes, particularly in ATM, CDH1, and MSH6. The highest fraction of VUSs relative to the overall gene-specific variant yield was found for STK11. In contrast, both the TP53 and PTEN genes showed very few sequence variations, which were all classified as (likely) benign. Among the total of 155 VUSs, 14.2% (22/155) were novel (previously unreported in LSDBs and reference populations), whereas 8.4% (13/155) were viewed as a VUS by international expert panels; an additional 36.8% (57/155) were consistently classified as VUSs by multiple submitters to the NCBI ClinVar database (“two-star” review status). Moreover, 117 (34.5%) different variants were known to the Human Gene Mutation Database (HGMD), among which 19 (5.6%) and 90 (26.5%) variants were tagged as “DM” (disease-causing mutations) and “DM?” (questionable disease-causing mutations), respectively.
Among the eight LP/P findings, half of the variants were likely gene-disrupting and half were missense (table 1). Apart from a novel ATM Glu2156* nonsense variant found in a 39-year-old Czech female, all variants had been previously described in the literature and/or LSDBs. For the BRCA1/2 genes, these included the recurrent high-risk BRCA1 c.181T>G European founder allele [36, 37] and two BRCA2 single nucleotide duplications, both of which are annotated as pathogenic in the BRCA Exchange or BRCA Share database. Interestingly, all three BRCA1/2 pathogenic variants were identified in middle-aged females of Italian ancestry, each of whom had no evidence of personal or family history of cancer. The non-BRCA-genes included the recurrent Caucasian CHEK2 c.349A>G [p.(Arg117Gly)] variant known as a moderate risk allele for breast cancer [38, 39], which was carried by two unrelated subjects originating from the German-speaking part of Switzerland. As for BRCA1/2, in none of the non-BRCA mutation carriers any personal and family history of cancer was reported. Of note, four of these LP/P variants were transmitted to the index offspring, for all of whom an unrelated genetic lesion underlying the particular NDD phenotype had been previously elucidated. The genetic consultants of the adult carriers of a LP/P variant were informed in accordance with the given consent for genetic counselling.
Regarding the HM variants, the recurrent Lys3326* nonsense variant within the last BRCA2 exon, for which case-control and genome-wide association studies suggested a mildly increased risk for HBOC [40, 41] and various other cancer entities [42], had the highest carrier rate of 2.3% in our sample (table 2). Of note, two thirds of the carriers were of autochthonous Swiss descent, which may reflect a high local prevalence in comparison with the 0.65% overall MAF. An additional 12 individuals (2.3%) harboured six further different HM sequence alterations in the BRIP1, CHEK2, NBN, and RAD51C genes. Both of the CHEK2 variants detected [c.320-5T>A, p.(Arg180Cys)] have been considered as low penetrant breast cancer risk alleles among white European females [39, 45, 46]. Likewise, the RAD51C Gly264Ser variant has been supposed to confer low ovarian cancer risk elevation [47, 48], which was corroborated by a partial homology-directed repair deficiency in vivo [49, 50]. For the detected hypomorphic NBN missense variants [p.(Ile171Val), p.(Arg215Trp)], minor pleiotropic cancer risks have been suggested by a meta-analysis conducted by Gao et al. [51]. Table 3 summarises all hypomorphic variants detected.
Table 2 Summary of all detected (likely) pathogenic variants.
|
Gene
|
HGVS
cDNA change*
|
Predicted AA change
|
rs number
dbSNP 147
|
Function
|
Founder mutation?
|
Evidence (ACMG evidence category, if meeting) [
25
]
|
Final classification
|
Carrier ID
|
Age, y
|
Sex
|
Ancestry
|
Variant transmitted to index?
|
NDD diagnosis established for affected offspring†
|
|
BRCA1
|
c.181T>G |
p.(Cys61Gly) |
rs28897672 |
Missense |
Europe [36] |
Classified as pathogenic by the ENIGMA consortium; one of the most recurrent founder BRCA1 pathogenic variants in multiple European populations [1] |
Pathogenic |
67202 |
36 |
f |
South Italian |
N |
N (NDD, primary microcephaly, speech delay, short stature) |
|
BRCA2
|
c.4284dup |
p.(Gln1429Serfs*9) |
rs80359439 |
Frameshift |
Greece / South-East Europe (?) [43, 44] |
Classified as pathogenic by the ENIGMA consortium |
Pathogenic |
47478 |
44 |
f |
South Italian (Sicily) |
N |
N (NDD, ataxia, epilepsy) |
|
BRCA2
|
c.4719dup |
p.(Lys1574*) |
None |
Nonsense |
n.k. to ENIGMA |
Predicted LoF-variant (PVS1); unreported (PM2); known to the UMD Database with one submission as causal (PP5) |
Pathogenic |
75475 |
39 |
f |
Italian (Sardinia) |
Y |
Y (MIM #300243, SLC9A6) |
|
ATM
|
c.6466G>T |
p.(Glu2156*) |
None |
Nonsense |
Novel |
Predicted LoF-Variant (PVS1); unreported (PM2) |
Likely pathogenic |
67514 |
39 |
f |
Czech |
N |
N (NDD, microcephaly, short stature, hypotonia) |
|
ATM
|
c.6385T>G |
p.(Tyr2129Asp) |
None |
Missense |
Founder effect n.k. |
Mutational hot spot (PIK-related kinase/FAT domain) (PM1); absent in population databases (PM2); detected in trans with a second pathogenic variant in an A-T patient (ClinVar submission SCV000537715.1, HUG) (PM3); highly deleterious in silico predictions (PP3) |
Likely pathogenic |
62542 |
40 |
m |
Turkish (Kurd) |
N |
Y (MIM #610536, EFTUD2) |
|
BRIP1
|
c.2684_2687del |
p.(Ser895*) |
rs760551339 |
Frameshift |
Founder effect n.k. |
Predicted LoF-variant (PVS1); MAF in population databases within pathogenic range (PM2); known to HGMD and ClinVar databases as (likely) pathogenic (PP5). |
Likely pathogenic |
68160 |
55 |
m |
Swiss, German-speaking part |
Y |
Y (MTATP6) |
|
CHEK2
|
c.349A>G |
p.(Arg117Gly) |
rs28909982 |
Missense |
Recurrent among Caucasians,but founder effect n.k. [38] |
Established recurrent pathogenic variant with statistically confirmed association with BC [39] |
Pathogenic |
73281 |
41 |
f |
Swiss, German-speaking part |
Y |
Y (MIM #121050, FBN2) |
|
CHEK2
|
c.349A>G |
p.(Arg117Gly) |
rs28909982 |
Missense |
73184 |
59 |
m |
Swiss, German-speaking part |
N |
N (severe NDD, spastic tetraplegia, epileptic encephalopathy) |
Table 3 Summary of all detected hypomorphic variants.
|
Gene
|
HGVS cDNA change*
|
Predicted AA change
|
rs number
dbSNP 147
|
Function
|
Selected case-control and functional studies (OR, Odds ratio [95%CI])
|
Carrier ID
|
Age, y
|
Sex
|
Ancestry
|
|
BRCA2
|
c.9976A>T |
p.(Lys3326*) |
rs11571833 |
Nonsense |
2634 BC kindreds: OR = 1.53 (1.00–2.34) [40]; 41,081 BCAC BC cases: ORw = 1.28 (1.17–1.40), 14,514 OCAC invasive OC cases: ORw = 1.26 (1.10–1.43) [41]; 43,641 genotyped cancer patients: OR (small cell lung cancer) = 2.06 (1.35–3.16), OR (squamous cell carcinoma of the skin) = 1.69 (1.26–2.26) [42]; minor reduction of HDR activity in [52] |
12 indivi-duals |
|
|
among these, eight Swiss individuals |
|
BRIP1
|
c.139C>G |
p.(Pro47Ala) |
rs28903098 |
Missense |
1882 glioma cases: OR = 3.83 (1.01–14.5) [53]; initially reported in an individual with early-onset BC and strong BC/OC family history [54, 55]; destabilisation and abolishment of the BRIP1 helicase activity in [55, 56] |
71230 |
44 |
F |
Dutch? |
|
BRIP1
|
c.139C>G |
p.(Pro47Ala) |
rs28903098 |
Missense |
62926 |
51 |
F |
Swiss |
|
CHEK2
|
c.320-5T>A |
p.(?) |
rs121908700 |
Noncoding |
13,087 ECRIC BC cases: OR = 13.9 (1.89–101) [45]; leaky splicing defect implied by [46] |
67184 |
48 |
M |
Swiss |
|
CHEK2
|
c.320-5T>A |
p.(?) |
rs121908700 |
Noncoding |
75475 |
nk |
F |
Italian |
|
CHEK2
|
c.538C>T |
p.(Arg180Cys) |
rs77130927 |
Missense |
34,488 unselected white European BC patients: OR = 1.34 (1.06–1.70) [39]; intermediate DNA damage response in [57] |
68159 |
53 |
F |
Dutch |
|
NBN
|
c.511A>G |
p.(Ile171Val) |
rs61754966 |
Missense |
Meta-analysis of 39,731 cancer cases: OR = 3.93 (1.68–9.20) [51]; impaired DSB repair activity and increased chromosomal instability in [58] |
77074 |
44 |
F |
Swiss |
|
NBN
|
c.511A>G |
p.(Ile171Val) |
rs61754966 |
Missense |
77745 |
32 |
F |
Serbian |
|
NBN
|
c.511A>G |
p.(Ile171Val) |
rs61754966 |
Missense |
57429 |
10 |
M |
Croatian |
|
NBN
|
c.643C>T |
p.(Arg215Trp) |
rs34767364 |
Missense |
Meta-analysis of 39,731 cancer cases: OR = 1.77 (1.07–2.91) [51]; impaired DSB repair activity in [59, 60] |
66310 |
38 |
M |
Portuguese |
|
RAD51C
|
c.790G>A |
p.(Gly264Ser) |
rs147241704 |
Missense |
620 BC cases, 480 BC/OC cases: OR (BC/OC) = 3.44 (1.51–7.80) [48]; intermediate HDR activity [49, 50] |
70861 |
51 |
F |
Swiss |
|
RAD51C
|
c.790G>A |
p.(Gly264Ser) |
rs147241704 |
Missense |
68968 |
36 |
F |
Armenian/Russian |
|
RAD51C
|
c.790G>A |
p.(Gly264Ser) |
rs147241704 |
Missense |
60577 |
39 |
F |
Swiss |
For the individual variant burden, the carrier prevalence for at least one rare variant was 28.8 vs 60.8% (BRCA1/2 vs non-BRCA genes) summing up to an overall carrier rate of 71% (fig. 2). In total, 2.0% of the individuals carried a LP/P variant (0.8 vs 1.3%); if broken down by sex, in the 202 women of the present cohort, the LP/P variant carrier rate was 2.5% (1.5 vs 1.0%). In 5.5% at least one HM variant was observed. The carrier frequency of at least one VUS was 32.0% (7.0 vs 27.3%), of which the vast majority (overall frequency 30.3%) harboured no additional HM or LP/P variants. Notably, this frequency included two VUS which were shared by consanguineous parental couples. In a further third of individuals (33.5%), single or multiple LB/B variants were found as the only rare sequence alteration.
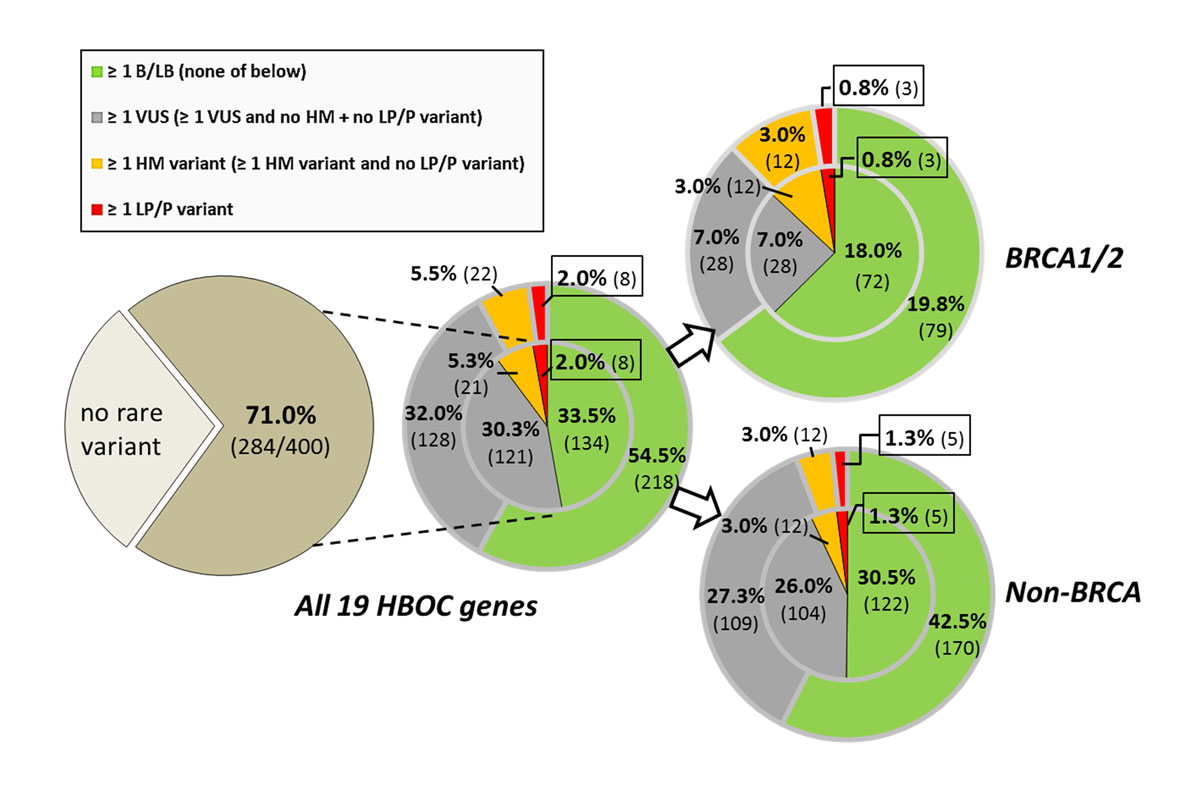
Figure 2 Carrier frequencies in non-cancer related individuals (n = 400) for at least one rare variant (overall MAF ≤0.65) in the HBOC-genes (dark brown), stratified to the BRCA1/2 and non-BRCA genes and the different pathogenicity classes (inset). Frequencies for mutually exclusive stratification categories (single count of the "highest" applicable pathogenicity category per individual) are given in the inner circle; the frequencies for non-exclusive categories (multiple categories were counted per individual if applicable) in the outer circle. BRCA1/2- and non-BRCA-specific carrier frequencies are determined separately and hence non-additive. The absolute counts are given in brackets. B = benign; HBOC = hereditary breast and ovarian cancer; HM = hypomorphic; LB = likely benign; LP = likely pathogenic; P = pathogenic; VUS = variant of unclear significance
Most strikingly, when considering the LP/P and HM variant burden as a whole, 7.3% of the individuals were identified to be carrier of HBOC-related germline alteration. Of note, these carrier individuals were not related to each other. Since none of the 13 NDD index patients exhibited any LP/P variant, no relevant difference in the carrier percentages existed when the entire study population was compared to the non-index proband group. Likewise, the carrier frequency did not significantly change if the analysis was restricted to the individuals of Swiss descent.
Discussion
Approaches to comprehensive population-wide genetic screening are being increasingly discussed since the advent of early high-throughput sequencing technologies. However, routine BRCA1/2 assessment for the entire or female population is currently not recommended [8, 9, 61, 62]. Nevertheless, according to the HBOC guideline by the Swiss Group for Clinical Cancer Research (SAKK), individuals of Ashkenazi Jewish heritage should be referred for genetic testing, irrespective of their personal or family cancer history [9]. For the expansion to further ethnicities, as recently proposed by Lasker awardee Mary-Claire King [61], knowledge about expected carrier frequencies and possible mutation recurrences among the population of interest is critically required [19].
In contrast to the traditional estimates of BRCA1/2 mutation carrier prevalence, which range from 0.2% to 0.3% (1:300 to 1:500) in the general female population [62], we found in our total cohort a significantly higher carrier rate of 0.8%. However, recent empirical data from the ExAC database indicate a BRCA1/2 mutation population frequency of 0.41% in total and of 0.51% in the European population (exclusion of data from The Cancer Genome Atlas [TCGA], considering pathogenic missense mutations known to the ENIGMA database and all protein-truncating variants [PTVs] of the respective canonical transcript outside the regions distal to the last bona-fide pathogenic PTV and sequencing error-prone repeats) [63]. This finding has very recently been confirmed by the Geisinger MyCode initiative for precision medicine, who reported in >50,000 mostly Caucasians a prevalence of 1:180 (0.56%) for Sanger-confirmed BRCA1/2 LP/P variants when controlled for relatedness up to the third degree [64, 65].
Surprisingly, all the three BRCA1/2 carriers in our cohort were women of Italian descent (South Italy and Sardinia). When restricted to this subpopulation the carrier rate would be as extraordinary high as 7.1% (3/42 individuals of Italian descent), which significantly differs from that of the non-Italian ancestry subgroup (0/353 with available ancestry information; p = 0.0011, Fisher's exact test). Of note, neither the BRCA1/2 carriers, nor the carriers of LP/P variants in non-BRCA genes had a documented personal or family history of cancer and therefore they are not listed in table S1.
This unprecedentedly high pathogenic variant rate in BRCA1/2 might be explained by the small sample size, but could reflect an actually higher BRCA1/2 mutation prevalence among subjects originating from Italy. In accordance with the latter assumption, in larger cohorts of Italian patients with familial breast/ovarian cancer, consistently high BRCA1/2 carrier rates of up to 33.6% have been reported, with multiple, partially regionally recurring, founder mutations accounting for more than 10% of all BRCA1/2 pathogenic variants [66–69]. In particular, in a large unselected invasive ovarian cancer series from Ontario, Canada, patients of Italian origin carried the most BRCA1/2 pathogenic variants (20/46, 43.5%), owing in part to the recurrence of three founder mutations, which caused the authors to discuss more widespread screening among Italians [70, 71]. In line with this, the significantly higher breast/ovarian cancer incidence in Western Switzerland and Canton of Ticino (Italian-speaking) compared with the German-speaking part of Switzerland [1] gives reason to speculate about a higher BRCA1/2 mutation burden among Italian-descent individuals. Accordingly, two of the three BRCA1/2 pathogenic variants observed in our patients are recurrently seen in Italian breast/ovarian cancer patients. First, we found in a healthy woman from Southern Italy the BRCA1 c.181T>G [p.(Cys61Gly)] variant, which represents the second most common pathogenic BRCA1 allele among Italian BRCA1 families (prevalence of 3.9%) [37]. Nevertheless, this variant ubiquitously recurs in Central Europe and had been identified on a common Polish and Ashkenazi Jewish haplotype, suggesting it is an Eastern European founder mutation [72]. The second recurrent Italian variant (BRCA2: c.4284dup) was previously reported in a North-Italian patient with synchronous breast and ovarian cancer with a frameshift BRCA1 mutation in double heterozygosis [73] as well as in two other Italian ovarian cancer patients [71, 74].
With regard to the non-BRCA genes, the comparability of our carrier rate of 1.3% is limited, because, to our knowledge, only screenings including subpanels of the 19 NCCN HBOC genes have been published so far. Most published WES screenings were for the detection of secondary findings across the ACMG-designated minimum list of 56, or nowadays 59, medically actionable genes (ACMG-56/59) [75, 76], among which nine genes only are shared with the NCCN gene panel, with important genes such as the ATM, CHEK2, and PALB2 genes missing. These core genes show the highest mutation prevalence (2.5, 1.5 and 1.2%) in a very recent series of >5500 German BRCA1/2-negative breast cancer index patients [77] and also account for 1.0% of the LP/P variant yield in the present cohort of individuals without cancer. Nevertheless, ACMG-56/59-based studies in predominantly Caucasian cohorts found that the carrier rates in nine shared HBOC genes ranged from 0.3 to 2.5% [78–83], with variant classification inconsistencies and other confounding factors. In a couple of pioneer papers [23, 84–86], large cohorts were screened for more expanded gene panels, including up to 16 genes [87] out of the present HBOC panel, but these studies also failed to consider, particularly, the ATM and CHEK2 core genes.
Thompson et al. [88], however, screened almost 2000 cancer-free Australian women for secondary findings in a panel of 18 breast cancer-associated genes covering the non-BRCA core genes ATM, BRIP1, CDH1, CHEK2, PALB2 and TP53, which is more suitable for the comparison with the data from our cohort. With regard to the 12 overlapping genes, the resulting prevalence was somewhat lower in their study population (1.8%) as compared with our cohort (2.0%). This also holds true when comparison is made for BRCA1/2 (0.65 vs 0.8% in our cohort) and non-BRCA genes (1.1 vs 1.3%). This difference may be partly explained by the inclusion of cancer-free women exclusively aged >60 by Thompson et al., which may result in a partial depletion of highly penetrant mutations in their series (mean age in our cohort 43 years). Similarly, the prevalence of PTVs in eight non-BRCA core genes in control datasets derived from ExAC was 1.4% (27,173 non-Finnish European [NFE] individuals), whereas it was 0.9%, in the hypernormal FLOSSIES populations (7325 cancer-free woman of European American ancestry over 70 years of age) [77]. However, in addition to inter-study variability in sample size, demographic structure and inclusion criteria, comparability is further hampered by considerable methodological inconsistencies in the stringency of variant filtering and pathogenicity assessment together with technical sequencing setups, and therefore data quality [31, 80–83]. As an illustration, the BRCA1/2 mutation carrier frequency among the Finnish ExAC cohort was considerably lower than that of NFE subpopulation (0.08 vs 0.24%); inter-population differences, however, may be confounded by the contribution of Ashkenazi Jewish founder mutations present only in the NFE cohort. Likewise, there is substantial variability between the Africans, Latinos, East and South Asians with ExAC frequencies ranging from 0.16% to 0.41% among Africans to Latinos [63], which may be influenced by limitations in sample size and the fact that the existing literature is biased towards Caucasian cohorts. The latter may mean that there is insufficient evidence to classify missense variants recurring among non-Caucasians as definitely pathogenic [89].
Moreover, Maxwell et al. [63], among others, rightly pointed out that true population prevalence might be underestimated in available studies, owing to the lack of consideration of copy number variants (CNVs), which account for ~10% of all BRCA1/2 mutations [90], and the disregard of uncharacterised but (potentially) pathogenic missense variants. Even though the present study also did not include CNVs and structural rearrangements, we assessed the clinical significance of missense variants by extensive manual curation without any database-driven pre-selection variant filtering strategy, allowing us to give a more representative reflection of the true mutation burden. Furthermore, we also incorporated into our evaluation hypomorphic variants, which were found in the considerable percentage of 5.5% of the studied individuals.
In contrast to many other populations, for Switzerland as a whole, the pathogenic variant spectrum and prevalence for the HBOC-related genes even in cancer patients is far from being established. To our knowledge, hitherto only three Swiss retrospective series with up to 350 breast/ovarian cancer patients meeting stringent clinical criteria have been published, in which typical BRCA1/2 pathogenic variant carrier rates of about 25% were detected [5, 84, 85]. Notably, these studies solely addressed BRCA1/2 and, regardless of the heterogeneity and peculiarities of the Swiss population [86, 91], provided no specific inclusion or stratification criteria related to the ancestry of the enrolled patients. In our cohort, two unrelated individuals originating from the German-speaking part of Switzerland shared the pathogenic CHEK2 Arg117Gly variant, which is one of the most recurrent non-founder CHEK2 mutations among Caucasians [38]. To our knowledge, this variant has not yet been characterised in Swiss individuals. Exploratory examinations on the basis of the trio-WES data argue against a shared CHEK2 haplotype, though only a limited number of WES covered single-nucleotide polymorphic (SNP) sites of the CHEK2 haplotype-constituting SNP ensembles provided by Einarsdottir et al. [92] and Kaufman et al. [93] could be analysed.
In conclusion, our pioneer analyses indicate a substantial high burden of HBOC-related breast and ovarian cancer susceptibility in the heterogeneous Swiss general population and relevant subpopulations, especially in subjects of Italian descent. Whereas the LP/P variants found in 2.0% of the studied local population and 7.1% of the Italian subpopulation would warrant clinical action, the clinical implications of the hypomorphic variants detected in 5.5% of the population are currently not defined. Especially since all the LP/P variants were detected in individuals without personal or family cancer history, our data may corroborate further considerations about population-based HBOC screenings in Switzerland, particularly for individuals of Italian ancestry, and may serve as a starting point for screening in larger cohorts, preferably with genetically inferred ancestry. Organised screening may contribute decisively to the identification of the many mutation carriers who are presently undiagnosed as a result of the current restrictive testing practice and therefore excluded from referral to special HBOC management programmes at an early age. Nevertheless, the high rate of VUS detected, especially in non-BRCA genes, would require strict policies for non-disclosure of VUS, as well as regular and systematic reassessments.
Web resources
Population databases and datasets:
Database of Single Nucleotide Polymorphism (dbSNP), https://www.ncbi.nlm.nih.gov/SNP
Exome Aggregation Consortium (ExAC) Browser, http://exac.broadinstite.org
Fabulous Ladies over Seventy (FLOSSIES) Database, https://whi.color.com
Genome Aggregation Database (gnomAD), http://gnomad.broadinstitute.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS
1000 Genomes (1000G) Project, http://www.1000genomes.org
Locus-specific databases (LSDBs):
ARUP BRCA Mutation Database, http://www.arup.utah.edu/database
BRCA Exchange Database, http://brcaexchange.org
Zhejiang University Center for Genetic and Genomic Medicine Database, http://www.genomed.org/lovd2/home.php
Global Variome Shared Leiden Open Variation Database (LOVD), https://databases.lovd.nl/shared/genes
Human Gene Mutation Database (HGMD), https://www.hgmd.org
International Agency for Research on Cancer (IARC) TP53 Database, http://p53.iarc.fr
International Society for Gastrointestinal Hereditary Tumours (InSight) Databases, http://www.insight-database.org/genes
Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Database, http://www.kconfab.org
LOVD - Human Mismatch Repair Genes, http://HCI-LOVD.hci.utah.edu/home.php
Mismatch Repair Genes Variant Database, http://www.med.mun.ca/mmrvariants
NCBI ClinVar Database, https://www.ncbi.nlm.nih.gov/clinvar
NHGRI Breast Cancer Information Core (BIC) Database, https://research.nhgri.nih.gov/bic
Universal Mutation Databases (UMD), http://www.umd.be
In-silico functional prediction algorithms:
Align-Grantham Variation Grantham Deviation (GVGD), http://agvgd.hci.utah.edu
Combined Annotation Dependent Depletion (CADD) score, http://cadd.gs.washington.edu/score
EMBL-EBI InterPro (v.67.0), https://www.ebi.ac.uk/interpro
Genomic Evolutionary Rate Profiling (GERP++)_Rejected Substitution (RS), http://mendel.standford.edu/SidowLab/downloads/gerp
MutationTaster (MutatTaster), http://www.mutationtaster.org
Polymorphism Phenotyping (PolyPhen)-2, http://genetics.bwh.harvard.edu/pph2
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org
Universal Mutation Database (UMD) predictor, http://umd-predictor.eu/
In-silico tools for splicing defect prediction (assessed by Alamut Visual v.2.10 software):
SpliceSiteFinder (SSF)-like, http://www.interactive-biosoftware.com
MaxEntScan (MES), http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
Splice Site Prediction by Neural Network (NNS), http://www.fruitfly.org/seq_tools/splice.html
Human Splicing Finder (HSF), http://www.umd.be/HSF
Appendices
Appendix 1
Table S1: Individuals with documentation of family history of cancer.
Appendix 2A and 2B
Table S2: Synopsis of all detected secondary variants including annotations and criteria for pathogenicity assessment.
Appendix 3
Table S3: Synopsis of the in-silico predictions of potentially splice-affecting variants.
Appendix 4
Figure S1: Box plot of the coverage data of all individual WES analyses.
Appendix 5
Reference list for the supplementary data appendices.
The appendices are available in separate files for downloading at https://smw.ch/en/article/doi/smw.2019.20092/
References
1Arndt V, Feller A, Hauri D, Heusser R, Junker C, Kuehni C, et al. Swiss Cancer Report 2015 - Current situation and developments. Neuchâtel: Federal Statistical Office (FSO); 2016
2
Walsh
T
,
Casadei
S
,
Lee
MK
,
Pennil
CC
,
Nord
AS
,
Thornton
AM
, et al.
Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032–7. doi:.https://doi.org/10.1073/pnas.1115052108
3
Katapodi
MC
,
Viassolo
V
,
Caiata-Zufferey
M
,
Nikolaidis
C
,
Bührer-Landolt
R
,
Buerki
N
, et al.
Cancer Predisposition Cascade Screening for Hereditary Breast/Ovarian Cancer and Lynch Syndromes in Switzerland: Study Protocol. JMIR Res Protoc. 2017;6(9):e184. doi:.https://doi.org/10.2196/resprot.8138
4
Bouchardy
C
,
Lorez
M
,
Arnst
V
, NICER Working group. Effects of age and stage on breast cancer survival in Switzerland. Swiss Cancer Bull.
2015;35(2):152–7.
5
Schoumacher
F
,
Glaus
A
,
Mueller
H
,
Eppenberger
U
,
Bolliger
B
,
Senn
HJ
. BRCA1/2 mutations in Swiss patients with familial or early-onset breast and ovarian cancer. Swiss Med Wkly. 2001;131(15-16):223–6.
6
Schroeder
C
,
Faust
U
,
Sturm
M
,
Hackmann
K
,
Grundmann
K
,
Harmuth
F
, et al.
HBOC multi-gene panel testing: comparison of two sequencing centers. Breast Cancer Res Treat. 2015;152(1):129–36. doi:.https://doi.org/10.1007/s10549-015-3429-9
7
Kast
K
,
Rhiem
K
,
Wappenschmidt
B
,
Hahnen
E
,
Hauke
J
,
Bluemcke
B
, et al.; German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC). Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016;53(7):465–71. doi:.https://doi.org/10.1136/jmedgenet-2015-103672
8
Daly
MB
,
Pilarski
R
,
Berry
M
,
Buys
SS
,
Farmer
M
,
Friedman
S
, et al.
NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Canc Netw. 2017;15(1):9–20. doi:.https://doi.org/10.6004/jnccn.2017.0003
9
Chappuis
POBB
,
Bürki
N
,
Buser
K
,
Heinimann
K
,
Monnerat
C
, et al.
Swiss guidelines for counselling and testing - Genetic predisposition to breast and ovarian cancer. Schweiz Arzteztg. 2017;98(2122):682–4. doi:.https://doi.org/10.4414/saez.2017.05502
10
Paluch-Shimon
S
,
Cardoso
F
,
Sessa
C
,
Balmana
J
,
Cardoso
MJ
,
Gilbert
F
, et al.; ESMO Guidelines Committee. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann Oncol. 2016;27(suppl 5):v103–10. doi:.https://doi.org/10.1093/annonc/mdw327
11Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF). Interdisziplinäre S3-Leitline für die Früherkennung, Diagnostik, Therapie und Nachsorge des Mammakarzinoms, Version 4.1; 2018. AWMF-Registernummer 032-045OL.
12
Rosenthal
ET
,
Bernhisel
R
,
Brown
K
,
Kidd
J
,
Manley
S
. Clinical testing with a panel of 25 genes associated with increased cancer risk results in a significant increase in clinically significant findings across a broad range of cancer histories. Cancer Genet. 2017;218-219:58–68. doi:.https://doi.org/10.1016/j.cancergen.2017.09.003
13
Tung
N
,
Battelli
C
,
Allen
B
,
Kaldate
R
,
Bhatnagar
S
,
Bowles
K
, et al.
Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121(1):25–33. doi:.https://doi.org/10.1002/cncr.29010
14
Couch
FJ
,
Hart
SN
,
Sharma
P
,
Toland
AE
,
Wang
X
,
Miron
P
, et al.
Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33(4):304–11. doi:.https://doi.org/10.1200/JCO.2014.57.1414
15
Kurian
AW
,
Sigal
BM
,
Plevritis
SK
. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol. 2010;28(2):222–31. doi:.https://doi.org/10.1200/JCO.2009.22.7991
16
Le-Petross
HT
,
Whitman
GJ
,
Atchley
DP
,
Yuan
Y
,
Gutierrez-Barrera
A
,
Hortobagyi
GN
, et al.
Effectiveness of alternating mammography and magnetic resonance imaging for screening women with deleterious BRCA mutations at high risk of breast cancer. Cancer. 2011;117(17):3900–7. doi:.https://doi.org/10.1002/cncr.25971
17
Kaufman
B
,
Shapira-Frommer
R
,
Schmutzler
RK
,
Audeh
MW
,
Friedlander
M
,
Balmaña
J
, et al.
Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50. doi:.https://doi.org/10.1200/JCO.2014.56.2728
18
Gabai-Kapara
E
,
Lahad
A
,
Kaufman
B
,
Friedman
E
,
Segev
S
,
Renbaum
P
, et al.
Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc Natl Acad Sci USA. 2014;111(39):14205–10. doi:.https://doi.org/10.1073/pnas.1415979111
19
Adams
MC
,
Evans
JP
,
Henderson
GE
,
Berg
JS
. The promise and peril of genomic screening in the general population. Genet Med. 2016;18(6):593–9. doi:.https://doi.org/10.1038/gim.2015.136
20
D’Andrea
E
,
Marzuillo
C
,
De Vito
C
,
Di Marco
M
,
Pitini
E
,
Vacchio
MR
, et al.
Which BRCA genetic testing programs are ready for implementation in health care? A systematic review of economic evaluations. Genet Med. 2016;18(12):1171–80. doi:.https://doi.org/10.1038/gim.2016.29
21
Thompson
ML
,
Finnila
CR
,
Bowling
KM
,
Brothers
KB
,
Neu
MB
,
Amaral
MD
, et al.
Genomic sequencing identifies secondary findings in a cohort of parent study participants. Genet Med. 2018;20(12):1635–43. Published online April 12, 2018. doi:.https://doi.org/10.1038/gim.2018.53
22
Weiner
C
. Anticipate and communicate: Ethical management of incidental and secondary findings in the clinical, research, and direct-to-consumer contexts (December 2013 report of the Presidential Commission for the Study of Bioethical Issues). Am J Epidemiol. 2014;180(6):562–4. doi:.https://doi.org/10.1093/aje/kwu217
23
Boonsawat
P
,
Joset
P
,
Steindl
K
,
Oneda
B
,
Gogoll
L
,
Azzarello-Burri
S
, et al.; Undiagnosed Diseases Network (UDN). Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet Med. 2019. Published online March 3, 2019 [Epub ahead of print]. doi:.https://doi.org/10.1038/s41436-019-0464-7
24
Papuc
SM
,
Abela
L
,
Steindl
K
,
Begemann
A
,
Simmons
TL
,
Schmitt
B
, et al.
The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Hum Genet. 2019;27(3):408–21. Published online December 14, 2018. doi:.https://doi.org/10.1038/s41431-018-0299-8
25
Richards
S
,
Aziz
N
,
Bale
S
,
Bick
D
,
Das
S
,
Gastier-Foster
J
, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23. doi:.https://doi.org/10.1038/gim.2015.30
26
Kobayashi
Y
,
Yang
S
,
Nykamp
K
,
Garcia
J
,
Lincoln
SE
,
Topper
SE
. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9(1):13. doi:.https://doi.org/10.1186/s13073-017-0403-7
27
Nykamp
K
,
Anderson
M
,
Powers
M
,
Garcia
J
,
Herrera
B
,
Ho
YY
, et al.; Invitae Clinical Genomics Group. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105–17. doi:.https://doi.org/10.1038/gim.2017.37
28
Spurdle
AB
,
Healey
S
,
Devereau
A
,
Hogervorst
FB
,
Monteiro
AN
,
Nathanson
KL
, et al.; ENIGMA. ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33(1):2–7. doi:.https://doi.org/10.1002/humu.21628
29
Szabo
C
,
Masiello
A
,
Ryan
JF
,
Brody
LC
. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16(2):123–31. doi:.https://doi.org/10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y
30
Plazzer
JP
,
Sijmons
RH
,
Woods
MO
,
Peltomäki
P
,
Thompson
B
,
Den Dunnen
JT
, et al.
The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam Cancer. 2013;12(2):175–80. doi:.https://doi.org/10.1007/s10689-013-9616-0
31
Olivier
M
,
Eeles
R
,
Hollstein
M
,
Khan
MA
,
Harris
CC
,
Hainaut
P
. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19(6):607–14. doi:.https://doi.org/10.1002/humu.10081
32
Béroud
C
,
Letovsky
SI
,
Braastad
CD
,
Caputo
SM
,
Beaudoux
O
,
Bignon
YJ
, et al.; Laboratory Corporation of America Variant Classification Group; Quest Diagnostics Variant Classification Group; UNICANCER Genetic Group BRCA Laboratory Network. BRCA Share: A Collection of Clinical BRCA Gene Variants. Hum Mutat. 2016;37(12):1318–28. doi:.https://doi.org/10.1002/humu.23113
33
Chandrasekharappa
SC
,
Chinn
SB
,
Donovan
FX
,
Chowdhury
NI
,
Kamat
A
,
Adeyemo
AA
, et al.
Assessing the spectrum of germline variation in Fanconi anemia genes among patients with head and neck carcinoma before age 50. Cancer. 2017;123(20):3943–54. doi:.https://doi.org/10.1002/cncr.30802
34
Tang
R
,
Prosser
DO
,
Love
DR
. Evaluation of Bioinformatic Programmes for the Analysis of Variants within Splice Site Consensus Regions. Adv Bioinforma. 2016;2016:5614058. doi:.https://doi.org/10.1155/2016/5614058
35
Baert
A
,
Machackova
E
,
Coene
I
,
Cremin
C
,
Turner
K
,
Portigal-Todd
C
, et al.
Thorough in silico and in vitro cDNA analysis of 21 putative BRCA1 and BRCA2 splice variants and a complex tandem duplication in BRCA2 allowing the identification of activated cryptic splice donor sites in BRCA2 exon 11. Hum Mutat. 2018;39(4):515–26. doi:.https://doi.org/10.1002/humu.23390
36
Janavičius
R
. Founder BRCA1/2 mutations in the Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010;1(3):397–412. doi:.https://doi.org/10.1007/s13167-010-0037-y
37
Rebbeck
TR
,
Friebel
TM
,
Friedman
E
,
Hamann
U
,
Huo
D
,
Kwong
A
, et al.; EMBRACE; GEMO Study Collaborators; HEBON. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018;39(5):593–620. doi:.https://doi.org/10.1002/humu.23406
38
Leedom
TP
,
LaDuca
H
,
McFarland
R
,
Li
S
,
Dolinsky
JS
,
Chao
EC
. Breast cancer risk is similar for CHEK2 founder and non-founder mutation carriers. Cancer Genet. 2016;209(9):403–7. doi:.https://doi.org/10.1016/j.cancergen.2016.08.005
39
Southey
MC
,
Goldgar
DE
,
Winqvist
R
,
Pylkäs
K
,
Couch
F
,
Tischkowitz
M
, et al.; Australian Ovarian Cancer Study Group. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. 2016;53(12):800–11. doi:.https://doi.org/10.1136/jmedgenet-2016-103839
40
Thompson
ER
,
Gorringe
KL
,
Rowley
SM
,
Li
N
,
McInerny
S
,
Wong-Brown
MW
, et al.; Lifepool Investigators. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci Rep. 2015;5(1):14800. doi:.https://doi.org/10.1038/srep14800
41
Meeks
HD
,
Song
H
,
Michailidou
K
,
Bolla
MK
,
Dennis
J
,
Wang
Q
, et al.; EMBRACE; kConFab Investigators; Australia Ovarian Cancer Study Group; HEBON; GEMO Study Collaborators; OCGN; PRostate cancer AssoCiation group To Investigate Cancer Associated aLterations in the genome. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J Natl Cancer Inst. 2016;108(2):djv315. doi:.https://doi.org/10.1093/jnci/djv315
42
Rafnar
T
,
Sigurjonsdottir
GR
,
Stacey
SN
,
Halldorsson
G
,
Sulem
P
,
Pardo
LM
, et al.
Association of BRCA2 K3326* With Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J Natl Cancer Inst. 2018;110(9):967–74. doi:.https://doi.org/10.1093/jnci/djy002
43
Karami
F
,
Mehdipour
P
. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. BioMed Res Int. 2013;2013:928562. doi:.https://doi.org/10.1155/2013/928562
44
Koumpis
C
,
Dimitrakakis
C
,
Antsaklis
A
,
Royer
R
,
Zhang
S
,
Narod
SA
, et al.
Prevalence of BRCA1 and BRCA2 mutations in unselected breast cancer patients from Greece. Hered Cancer Clin Pract. 2011;9(1):10. doi:.https://doi.org/10.1186/1897-4287-9-10
45
Decker
B
,
Allen
J
,
Luccarini
C
,
Pooley
KA
,
Shah
M
,
Bolla
MK
, et al.
Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J Med Genet. 2017;54(11):732–41. doi:.https://doi.org/10.1136/jmedgenet-2017-104588
46
Kraus
C
,
Hoyer
J
,
Vasileiou
G
,
Wunderle
M
,
Lux
MP
,
Fasching
PA
, et al.
Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int J Cancer. 2017;140(1):95–102. doi:.https://doi.org/10.1002/ijc.30428
47
Kushnir
A
,
Laitman
Y
,
Shimon
SP
,
Berger
R
,
Friedman
E
. Germline mutations in RAD51C in Jewish high cancer risk families. Breast Cancer Res Treat. 2012;136(3):869–74. doi:.https://doi.org/10.1007/s10549-012-2317-9
48
Meindl
A
,
Hellebrand
H
,
Wiek
C
,
Erven
V
,
Wappenschmidt
B
,
Niederacher
D
, et al.
Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–4. doi:.https://doi.org/10.1038/ng.569
49
Somyajit
K
,
Mishra
A
,
Jameei
A
,
Nagaraju
G
. Enhanced non-homologous end joining contributes toward synthetic lethality of pathological RAD51C mutants with poly (ADP-ribose) polymerase. Carcinogenesis. 2015;36(1):13–24. doi:.https://doi.org/10.1093/carcin/bgu211
50
Somyajit
K
,
Subramanya
S
,
Nagaraju
G
. Distinct roles of FANCO/RAD51C protein in DNA damage signaling and repair: implications for Fanconi anemia and breast cancer susceptibility. J Biol Chem. 2012;287(5):3366–80. doi:.https://doi.org/10.1074/jbc.M111.311241
51
Gao
P
,
Ma
N
,
Li
M
,
Tian
QB
,
Liu
DW
. Functional variants in NBS1 and cancer risk: evidence from a meta-analysis of 60 publications with 111 individual studies. Mutagenesis. 2013;28(6):683–97. doi:.https://doi.org/10.1093/mutage/get048
52
Shimelis
H
,
Mesman
RLS
,
Von Nicolai
C
,
Ehlen
A
,
Guidugli
L
,
Martin
C
, et al.; for kConFab/AOCS Investigators; for NBCS Collaborators.
BRCA2 Hypomorphic Missense Variants Confer Moderate Risks of Breast Cancer. Cancer Res. 2017;77(11):2789–99. doi:.https://doi.org/10.1158/0008-5472.CAN-16-2568
53
Kinnersley
B
,
Kamatani
Y
,
Labussière
M
,
Wang
Y
,
Galan
P
,
Mokhtari
K
, et al.
Search for new loci and low-frequency variants influencing glioma risk by exome-array analysis. Eur J Hum Genet. 2016;24(5):717–24. doi:.https://doi.org/10.1038/ejhg.2015.170
54
Seal
S
,
Thompson
D
,
Renwick
A
,
Elliott
A
,
Kelly
P
,
Barfoot
R
, et al.; Breast Cancer Susceptibility Collaboration (UK). Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38(11):1239–41. doi:.https://doi.org/10.1038/ng1902
55
Cantor
SB
,
Bell
DW
,
Ganesan
S
,
Kass
EM
,
Drapkin
R
,
Grossman
S
, et al.
BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105(1):149–60. doi:.https://doi.org/10.1016/S0092-8674(01)00304-X
56
Cantor
S
,
Drapkin
R
,
Zhang
F
,
Lin
Y
,
Han
J
,
Pamidi
S
, et al.
The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101(8):2357–62. doi:.. Correction in: Natl Acad Sci USA. 2004;101(17):6834. doi:https://doi.org/10.1073/pnas.0308717101
57
Roeb
W
,
Higgins
J
,
King
MC
. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum Mol Genet. 2012;21(12):2738–44. doi:.https://doi.org/10.1093/hmg/dds101
58
Yamamoto
Y
,
Miyamoto
M
,
Tatsuda
D
,
Kubo
M
,
Nakagama
H
,
Nakamura
Y
, et al.
A rare polymorphic variant of NBS1 reduces DNA repair activity and elevates chromosomal instability. Cancer Res. 2014;74(14):3707–15. doi:.https://doi.org/10.1158/0008-5472.CAN-13-3037
59
di Masi
A
,
Viganotti
M
,
Polticelli
F
,
Ascenzi
P
,
Tanzarella
C
,
Antoccia
A
. The R215W mutation in NBS1 impairs gamma-H2AX binding and affects DNA repair: molecular bases for the severe phenotype of 657del5/R215W Nijmegen breakage syndrome patients. Biochem Biophys Res Commun. 2008;369(3):835–40. doi:.https://doi.org/10.1016/j.bbrc.2008.02.129
60
Schröder-Heurich
B
,
Bogdanova
N
,
Wieland
B
,
Xie
X
,
Noskowicz
M
,
Park-Simon
TW
, et al.
Functional deficiency of NBN, the Nijmegen breakage syndrome protein, in a p.R215W mutant breast cancer cell line. BMC Cancer. 2014;14(1):434. doi:.https://doi.org/10.1186/1471-2407-14-434
61
King
MC
,
Levy-Lahad
E
,
Lahad
A
. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA. 2014;312(11):1091–2. doi:.https://doi.org/10.1001/jama.2014.12483
62
Nelson
HD
,
Pappas
M
,
Zakher
B
,
Mitchell
JP
,
Okinaka-Hu
L
,
Fu
R
. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: a systematic review to update the U.S. Preventive Services Task Force recommendation. Ann Intern Med. 2014;160(4):255–66. doi:.https://doi.org/10.7326/M13-1684
63
Maxwell
KN
,
Domchek
SM
,
Nathanson
KL
,
Robson
ME
. Population Frequency of Germline BRCA1/2 Mutations. J Clin Oncol. 2016;34(34):4183–5. doi:.https://doi.org/10.1200/JCO.2016.67.0554
64
Linderman
MD
,
Nielsen
DE
,
Green
RC
. Personal Genome Sequencing in Ostensibly Healthy Individuals and the PeopleSeq Consortium. J Pers Med. 2016;6(2):14. doi:.https://doi.org/10.3390/jpm6020014
65
Manickam
K
,
Buchanan
AH
,
Schwartz
MLB
,
Hallquist
MLG
,
Williams
JL
,
Rahm
AK
, et al.
Exome Sequencing-Based Screening for BRCA1/2 Expected Pathogenic Variants Among Adult Biobank Participants. JAMA Netw Open. 2018;1(5):e182140. doi:.https://doi.org/10.1001/jamanetworkopen.2018.2140
66
Azzollini
J
,
Scuvera
G
,
Bruno
E
,
Pasanisi
P
,
Zaffaroni
D
,
Calvello
M
, et al.
Mutation detection rates associated with specific selection criteria for BRCA1/2 testing in 1854 high-risk families: A monocentric Italian study. Eur J Intern Med. 2016;32:65–71. doi:.https://doi.org/10.1016/j.ejim.2016.03.010
67
Cipollini
G
,
Tommasi
S
,
Paradiso
A
,
Aretini
P
,
Bonatti
F
,
Brunetti
I
, et al.
Genetic alterations in hereditary breast cancer. Ann Oncol. 2004;15(Suppl 1):i7–13. doi:.https://doi.org/10.1093/annonc/mdh651
68
Santonocito
C
,
Scapaticci
M
,
Guarino
D
,
Bartolini
A
,
Minucci
A
,
Concolino
P
, et al.
Identification of twenty-nine novel germline unclassified variants of BRCA1 and BRCA2 genes in 1400 Italian individuals. Breast. 2017;36:74–8. doi:.https://doi.org/10.1016/j.breast.2017.09.007
69
Nedelcu
R
,
Liede
A
,
Aubé
J
,
Finch
A
,
Kwan
E
,
Jack
E
, et al.
BRCA mutations in Italian breast/ovarian cancer families. Eur J Hum Genet. 2002;10(2):150–2. doi:.https://doi.org/10.1038/sj.ejhg.5200755
70
Zhang
S
,
Royer
R
,
Li
S
,
McLaughlin
JR
,
Rosen
B
,
Risch
HA
, et al.
Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol Oncol. 2011;121(2):353–7. doi:.https://doi.org/10.1016/j.ygyno.2011.01.020
71
Risch
HA
,
McLaughlin
JR
,
Cole
DE
,
Rosen
B
,
Bradley
L
,
Kwan
E
, et al.
Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68(3):700–10. doi:.https://doi.org/10.1086/318787
72
Kaufman
B
,
Laitman
Y
,
Gronwald
J
,
Lubinski
J
,
Friedman
E
. Haplotype of the C61G BRCA1 mutation in Polish and Jewish individuals. Genet Test Mol Biomarkers. 2009;13(4):465–9. doi:.https://doi.org/10.1089/gtmb.2009.0001
73
Zuradelli
M
,
Peissel
B
,
Manoukian
S
,
Zaffaroni
D
,
Barile
M
,
Pensotti
V
, et al.
Four new cases of double heterozygosity for BRCA1 and BRCA2 gene mutations: clinical, pathological, and family characteristics. Breast Cancer Res Treat. 2010;124(1):251–8. doi:.https://doi.org/10.1007/s10549-010-0853-8
74
Marchetti
C
,
De Leo
R
,
Musella
A
,
D’Indinosante
M
,
Capoluongo
E
,
Minucci
A
, et al.
BRCA Mutation Status to Personalize Management of Recurrent Ovarian Cancer: A Multicenter Study. Ann Surg Oncol. 2018;25(12):3701–8. doi:.https://doi.org/10.1245/s10434-018-6700-6
75
Green
RC
,
Berg
JS
,
Grody
WW
,
Kalia
SS
,
Korf
BR
,
Martin
CL
, et al.; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–74. doi:.https://doi.org/10.1038/gim.2013.73
76
Kalia
SS
,
Adelman
K
,
Bale
SJ
,
Chung
WK
,
Eng
C
,
Evans
JP
, et al.
Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55. doi:.. Correction in: Genet Med. 2017;19:484. doi:https://doi.org/10.1038/gim.2016.190
77
Hauke
J
,
Horvath
J
,
Groß
E
,
Gehrig
A
,
Honisch
E
,
Hackmann
K
, et al.
Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018;7(4):1349–58. doi:.https://doi.org/10.1002/cam4.1376
78
Natarajan
P
,
Gold
NB
,
Bick
AG
,
McLaughlin
H
,
Kraft
P
,
Rehm
HL
, et al.
Aggregate penetrance of genomic variants for actionable disorders in European and African Americans. Sci Transl Med. 2016;8(364):364ra151. doi:.https://doi.org/10.1126/scitranslmed.aag2367
79
Lawrence
L
,
Sincan
M
,
Markello
T
,
Adams
DR
,
Gill
F
,
Godfrey
R
, et al.
The implications of familial incidental findings from exome sequencing: the NIH Undiagnosed Diseases Program experience. Genet Med. 2014;16(10):741–50. doi:.https://doi.org/10.1038/gim.2014.29
80
Jurgens
J
,
Ling
H
,
Hetrick
K
,
Pugh
E
,
Schiettecatte
F
,
Doheny
K
, et al.
Assessment of incidental findings in 232 whole-exome sequences from the Baylor-Hopkins Center for Mendelian Genomics. Genet Med. 2015;17(10):782–8. doi:.https://doi.org/10.1038/gim.2014.196
81
Yang
Y
,
Muzny
DM
,
Xia
F
,
Niu
Z
,
Person
R
,
Ding
Y
, et al.
Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–9. doi:.https://doi.org/10.1001/jama.2014.14601
82
Olfson
E
,
Cottrell
CE
,
Davidson
NO
,
Gurnett
CA
,
Heusel
JW
,
Stitziel
NO
, et al.
Identification of Medically Actionable Secondary Findings in the 1000 Genomes. PLoS One. 2015;10(9):e0135193. doi:.https://doi.org/10.1371/journal.pone.0135193
83
Retterer
K
,
Juusola
J
,
Cho
MT
,
Vitazka
P
,
Millan
F
,
Gibellini
F
, et al.
Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704. doi:.https://doi.org/10.1038/gim.2015.148
84
Maillet
P
,
Chappuis
PO
,
Khoshbeen-Boudal
M
,
Sciretta
V
,
Sappino
AP
; SIAK Network for Cancer Predisposition Testing and Counseling. Twenty-three novel BRCA1 and BRCA2 sequence variations identified in a cohort of Swiss breast and ovarian cancer families. Cancer Genet Cytogenet. 2006;169(1):62–8. doi:.https://doi.org/10.1016/j.cancergencyto.2006.03.010
85
Garvin
AM
,
Attenhofer-Haner
M
,
Scott
RJ
. BRCA1 and BRCA2 mutation analysis in 86 early onset breast/ovarian cancer patients. J Med Genet. 1997;34(12):990–5. doi:.https://doi.org/10.1136/jmg.34.12.990
86
Lao
O
,
Lu
TT
,
Nothnagel
M
,
Junge
O
,
Freitag-Wolf
S
,
Caliebe
A
, et al.
Correlation between genetic and geographic structure in Europe. Curr Biol. 2008;18(16):1241–8. doi:.https://doi.org/10.1016/j.cub.2008.07.049
87
Bodian
DL
,
McCutcheon
JN
,
Kothiyal
P
,
Huddleston
KC
,
Iyer
RK
,
Vockley
JG
, et al.
Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PLoS One. 2014;9(4):e94554. doi:.https://doi.org/10.1371/journal.pone.0094554
88
Thompson
ER
,
Rowley
SM
,
Li
N
,
McInerny
S
,
Devereux
L
,
Wong-Brown
MW
, et al.
Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J Clin Oncol. 2016;34(13):1455–9. doi:.https://doi.org/10.1200/JCO.2015.63.7454
89
Tang
CS
,
Dattani
S
,
So
MT
,
Cherny
SS
,
Tam
PKH
,
Sham
PC
, et al.
Actionable secondary findings from whole-genome sequencing of 954 East Asians. Hum Genet. 2018;137(1):31–7. doi:.https://doi.org/10.1007/s00439-017-1852-1
90
Judkins
T
,
Rosenthal
E
,
Arnell
C
,
Burbidge
LA
,
Geary
W
,
Barrus
T
, et al.
Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer. 2012;118(21):5210–6. doi:.https://doi.org/10.1002/cncr.27556
91
Novembre
J
,
Johnson
T
,
Bryc
K
,
Kutalik
Z
,
Boyko
AR
,
Auton
A
, et al.
Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. doi:.https://doi.org/10.1038/nature07331
92
Einarsdóttir
K
,
Humphreys
K
,
Bonnard
C
,
Palmgren
J
,
Iles
MM
,
Sjölander
A
, et al.
Linkage disequilibrium mapping of CHEK2: common variation and breast cancer risk. PLoS Med. 2006;3(6):e168. doi:.https://doi.org/10.1371/journal.pmed.0030168
93
Kaufman
B
,
Laitman
Y
,
Gronwald
J
,
Winqvist
R
,
Irmejs
A
,
Lubinski
J
, et al.
Haplotypes of the I157T CHEK2 germline mutation in ethnically diverse populations. Fam Cancer. 2009;8(4):473–8. doi:.https://doi.org/10.1007/s10689-009-9269-1