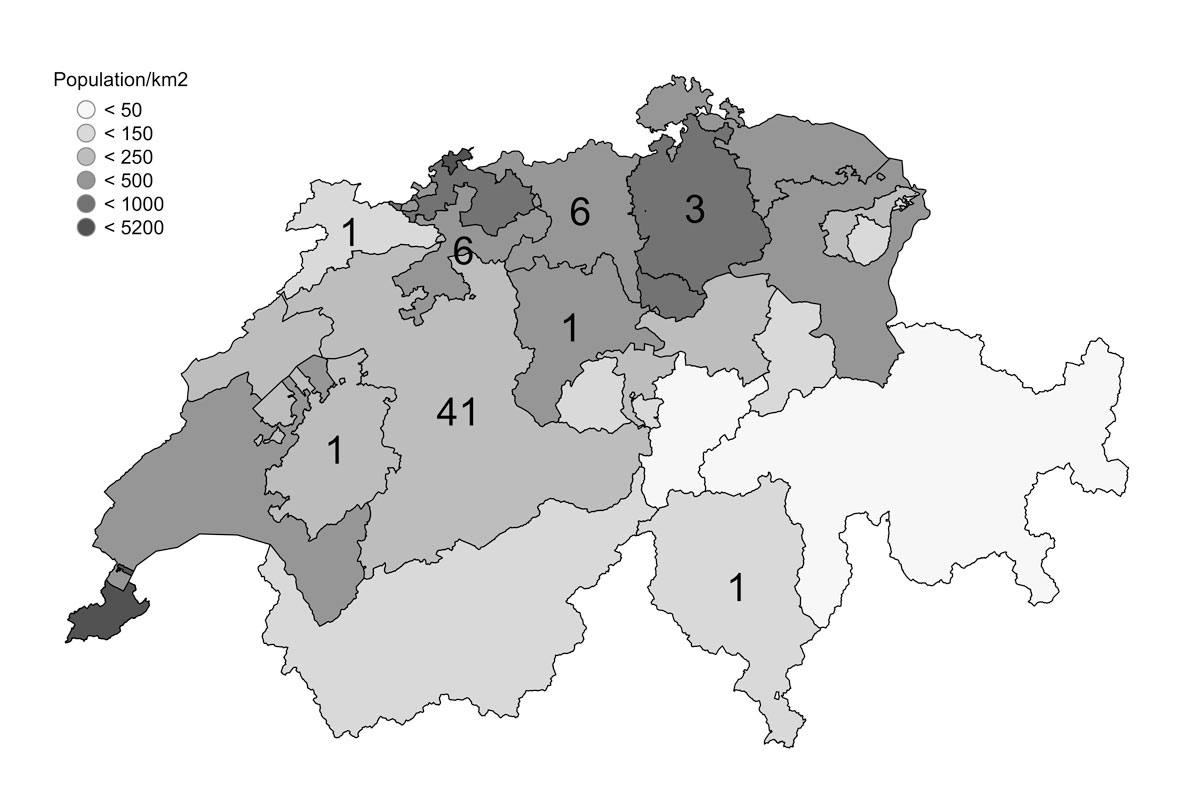
Figure 1a Cantons of Switzerland and their populations per km2 (grey), with the number of patients with Behçet’s syndrome per canton.
DOI: https://doi.org/10.4414/smw.2019.20072
Behçet’s syndrome is a chronic relapsing systemic autoimmune/autoinflammatory disease with a genetic background and inflammatory features. It was described by the Turkish dermatologist Hulusi Behçet in 1937 [1]. He suggested that the symptom triad of recurrent oral aphthae, genital ulcers and eye inflammation is a distinctive disease. Soon it was recognised that Behçet’s syndrome also affects the skin, joints, central nervous system, gastrointestinal tract and blood vessels. The most recent version of the Chapel Hill Classification for vasculitides includes Behçet’s syndrome [2]. In contrast to other vasculitides, Behçet’s syndrome affects all types and sizes of blood vessels.
In 1990, the International Study Group (ISG) published the first broadly accepted criteria for diagnosing Behçet’s syndrome [3]. These were revised in 2006 and in 2010, which resulted in the current “international criteria for Behçet’s disease” which require a score of ≥4 points, based on the following: oral aphthae, genital aphthae and eye involvement (each counting two points); skin, vascular, neurological manifestations and a positive Pathergy test (each counting one point) [4].
It is recognised that human leucocyte antigen B51 (HLA-B51) and other genetic markers are associated with Behçet’s syndrome [5]. In 1997, the concept of a T-helper cell 1 (Th1) driven disease was introduced [6], and in 2011 the role of Th17 lymphocytes was discussed [7]. In addition to these autoimmune features, many disease characteristics argue for a dysregulated inflammation. This is fuelled by an important role of proinflammatory cytokines [8] and by the beneficial therapeutic effect of tumour necrosis factor (TNF) blocking agents [9].
The prevalence of the disease shows a strong geographic variation. It is highest in the countries between the Mediterranean Sea and East Asia, the so-called silk road (up to 420/100,000) [10], but low in non-Mediterranean and non-Asian countries. A recent meta-analysis showed a prevalence of 3.3/100,000 for European countries [11].
Organ involvement and prognosis vary widely. The most prevalent mucocutaneous lesions are painful, but have a good prognosis and often heal without scarring. In contrast, eye involvement remains one of the leading causes of blindness in south-eastern countries. Vascular involvement, such as pulmonary aneurysm, is a life-threatening manifestation in young males and central nervous system involvement is a characteristic vascular or encephalitic complication in young females [12, 13].
As a result of the low prevalence of Behçet’s syndrome, few randomised controlled trials of immunosuppressive drugs have been performed. The current knowledge has been condensed in the first consensus paper of The European League Against Rheumatism (EULAR) published in 2008 [14]. An update is awaiting publication.
In Switzerland there are no published data regarding prevalence of Behçet’s syndrome, its manifestations or its management. Our cohort study addressed these issues.
The catchment area of the University Hospital is the canton of Bern and areas of neighbouring cantons adding up to 1.5–2 millions of inhabitants. In an attempt to reach all Behçet patients of our catchment area, we contacted the rheumatologists of the Swiss Society of Rheumatology and the heads of ophthalmology, dermatology, angiology, gastroenterology, neurology and gynaecology of the University Hospital of Bern.
Clinical and serological data were extracted from the patient charts of the outpatient clinic, the day clinic and the rheumatology ward of the University Hospital of Bern, Switzerland. In addition, patients were asked to answer a questionnaire, 5 refused to show up for a further clinical examination. The database was established 8 years ago and is run by the Clinical Trial Unit (CTU) of the University of Bern, Switzerland.
Behçet’s syndrome was categorised as “diagnosed” if the patient fulfilled the international criteria for Behçet’s syndrome [4]. “Suspicion” of Behçet’s syndrome was defined by clinical manifestations characteristic of Behçet’s syndrome but do not fulfil the criteria. Before inclusion into the study diagnosis were assessed and confirmed by the supervisors S. Adler or P. Villiger.
For calculation of prevalence, an anonymised list of the patients fulfilling the criteria (including the patients who refused to have their data analysed) was used. Based on the postal code, municipality was identified [15]. The population density of each municipality/canton was extracted from tables of the Bundesamt für Statistik (the Swiss Federal Office of Statistics) [16, 17]. Boundaries of each municipality/canton were retrieved from the Bundesamt für Landestopografie swisstopo [18]. The spatial distribution of patients was visualised on maps: (i) whole of Switzerland, (ii) Bern only, (iii) with population, and (iv) with population density. Additionally, we calculated the prevalence of non-Swiss patients originating from high-prevalence countries.
Regarding extrapolation of the findings of the Canton of Bern to Switzerland, the following facts need to be taken into account. The Canton of Bern lays in the geographic centre of the country, it covers areas of the Jura, the Midland and the Alps, it represents cities and rural areas, and the number of inhabitants represents one seventh of the whole population of Switzerland. On the other hand, the percentage of non-Swiss inhabitants is 40% lower in the Canton of Bern than in the country as a whole.
Data were exported from RedCAP. Data clearance and statistical analysis were performed by Lukas Bütikofer, statistician of the CTU of the University of Bern Switzerland. Continuous and categorical variables are presented as median (lower quartile, upper quartile) or number and percentage of patients. The number of patients for a specific variable refers to the number of non-missing observations. The difference between groups is indicated as a probabilistic index (PrI) for continuous and as a risk difference (RD) for categorical variables. The former refers to the probability that the variable of interest is larger in one group than in the other (50% indicates no difference between the groups).
The study was approved by the ethical commission of Bern, Switzerland. The study was performed in accordance with the Declaration of Helsinki. All patients gave written informed consent.
Patient characteristics are summarised in table 1. Of 52 patients, 46 (88%) fulfilled current criteria for Behçet’s syndrome, whereas 6 (12%) were diagnosed with Behçet’s syndrome but did not fulfil criteria. The median age on the day of analysis was 47.8 years (33.7, 56.9). Of the 52 patients, 28 (54%) were male and 31 (61%) were of Swiss origin. The median duration of the disease was 19 years (14.1, 27.7), the median age at symptom onset was 21.4 years (12.7, 35.6). The mean diagnosis delay (time between first symptom and diagnosis) was 8 years; (2.17, 17.00). The diagnostic delay was shorter in non-Swiss patients (median 7 years. 2.00, 17.00) than in patients with Swiss ethnicity (median 9 years; 2.17, 19,00; Prl 43.6%, 95% confidence interval [CI] 26.1–61.1; p = 0.45).
Table 1 Baseline characteristics of the patients.
| Variable | n (%) or mean | Number of subjects analysed |
|---|---|---|
| Mean age, years | 47.8 | 52 |
| Male sex, n (%) | 28 (54) | 52 |
| Diagnose confirmed, n (%) | 46 (88) | 52 |
| Ethnic origin Swiss, n (%) | 31 (61) | 51 |
| Years since symptom onset | 19 | 49 |
| Age at symptom onset | 21.4 | 49 |
| Years since diagnosis | 7.91 | 52 |
| Age at diagnosis | 36.1 | 52 |
| Years from symptom onset to diagnosis | 8 | 49 |
Sixty patients fulfilled the criteria for Behçet’s syndrome, 41 in the Canton of Bern and 19 in neighbouring cantons. Based on the geographical distribution of the patients, we calculated a prevalence of 4.03/100,000 inhabitants for the Canton of Bern. Figure 1 displays the number of patients per canton (a) and per community in the Canton of Bern (b).
Figure 1a Cantons of Switzerland and their populations per km2 (grey), with the number of patients with Behçet’s syndrome per canton.
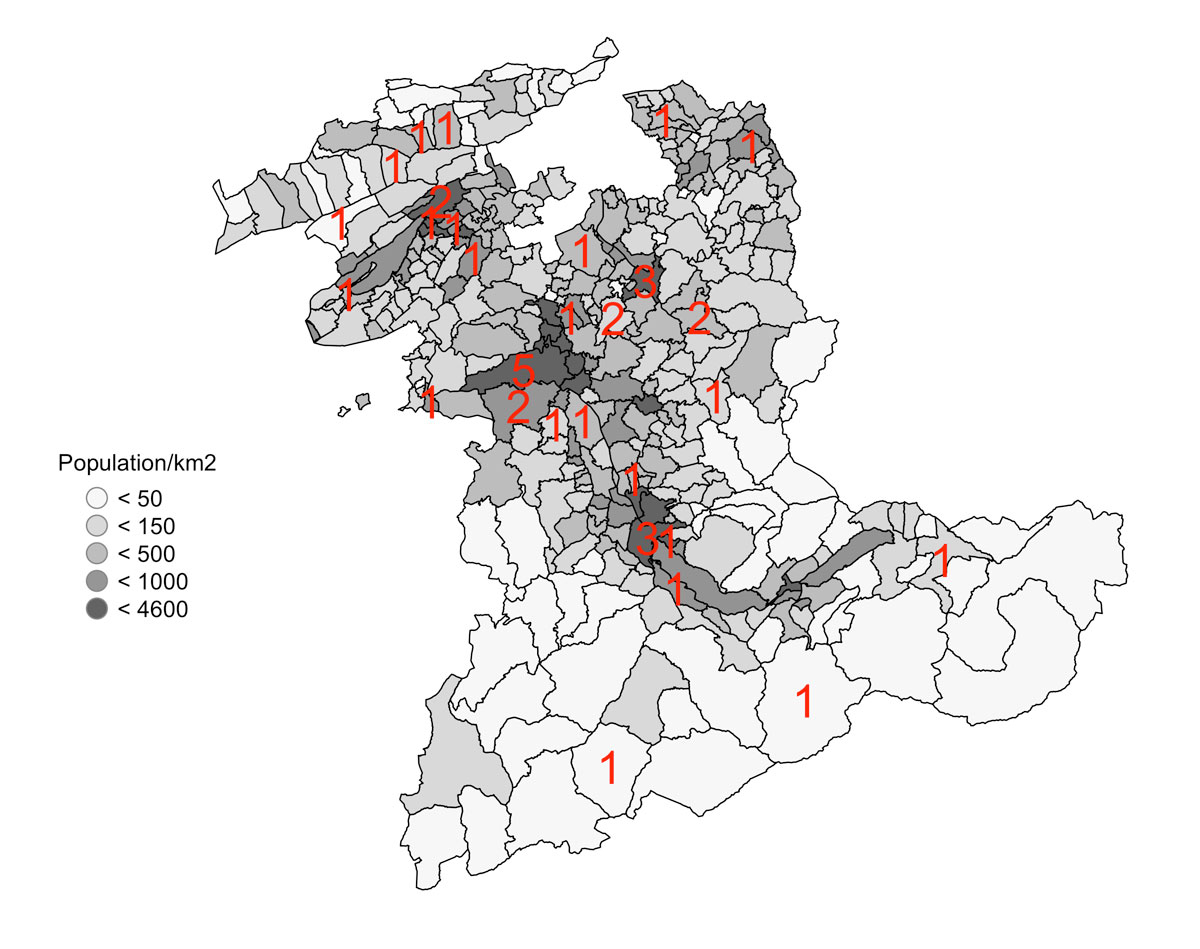
Figure 1b Communities of the Canton of Bern and their populations per km2 (grey), with the number of patients with Behçet’s syndrome per community in red.
As displayed in table 2, the inhabitants of Bern represent one seventh of the population of Switzerland. The percentage of foreigners is 37% lower in the Canton of Bern than in the whole country (15.5 and 24.6%, respectively), but the percentage of foreigners of high-risk countries is nearly identical (48.7 and 50.9%, respectively). The prevalence of Behçet’s syndrome in non-Swiss individuals originating from high-prevalence countries is 19.54/100,000.
Table 2 Prevalence of Behçet’s syndrome.
| Canton Bern | Switzerland | ||
|---|---|---|---|
| Number of residents | Total | 1,017,483 | 8,327,126 |
| Swiss | 860,017 | 6,278,459 | |
| Foreign | 157,466 | 2,048,667 | |
| High-risk countries* | 76,758 | 1.042,783 | |
| Foreign national / residents (%) | 15.5 | 24.6 | |
| High-risk countries / foreign nationals (%) | 48.7 | 50.9 | |
| Patients with Behçet’s syndrome | All | 41 | – |
| High-risk countries* | 15 | – | |
| Prevalence (/100,000 inhabitants) | All | 4.03 | – |
| High-risk countries* | 19.54 | – | |
* Countries along the silk road: Afghanistan, Albania, Andorra, Armenia, Azerbaijan, Bahrain, Bangladesh, Bhutan, Bosnia and Herzegovina, Cambodia, China, Croatia, Cyprus, Egypt, France, Georgia, Greece, Hungary, India, Indonesia, Iran, Iraq, Israel, Italy, Japan, Jordan, Kazakhstan, Korea (North), Kosovo, Kuwait, Kyrgyzstan, Laos, Lebanon, Libya, Macedonia, Malta, Moldova, Monaco, Montenegro, Myanmar, Oman, Pakistan, Palestine, Philippines, Qatar, Romania, San Marino, Saudi -Arabia, Serbia, Singapore, Spain, Sri Lanka, Syria, Taiwan (Chinese Taipei), Tajikistan, Tunisia, Turkey, Turkmenistan, United Arab Emirates, Uzbekistan, Vatican City, Vietnam, Yemen. High-risk countries of origin for patients in the cohort are in bold type. Data source: Ständige und nichtständige Wohnbevölkerung nach institutionellen Gliederungen, Geburtsort und Staatsangehörigkeit; 31 December 2015; BFS Reference code: px-x-0102010000_104; Availlable at: https://www.pxweb.bfs.admin.ch/pxweb/de/
Organ involvement and clinical characteristics are presented in table 3. The most frequent clinical manifestations were oral aphthae (including minor, major and herpetifom aphthae) (n = 48), skin involvement (including pseudo folliculitis, erythema nodosum, acneiform lesions, papulo-pustular lesions and positive Pathergy test) (n = 41), joint involvement (including arthralgia and arthritis) (n = 40) and genital aphthae (n = 35).
Table 3 Organ involvement (total n = 52).
| Clinical signs | n | % |
|---|---|---|
| Oral aphthae | 48 | 92 |
| Genital aphthae | 35 | 67 |
| Eye | 25 | 48 |
| Skin | 41 | 79 |
| Joint | 40 | 77 |
| Gastrointestinal tract | 16 | 31 |
| Central nervous system | 22 | 42 |
| Blood vessels | 27 | 52 |
| Pathergy phenomenon | 26 | 65* |
* tested in 40 patients
Vascular disorders (including deep vein thrombosis, thrombophlebitis, arterial thrombosis, acute arterial occlusion and aneurysm) were reported by 27 patients. Twent-five persons suffered from eye inflammation (uveitis, retinitis, episkleritis, skleritis and conjunctivitis, glaucoma and cataract). Neurological manifestations such as headache, peripheral neuropathy, thrombosis of the sinus vein, aseptic meningitis, optic neuritis, stroke and transient ischaemic attack, epileptic seizure and other symptoms were reported in 22 cases. Sixteen patients suffered from gastrointestinal symptoms such as abdominal pain, diarrhoea, melaena / blood in stool, perforation/surgery.
The interval between occurrence of a given organ manifestation and diagnosis of Behçet’s syndrome varied between −8 and +11 years (fig. 2). Whereas oral aphthae preceded diagnosis of Behçet’s syndrome for up to 8 years, vessel or central nervous affection occurred up to 11 years after diagnosis of Behçet’s syndrome.
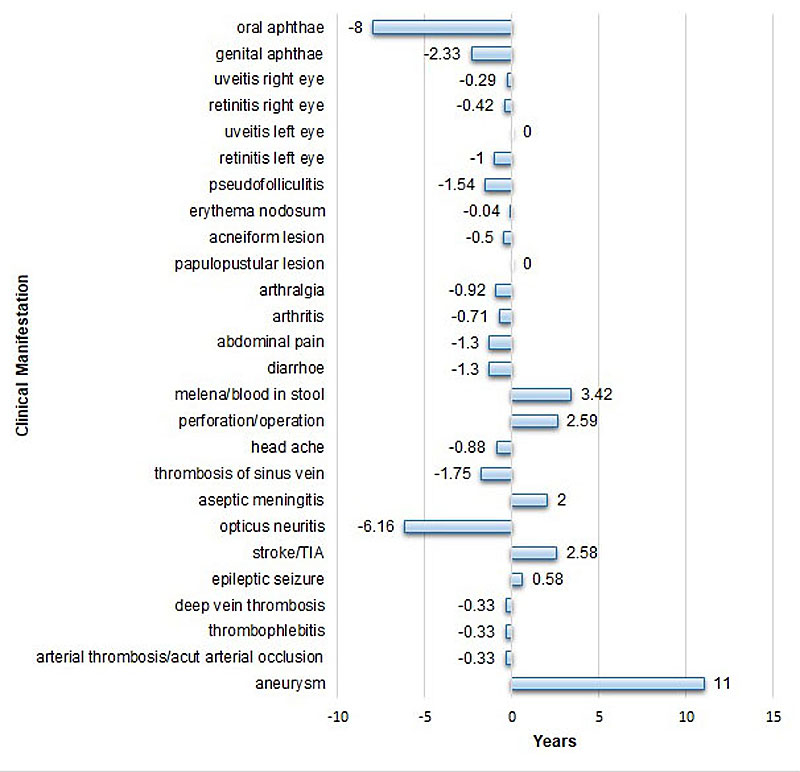
Figure 2 Time (in years) between diagnosis (0) and occurrence of disease manifestation. Negative values represent the delay of diagnosis after occurrence of disease manifestation; positive values indicate the occurrence of manifestation after diagnosis. Values represent the median time between diagnosis and the respective disease manifestation.
The levels of inflammatory parameters help to quantify disease activity; immunoglobulin levels increase in chronically active disease. The erythrocyte sedimentation rate (ESR) and of C-reactive protein levels (CRP) were normal: median values 8 mm/h (4.00, 12.00) and 3 mg/l (1.00, 4.00), respectively; also the median of the total immunoglobulin G (IgG) concentration in serum was normal at 10.4 g/l (8.6, 11.9). HLA B51 was positive in 18 of 40 analysed cases.
Fifty of the 52 patients had received medication during their disease course (table 4). Systemic glucocorticoids were the most commonly used drugs (80%), followed by TNF inhibitors (64%) and topical glucocorticoids (60%), azathioprine (60%), colchicine (52%) and nonsteroidal antirheumatic drugs (NSARs; 50%). Methotrexate was used by 20 patients (40%), ciclosporin by 12 patients (24%). Dapsone, thalidomide, cyclophosphamide, chlorambucil and sulfasalazine were prescribed for a few patients only. Anticoagulant medications were prescribed for almost half of the cases (48%).
Table 4 Medication (total n = 50).
| Medication | n | % |
|---|---|---|
| Topical glucocorticoids | 30 | 60 |
| Systemic glucocorticoids | 40 | 80 |
| Colchicine | 26 | 52 |
| Thalidomide | 2 | 4 |
| Dapsone | 1 | 2 |
| Azathioprine | 30 | 60 |
| Ciclosporin | 12 | 24 |
| Cyclophosphamide | 2 | 4 |
| Methotrexate | 20 | 40 |
| TNF inhibitors | 32 | 64 |
| Chlorambucil | 1 | 2 |
| Sulfasalazine | 1 | 2 |
Of the five currently available TNF-inhibitors, infliximab was prescribed for 21 patients, adalimumab for 10, etanercept for 5, golimumab for 3 and certolizumab for 1. TNF inhibitors were switched over time in seven cases (with two switches in one case), and could be stopped in 10 patients. TNF inhibitors were switched because of recurrent thrombophlebitis, heart insufficiency, insufficient efficacy and in one case because of an allergic reaction to infliximab. The TNF inhibitors were stopped because of lasting remission, pregnancy and insufficient effect, and once because of a complement-induced reaction to infliximab.
This is the first study analysing in detail the disease characteristics and the disease management of Behçet syndrome in Switzerland, as well as assessing its prevalence.
Based on the collected data of the Canton of Bern, the prevalence of Behçet’s syndrome in Switzerland is 4.03/100,000 inhabitants. This corresponds to the recently published data for Europe [11] and equals the figure published for southern Sweden [19]. Several arguments have to be taken into account when interpreting these figures: As other disciplines involved in diagnosing and treating Behçet’s syndrome were asked to share information about patients, it appears unlikely that many cases were missed. As all patients were reassessed at study inclusion, there should not be misdiagnoses. However, based on the long diagnostic delay there will exist not yet diagnosed cases. In summary, the true prevalence will be slightly higher than the calculated 4.03/100,000 inhabitants.
When extrapolating from the Canton of Bern to the whole country, one has to consider that the Canton of Bern lies in the middle of the country and it is large, representing approximately one seventh of the whole population. Remarkably, however, the percentage of non-Swiss inhabitants originating from high-prevalence countries is substantially lower in the Canton of Bern than in the whole country. This indicates that the prevalence of Behçet’s syndrome in Switzerland is higher than calculated. Further support for this interpretation is the fact that the calculated prevalence of Behçet’s syndrome in individuals from high-prevalence countries is 19.54/100,000.
The patient characteristics of this Swiss cohort resemble the characteristics of the cohorts of Western Europe and the United States. Cohorts in high-prevalence countries such as Turkey and Iran, however, are reported to have lower percentages of vascular and neurological involvement [12, 13]. Interestingly, disease pattern and disease severity are different in Turkish patients living in Turkey compared with Turkish patients living in Germany [20]. Indeed, the difference between German and Turkish patients living in Germany is small, pointing to the importance of environmental factors regarding disease phenotype.
A major issue of our findings is the diagnostic delay of 8 years. Remarkably, the difference in delay between immigrants and Swiss did not reach significance. This contrasts to Germany, where immigrant patients are diagnosed significantly earlier than German patients [20]. This difference was explained by the fact that physicians are aware of Behçet’s syndrome in patients from the countries of the Silk Road. It appears likely that the difference in Switzerland did not reach significance because of the lower patient numbers. The delay in diagnosis of disease manifestations that eventually lead to severe damage or even to sudden death is of particular concern. This is all the more unacceptable as, as a result of recent progress regarding pathogenesis and treatment, there is an increase in therapeutic options to control the disease and to prevent severe damage [14, 21, 22].
We observed an interesting difference in diagnostic delay as a function of disease manifestation. Patients with mild mucocutaneous disease may be misdiagnosed until more severe organ involvement occurs. On the other hand, severe vascular disease often follows milder disease manifestations, exemplified by a patient who had 11 years of known disease until arterial aneurysms developed. Thus, earlier recognition of Behçet’s syndrome would help to avoid potentially fatal disease complications. This is corroborated by our recent case series of patients with severe vascular Behçet [22]. It illustrates how ignorance of Behçet’s syndrome may lead to life-threatening complications. In conclusion, it supports the notion that patients at highest risk of severe organ involvement – young male patients with an established diagnosis – should be informed about the signs of major organ involvement and should be followed up on a regular basis by physicians experienced with this form of vasculitis.
Several findings are of interest regarding treatment. Colchicine, which is largely used in south-eastern and Asian countries [23], was prescribed for half of the patients only. This may be explained by the off-label status of this drug in Switzerland, and also by the fear of patients, physicians and pharmacists of potential (but rare) side effects. Interferon α, a drug mainly proposed for treatment of ocular manifestations [24, 25], is used rarely in Switzerland and in none of the patients of our cohort. On the other hand, TNF blocking agents are prescribed liberally. There was concern that TNF inhibition, in contrast to interferon therapy, would need to be continued for years, leading to substantial costs and risks. It is remarkable that TNF inhibition was stopped in almost 30% of our cases, in several because of lasting remission. A further example is a patient with potentially fatal pulmonary aneurysms [26]. After achieving complete remission, infliximab was stopped and azathioprine was introduced to successfully maintain remission. Another surprise regarding medication is the frequent prescription of methotrexate, which is not listed in the EULAR recommendations at all [14]. As referring physicians are informed at inclusion of patients into the registry about our conclusions and get therapeutic recommendations, and also because many patients are now managed in our out-patient clinic, treatment decisions largely reflect the management by our own senior physicians. Thus, for TNF inhibitors and methotrexate, rheumatologists appear to extrapolate from positive experience in the treatment of autoimmune arthritides.
In summary, the data show a prevalence of Behçet’s syndrome of 4.03/100,000 inhabitants in Switzerland. They document an alarming diagnostic delay, which suggests a need for measures to increase awareness of this disease. Furthermore, they report disease management that only partially follows current EULAR guidelines. Taken together, the data suggest that establishing a national registry for Behçet’s syndrome might help to improve diagnosis and management and to address unmet needs of patients.
We thank Leonie Villiger, Institute of Geography, University of Bern and Luka Bütikofer, Clinical Trial Unit (CTU), University of Bern, for statistical assistance.
The study was financed by the research funds of the Division of Rheumatology of the Department of Rheumatology, Immunology and Allergology, University Hospital of Bern, Bern, Switzerland
The authors declare no conflict of interest relevant to this article.
1 Behçet H . Über rezidivierende aphtöse, durch Virus verursachte Geschwüre am Mund, am Auge und an den Genitalien. Dermatol Monatsschr. 1937;105:1152–7.
2 Jennette JC , Falk RJ , Bacon PA , Basu N , Cid MC , Ferrario F , et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11. doi:.https://doi.org/10.1002/art.37715
3 Wechsler B , Davatchi F , Mizushima Y , et al.; International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet. 1990;335(8697):1078–80.
4 Davatchi F , Assaad-Khalil S , Calamia KT , Crook JE , Sadeghi-Abdollahi B , Schirmer M , et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–47 https://doi.org/10.1111/jdv.12107
5 Meguro A , Inoko H , Ota M , Katsuyama Y , Oka A , Okada E , et al. Genetics of Behçet disease inside and outside the MHC. Ann Rheum Dis. 2010;69(4):747–54. doi:.https://doi.org/10.1136/ard.2009.108571
6 Turan B , Gallati H , Erdi H , Gürler A , Michel BA , Villiger PM . Systemic levels of the T cell regulatory cytokines IL-10 and IL-12 in Bechçet’s disease; soluble TNFR-75 as a biological marker of disease activity. J Rheumatol. 1997;24(1):128–32.
7 Geri G , Terrier B , Rosenzwajg M , Wechsler B , Touzot M , Seilhean D , et al. Critical role of IL-21 in modulating TH17 and regulatory T cells in Behçet disease. J Allergy Clin Immunol. 2011;128(3):655–64. doi:.https://doi.org/10.1016/j.jaci.2011.05.029
8 Lopalco G , Lucherini OM , Vitale A , Talarico R , Lopalco A , Galeazzi M , et al. Putative Role of Serum Amyloid-A and Proinflammatory Cytokines as Biomarkers for Behcet’s Disease. Medicine (Baltimore). 2015;94(42):e1858. doi:.https://doi.org/10.1097/MD.0000000000001858
9 Desbois AC , Vallet H , Domont F , Comarmond C , Cacoub P , Saadoun D . Management of severe complications in Behçet’s disease with TNF inhibitors. Expert Opin Biol Ther. 2017;17(7):853–9. doi:.https://doi.org/10.1080/14712598.2017.1328496
10 Davatchi F , Chams-Davatchi C , Shams H , Shahram F , Nadji A , Akhlaghi M , et al. Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol. 2017;13(1):57–65. doi:.https://doi.org/10.1080/1744666X.2016.1205486
11 Maldini C , Druce K , Basu N , LaValley MP , Mahr A . Exploring the variability in Behçet’s disease prevalence: a meta-analytical approach. Rheumatology (Oxford). 2018;57(1):185–95. doi:.https://doi.org/10.1093/rheumatology/kew486
12 Tursen U , Gurler A , Boyvat A . Evaluation of clinical findings according to sex in 2313 Turkish patients with Behçet’s disease. Int J Dermatol. 2003;42(5):346–51. doi:.https://doi.org/10.1046/j.1365-4362.2003.01741.x
13 Alpsoy E , Dönmez L , Önder M , Gunasti S , Usta A , Karincaoglu Y , et al. Clinical features and natural course of Behçet’s disease in 661 cases: a multicentre study. Br J Dermatol. 2007;157(5):901–6. doi:.https://doi.org/10.1111/j.1365-2133.2007.08116.x
14 Hatemi G , Silman A , Bang D , Bodaghi B , Chamberlain AM , Gul A , et al.; EULAR Expert Committee. EULAR recommendations for the management of Behçet disease. Ann Rheum Dis. 2008;67(12):1656–62. doi:.https://doi.org/10.1136/ard.2007.080432
15Table of the Bundesamt für Statistik, as of 31 December 2016: Bilanz der ständigen Wohnbevölkerung nach institutionellen Gliederungen, Staatsangehörigkeit (Kategorie), Geschlecht und demographischen Komponenten. Published 30 August 2017; BFS-Nummer: px-x-0102020000_201; Available at: https://www.bfs.admin.ch/bfs/de/home/statistiken/kataloge-datenbanken/tabellen.assetdetail.3262106.html
16Table of the Bundesamt für Statistik, as of 31 December 2015; Regionalporträts 2017: Kennzahlen aller Gemeinden. Published 18 May 2017; BFS-Nummer: je-d-21.03.01; Available at: https://www.bfs.admin.ch/bfs/de/home/statistiken/regionalstatistik/regionale-portraets-kennzahlen/gemeinden/daten-erlaeuterungen.assetdetail.2422865.html
17Table of the Bundesamt für Statistik. as of 31 December 2015; Ausgewählte Indikatoren im regionalen Vergleich, 2017 (Kantone). Published 28 March 2017; BFS-Nummer: je-d-21.03.02; Available at: https://www.bfs.admin.ch/bfs/de/home/statistiken/regionalstatistik/regionale-portraets-kennzahlen/kantone.assetdetail.1922812.html
18Shapefiles of the Bundesamt für Landestopografie swisstopo; „swissBOUNDARIES3D“, as of January 2018; Link: https://shop.swisstopo.admin.ch/de/products/landscape/boundaries3D
19 Mohammad A , Mandl T , Sturfelt G , Segelmark M . Incidence, prevalence and clinical characteristics of Behcet’s disease in southern Sweden. Rheumatology (Oxford). 2013;52(2):304–10. doi:.https://doi.org/10.1093/rheumatology/kes249
20 Kötter I , Vonthein R , Müller CA , Günaydin I , Zierhut M , Stübiger N . Behçet’s disease in patients of German and Turkish origin living in Germany: a comparative analysis. J Rheumatol. 2004;31(1):133–9.
21 Hatemi G , Melikoglu M , Tunc R , Korkmaz C , Turgut Ozturk B , Mat C , et al. Apremilast for Behçet’s syndrome--a phase 2, placebo-controlled study. N Engl J Med. 2015;372(16):1510–8. doi:.https://doi.org/10.1056/NEJMoa1408684
22 Adler S , Baumgartner I , Villiger PM . Behçet’s disease: successful treatment with infliximab in 7 patients with severe vascular manifestations. A retrospective analysis. Arthritis Care Res (Hoboken). 2012;64(4):607–11. doi:.https://doi.org/10.1002/acr.21557
23 Yurdakul S , Mat C , Tüzün Y , Ozyazgan Y , Hamuryudan V , Uysal O , et al. A double-blind trial of colchicine in Behçet’s syndrome. Arthritis Rheum. 2001;44(11):2686–92. doi:.https://doi.org/10.1002/1529-0131(200111)44:11<2686::AID-ART448>3.0.CO;2-H
24 Kötter I , Zierhut M , Eckstein AK , Vonthein R , Ness T , Günaydin I , et al. Human recombinant interferon alfa-2a for the treatment of Behçet’s disease with sight threatening posterior or panuveitis. Br J Ophthalmol. 2003;87(4):423–31. doi:.https://doi.org/10.1136/bjo.87.4.423
25 Alpsoy E , Durusoy C , Yilmaz E , Ozgurel Y , Ermis O , Yazar S , et al. Interferon alfa-2a in the treatment of Behçet disease: a randomized placebo-controlled and double-blind study. Arch Dermatol. 2002;138(4):467–71. doi:.https://doi.org/10.1001/archderm.138.4.467
26 Baki K , Villiger PM , Jenni D , Meyer T , Beer JH . Behcet’s disease with life-threatening haemoptoe and pulmonary aneurysms: complete remission after infliximab treatment. Ann Rheum Dis. 2006;65(11):1531–2. doi:.https://doi.org/10.1136/ard.2005.045195
The study was financed by the research funds of the Division of Rheumatology of the Department of Rheumatology, Immunology and Allergology, University Hospital of Bern, Bern, Switzerland
The authors declare no conflict of interest relevant to this article.