Chronic rhinosinusitis in unified airway disease: surfactant proteins as mediators of respiratory immunity
DOI: https://doi.org/10.4414/smw.2019.20104
George T.
Noutsiosa, Saurabh
Sharmab
aSchool of Mathematical and Natural Sciences, Arizona State University, Phoenix, USA
bDepartment of Head and Neck Surgery, Kaiser Permanente Medical Center, Roseville, USA
Summary
PURPOSE OF REVIEW
The aim of this review is to describe the co-occurrence of chronic rhinosinusitis (CRS) with other inflammatory illnesses of the lower respiratory system characterised by airway obstruction and hyperresponsiveness, such as asthma, cystic fibrosis (CF), and chronic obstructive pulmonary disease (COPD) in the context of the unified airway disease (UAD). We also sought to discuss the novel role of surfactant proteins as mediators of innate immunity in the sinonasal epithelium and their potential as therapeutic interventions.
RECENT FINDINGS
Different epidemiological and physiological studies in CRS and asthma have outlined that there are common clustering patterns in the phenotypes/endotypes of both diseases, reinforcing the notion of the UAD. Also, surfactant proteins A (SP-A) and SP-D have now emerged as novel innate immunity molecules in bacterial sinusitis and allergic fungal sinusitis patients, respectively.
SUMMARY
CRS and asthma coexist and are interconnected. Therefore, management of CRS and asthma must be jointly carried out as one functional entity. SP-A and SP-D bridge the innate and adaptive immunity mechanisms of the sinonasal epithelium to bring together a well-orchestrated mechanism that effectively fights pathogens. The use of SP-A to ameliorate the innate immune responses in CRS is a new concept and is likely to lead to new horizons in CRS therapeutic regimens.
Abbreviations:
- AM
-
alveolar macrophage
- AR
-
allergic rhinitis
- asialoGM1
-
asialoganglioside ganliotetraosylceramide
- CCL2
-
C-C motif chemokine Ligand 2
- CCL8
-
C-C motif chemokine Ligand 8
- CD4 T
-
T helper lymphocytes
- CD8 T
-
cytotoxic T cells
- CDHR3
-
cadherin-related family member 3
- CF
-
cystic fibrosis
- c-Jun
-
transcription factor Activator protein 1
- c-MET
-
tyrosine–protein kinase Met
- COPD
-
chronic obstructive pulmonary disease
- CRS
-
chronic rhinosinusitis
- CT
-
computed tomography
- CX3CR1
-
C-X3-C motif chemokine receptor 1
- CXCL5
-
C-X-C motif chemokine 5
- DCs
-
dendritic cells
- DC-SIGN
-
dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin
- ECP
-
eosinophilic cationic protein
- EGFR
-
epidermal growth factor receptor
- ENA-78
-
epithelial-derived neutrophil-activating peptide 78
- Foxp3
-
forkhead box P3
- GCP-2
-
granulocyte chemotactic protein 2
- G-CSF
-
granulocyte colony-stimulating factor
- GM-CSF
-
granulocyte–macrophage colony-stimulating factor
- GRO-α
-
growth-regulated protein/melanoma growth stimulatory activity
- HA
-
haemagglutinin
- HEF
-
haemagglutinin-esterase fusion
- HRV
-
human rhinovirus
- HSPG
-
heparan sulfate proteoglycan
- ICAM-1
-
intracellular adhesion molecule 1
- IFN
-
interferon
- IgE
-
immunoglobulin E
- IL
-
interleukin
- IL-2R
-
interleukin-2 receptor
- ILC2
-
type 2 innate lymphoid cells
- ILC3
-
type 3 innate lymphoid cells
- LDLDR
-
low-density lipoprotein receptor
- LP
-
lipoprotein
- LPS
-
lipopolysaccharides
- L-SIGN
-
liver/lymph node-specific ICAM-3-grabbing nonintegrin
- LTA
-
lipoteichoic acid
- MCP-1
-
monocyte chemoattractant protein-1
- MGL
-
macrophage galactose-type lectin
- MHC
-
major histocompatibility complex
- MMP
-
matrix metalloproteinases
- MMR
-
macrophage mannose receptor
- Mφ
-
macrophages
- NF-κB
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- NKCs
-
natural killer cells
- NOD1
-
nucleotide-binding oligomerisation domain-containing protein 1
- p50
-
transcription factor p50
- p65
-
transcription factor p65
- PAMPs
-
pathogen-associated molecular patterns
- PMNs
-
polymorphonuclear neutrophils
- PG
-
peptidoglycan
- RANTES
-
chemokine (C-C motif) ligand 5 (also CCL5)
- RSV
-
respiratory syncytial virus
- SIGN-R1
-
specific ICAM-3-grabbing nonintegrin-related 1
- SNE
-
sinonasal epithelium
- SP-A
-
surfactant protein A
- SP-D
-
surfactant protein D
- T-cell
-
T lymphocyte
- TF2
-
transcription factor 2
- TGF-β
-
transforming growth factor β
- Th cells
-
T helper cells
- TLR
-
toll-like receptor
- TNF-α R1
-
tumour necrosis factor receptor 1
- UAD
-
unified airway disease
- URIs
-
upper respiratory tract infections
Introduction
Chronic rhinosinusitis (CRS) and asthma are inflammatory, chronic disorders characterised by recurrent, often enervating symptoms leading to frequent physician visits, hospitalisations and high prescription costs. Disease management has a high societal cost, estimated around $5 billion per year for CRS [1] and $56 billion for asthma only in the USA [2]. CRS and asthma have high rates of co-occurring disease, with up to 50% of those with CRS having asthma and up to 80% of those with severe asthma suffering from CRS. Although the mortality risk from these inflammatory diseases is low, the morbidity is significant with a high impact in quality of life of individuals with CRS, often exceeding that of congestive heart failure, chronic obstructive pulmonary disease (COPD), and Parkinson’s disease. In addition, when these diseases co-occur, the societal burden is magnified, often leading to recalcitrant disease and increased medical costs.
Epidemiology of airway diseases
In the past few years, a connection between upper and lower airways has been recognised leading to treatment of the entire respiratory system from the nose to the distal bronchioles and the alveolar sacs as one integrated system. This is supported by the fact that patients with upper airway disease have a higher prevalence of lower tract disease and vice versa. In 1997, Corren et al. reviewed this relationship and found that 78% of asthmatics have nasal symptoms [3]. Thirty-eight percent of patients with rhinitis have asthma. Other studies have shown threefold increase in asthma patients with allergic rhinitis (AR) compared to nonallergic controls [4, 5]. In addition, the severity of nasal symptoms has been found to correlate with the severity of asthma symptoms and this again further stresses the link between the two conditions [6]. Asthmatic children with AR were shown to have a higher risk of hospital admissions and healthcare costs [7]. Effective management of AR symptoms was noted to help improve the asthma symptoms and vice versa [8]. Therefore, managements of rhinitis and asthma have to be jointly carried out as one functional entity – “the unified airway disease” (UAD).
Common pathophysiology of upper and lower airway diseases
Recently, it has been suggested that CRS and the lower respiratory disease are interconnected and arise from same underlying mechanisms (atopic or not). Although allergy status and age of onset of atopy have been shown to influence the linkage between asthma and rhinitis, atopy is not required for this relationship. Non-AR has been also recognised an independent risk factor for asthma [4, 9]. The exact basis for this connection and pathophysiological mechanism is still a matter of investigation. The sinuses filter, moisten, and warm the ambient air before it is delivered to the lung that is the primary organ of the respiratory system. With severe rhinitis leading to nasal obstruction, theoretically inspiration of unfiltered and unconditioned air could exacerbate any underlying lung disease. It is also suggested that this connection is systemic, with nasal or bronchial inflammation propagating through systemic mediators [5, 6]. As such, the molecular mechanisms and aetiology that unite these two diseases are not yet fully understood. Human rhinoviruses (HRV) are the most common causes of upper respiratory tract infections (URIs) and are implicated as one of the inciting factors for both CRS and asthma exacerbations [10, 11]. Additionally, several studies have identified genome–virome interactions that shed some light in our understating of the susceptibility or protection from HRV infections [12].
Upper and lower airway disease, same condition but different location
Clinical definitions of CRS and asthma
The clinical definition and histopathologic changes seen in CRS and asthma are remarkably similar with the major common element being that of inflammation of airways. Asthma is clinically defined as the state of chronic inflammation of the lower airways that can cause swelling and spasm of the airways leading to shortness of breath, chest tightness, coughing and wheezing. Apart from these patient-reported subjective findings, objective diagnostic criteria include chest radiography and spirometrical tests that access the pulmonary function by measuring airflow rates (i.e. volume of air that passes through lungs per unit time) and forced expiratory volumes (i.e. vital capacity or the volume of air that is expelled from the lung during a maximally forced expiratory effort after taking the deepest possible breath) [13]. In severe case of asthma, computed tomography (CT) findings may include bronchial wall thickening and bronchiectasis [14, 15].
CRS on the other hand clinically is defined as a sinonasal inflammation persisting for greater than 12 weeks despite treatment attempts, with a variety of symptoms such as nasal obstruction, mucopurulent drainage, facial pain, and olfactory malfunction (subjective findings). Objective findings include nasal endoscopy, which can provide extensive examination and visualisation of the nasal cavity including the oedema, mucus, and presence of polyps, and CT scan to assess the extent of mucosal thickening or sinus opacification.
CRS endotypes
CRS is currently phenotypically classified as CRS with polyps and CRS without polyps. However, this classification does not adequately represent the spectrum of disease as there is still heterogeneity within the two groups. Attempts have been made to differentiate CRS endotypes based on the expression levels of different inflammatory cell mediators [16, 17]. High levels of eosinophils, eotaxin, interleukin 5 (IL-5), immunoglobulin E (IgE), T-cell activation, IL-2 receptor α, and downregulation of transforming growth factor β (TGF-β) and forkhead box P3 (Foxp3) protein, and polarisation towards T helper cells (Th cells) 2 responses have been found for allergy-induced CRS with polyps in Caucasians (table 1). The inflammatory cell mediators are slightly different for CRS with polyps in Asians where neutrophilic infiltration and Th1/Th17 cell biased responses are observed (table 1). Nonallergy-induced CRS without polyps is associated with high levels of TGF-β, interferon γ (IFN-γ), decreased T-regulatory function, and Th1 polarisation [18]. Despite the fact that the clinical classifications of CRS with or without polyps reflects their differences at the molecular levels (Th2-mediated for polyps and Th1-mediated without polyps), these classifications continue to be broad and fail to account for the spectrum of clinical phenotypes present in CRS or help predict successful therapeutic interventions [19].
Table 1 Differentiation of the unified airway diseases based on the expression levels of inflammatory cell mediators
CRS with
nasal polyps
|
CRS without nasal polyps
|
Allergic
asthma
|
CF with nasal polyps
|
|
Caucasians
|
Asians
|
| T-cell activation |
T-cell activation |
|
|
T-cell activation |
| Plasma cells (↑) |
|
|
|
Plasma cells (↑) |
| Eosinophil infiltration |
Neutrophils infiltration |
|
|
Neutrophils infiltration |
| ECP |
|
|
|
|
| IL-2Rα (↑) |
IL-2Rα (↑) |
|
|
|
| Foxp3 (↓) |
Foxp3 (↓) |
|
|
|
| TGF-β (↓) |
TGF-β (↓) |
TGF-β (↑) |
|
|
|
|
IFN-γ (↑) |
|
|
| Th2 polarisation |
Th1/Th17 polarisation |
Th1 polarisation |
Th2 polarisation |
|
|
|
|
IL-4 (↑) |
|
| IL-5 (↑) |
|
|
IL-5 (↑) |
|
|
|
|
IL-8
(↑) |
|
|
|
|
IL-9 (↑) |
|
|
|
|
IL-13 (↑) |
|
| IgE (↑) |
|
|
IgE (↑) |
|
Asthma endotypes
Asthma is also a complex syndrome than has been differentiated into phenotypes and endotypes. Five different phenotypes include allergic early-onset, eosinophilic late-onset, exercise-induced, obesity-related, and neutrophilic asthma [20]. Four different endotypes identified: early-onset severe allergic asthma, late-onset persistent eosinophilic asthma, aspirin-exacerbated airway disease, and allergic bronchopulmonary mucosal asthma [21]. It has been reported that a number of environmental and genetic factors in early childhood can favour Th2 cell responses and select against Th1 responses leading to inflammation of the lower airways, production of IL-4, IL-5, IL-8 and IL-13, and increased levels of IgE, eosinophils and eosinophilic cationic protein, which in turn can lead to asthma exacerbations (table 1). In some cases, the environmental stimuli can lead to Th1 cell responses that produce anti-inflammatory cytokines and these render protection against asthma [22]. It is striking that both cases, in allergy-induced CRS with polyps and in allergic asthma, Th2 cell responses are involved. In nonallergic CRS without polyps, the Th1 responses are associated with a nonasthmatic phenotype. These reinforce further the notion that the two conditions CRS and asthma actually coexist and are interconnected.
CRS phenotypes and endotypes clustering efforts for stratified medical treatments
Considerable effort has been placed to better understand these underlying pathologies and separate the patients into individual “clusters” in order to investigate possible disease phenotypes and endotypes [23–25]. However, no consensus has been formed. Multiple different clusters have been suggested by different groups based on different clinical, histological, and cytokine biomarkers to provide insight into the underlying pathophysiology [26–28].
Tomassen et al. attempted to cluster CRS patients based on inflammatory markers using cytokines and found few of these clusters correlated with the already defined phenotypes, that is, CRS with and without polyps. Using unsupervised hierarchical clustering methods (i.e. dendrograms that do not take any of the experimental variables such as phenotype, tissue, and treatment into account while clustering), Soler et al. identified phenotypic subgroups within CRS based on just three clinical variables (productivity loss, sinonasal outcome test score, and age) that was shown to provide prognostic information regarding a potential surgical candidate versus continued medical management in their follow-up study [29]. This analysis helped characterised patients into different groups that can be used to assess their clinical response to surgical intervention. Although it failed to illustrate the underlying pathophysiological mechanisms, it has the potential to help guide treatments for patients with CRS. One major drawback with the current classification system is that it rarely helps prognosticate patient outcome after surgery or medical treatment. However, a significant gap continues to exist, and further research is required to identify and understand individual clusters. A better understanding of such endotypes and their relationship to distinct underlying disease mechanisms should enable identification of novel therapeutic targets and facilitate the aim of stratified medicine.
Common clustering patterns in CRS and asthma
Histopathologically, in both CRS and asthma, the normal airway epithelium of columnar ciliated cells gets damaged, demonstrating enhanced paracellular permeability, disrupted epithelial repair mechanisms and inflammation. The airway epithelia transform to a hypersecretory mucus state with increased proliferation rates of goblet cells, hypertrophy of submucosal glands, basement membrane thickening, smooth muscle hypertrophy, and a thick mucus layer on the apical surface [30].
It has been recently realised that the severity and chronic state of both CRS and asthma cannot be explained only by the upper and lower airway inflammatory state, so it has been postulated that the above-mentioned changes lead to airway remodelling of nasal, bronchial and lung tissues, which are not reversible and progress to loss of the overall respiratory function [31]. In severe cases of asthma, a number of cytokines (specifically, matrix metalloproteinase 9, serum soluble intracellular adhesion molecule 1 (ICAM-1), transforming growth factor β, tissue inhibitor of matrix metalloproteinase 1, IL-4, IL-5, IL-8, and IL-13) lead to increased collagen deposition within the lamina propia, thickening of lamina reticularis, generation of myofibroblasts, and smooth muscle hypertrophy [32]. We postulate a similar pattern takes place at the nasal epithelium. The exact mechanisms that lead to inflammation and the subsequent airway changes as well as the factors that predispose some patients to these conditions are still not fully understood. It is believed that it is the interplay of genetic factors, environmental stimuli, and epigenetic factors that can either favour Th2 biased cell responses leading to asthma and atopic CRS or select against it and render protection against these diseases.
Human rhinovirus in unified airway disease
HRV infections not only can cause sinusitis in both young children and adults but it is well established that it is the bio-agent that is the most associated with childhood asthma [33]. URI are associated with at least 80% of acute asthma exacerbations in children and adolescents and about 60% of exacerbations in adults. In each case, HRV is the dominant viral pathogen, accounting for some 60% of all virus-induced exacerbation [34]. There are several subtypes of HRV. HRV-A and HRV-B cause URI and it has been shown that ICAM-1 is the major receptor for the SNE [35], and the low-density lipoprotein receptor (LDLDR) is the secondary receptor [36]. HRV-C, which is the most aggressive subtype of HRV, enters the SNE through cadherin-related family member 3 (CDHR3) [37, 38]. Generally, it is well accepted that most subtypes of HRVs proliferate best at the SNE temperatures of 33°C–35°C and less favourably at the lower lung temperatures of 37°C [34]. In vivo mouse studies of Foxman et al. showed that this was in part explained by the fact that respiratory epithelial cells exert a heightened antiviral activity at 37°C versus 33°C [39]. Recently, the same group showed that regional differences exist between the upper and lower airway epithelial cells. Nasal epithelial cells were found to be primed to fight virus aggressors, whereas lower bronchial cells responded robustly to mitigate the effect of oxidative stress [40].
While HRV infections result to sinusitis and CRS, upregulation of ICAM-1 is observed in asthmatic children which in turn facilitates translocation of HRV into lower respiratory epithelial cells (bronchial and potentially alveolar) [41]. Furthermore, in asthmatic children after viral replication to the epithelial cells, the expected upregulation of INF-β that would normally cause apoptosis and clearance of the virus does not take place, and this inhibits HRV clearance leading to epithelial cell lysis and HRV propagation through the airways. Thus, as the HRV replicates and spreads, the infected epithelium releases cytokines and chemokines, which are the distress signals that in turn activate a cascade of inflammatory mediators. HRV infection has been shown to trigger increased epithelial expression of a number of growth factors and proteins linked to other aspects of airway remodelling including angiogenesis, subepithelial fibrosis, decreased ciliated cell numbers, and expression of epithelial mucin markers [42–44].
As far as the CRS is concerned, it has been shown that transforming growth factor-beta (TGF-β) and matrix metalloproteinases (MMP-9, ADAM-33) are involved in the nasal tissue remodelling process [45], although our knowledge about how HRV mediates tissue remodelling in CRS is still very limited. HRV has been detected in significantly higher infection rates in those with CRS compared to controls, suggesting either a higher prevalence or increased persistence of disease [46]. Few and smaller studies have reviewed histopathology specimens from patients with CRS compared to healthy controls and found those with CRS display epithelial damage, basement membrane thickening, and eosinophilia infiltration [47–49]. Further understanding of these changes is significant as they can help stratify patients in individual endotypes. Certain characteristics like tissue eosinophilia have been associated with significantly less improvement in symptoms, quality of life, and increased relapse after surgical intervention [49].
Immune responses in the unified airway disease
The well-established allergic asthma association with the adaptive immune system and in particular with the atopic Th2 biased cell responses and eosinophil infiltration has been recently challenged. The Th2-focused therapeutic regimens have little effect and the Th2 immune responses are loosely linked with inflammation of the airways, and they cannot explain nonallergic asthma [50]. It is now recognised that asthma is a highly heterogeneous syndrome that involves both innate and adaptive immune responses. The innate immune responses cover and explain the respiratory viruses like HRV and respiratory syncytial virus (RSV) and the possibility that these could drive the development of asthma and other upper respiratory diseases (CRS, CF and COPD). Thus, innate immunity cells like the airway epithelial cells (sinonasal, bronchial, alveolar), alveolar macrophages (AMs), natural killer cells (NKCs), and dendritic cells (DCs) have a potentially critical role in the pathogenesis of CRS and asthma [51].
The innate immune system recognises pathogen-associated molecular patterns (PAMPs), which include dsRNAs (viruses), bacterial DNAs, lipoteichoic acid of gram+ bacteria, lipopolysaccharides of gram- bacteria, peptidoglycans and mannans. Nasal, bronchial, and alveolar epithelial cells, macrophages (Mφ), DCs, NKCs and B cells contain receptors of the PAMPs and allow them to recognise a variety of pathogens and initiate innate response patterns [52].
For example, Mφ that are recognised as the front line of defence in the lower respiratory innate immunity express the Mφ mannose receptor and upon activation they mediate binding and engulfment of the pathogen, transportation to lysosomes for degradation, as well as a cascade of signalling pathways and transcription initiation of inflammatory cytokines. The same receptor also is found in DCs, which present the pathogen’s antigen to T cells, inducing adaptive immunity responses [53, 54]. SNE can also present antigens to the lymphocytes and initiate inflammatory processes at the nasal cavity area, since it has been shown that when isolated from middle turbinate and cultured at air–liquid interface (mimicking the physiological conditions), the SNE expresses the CD80 and CD86 markers, which are established as having antigen-presenting functions [55]. The above show that the two immune systems (innate and adaptive) are synergistic and cross talk to each other to combat viruses, bacteria, allergens and other intruders.
Immune responses in CRS
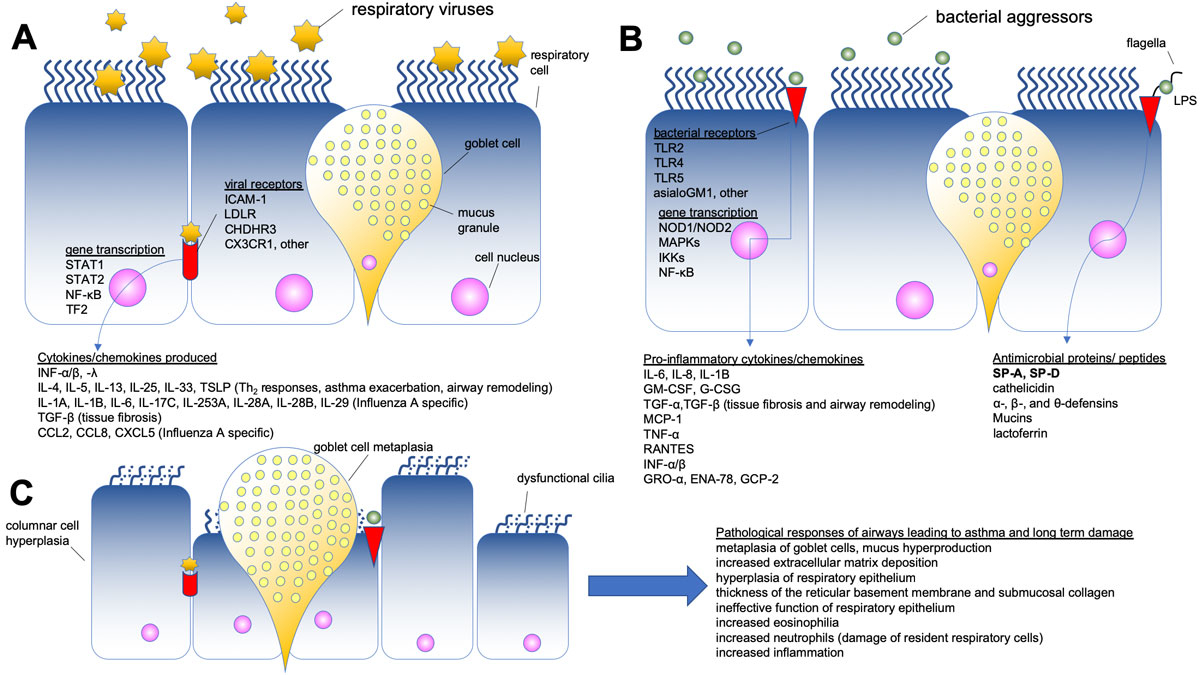
The nasal cavities constitute the first barrier of the whole airway system that has immunological functions against bacteria, viruses and other pathogens. Each aggressor employs different receptors to invade the sinonasal epithelium (table 2) and initiate transcriptional activation of genes driving innate and adaptive immune responses. These immune responses lead to production of pro- and anti-inflammatory cyto- and chemokines and antimicrobial agents (table 3) such as lactoferins [108], mucins, defensins [109, 110], interferons [111], and surfactant proteins A and D (SP-A and SP-D) aiming to combat respiratory aggressors (fig. 1, panels A and B). These mechanisms can rapidly and effectively clear pathogens and ensure the normal function of the airways.
Table 2 Mechanisms deployed by viral and bacterial aggressors to invade upper airway epithelium
|
Role
|
Molecule(s)
|
Function/Mechanism
|
References
|
|
Viral aggressors
|
| HRV-A |
ICAM-1 |
Intracellular receptors or apical (CDHR3, LDLR); replication viral RNA activates TLR-3 and -7 and activates IFN regulatory factors -3, -7, NF-kB, p65/p50, TF2, c-Jun |
[56–58] |
| HRV-B |
ICAM-1, LDLR |
[35, 36] |
| HRV-C |
CDHR3 |
[38, 59] |
| RSV |
TLR4, CX3CR1, HSPG |
Interferon signalling canonical pathway (IFN α/β) in both RSV and influenza A infections |
[60–62] |
| Influenza A H1N1 |
SIGN-R1 |
[63] |
| Influenza A H3N2 |
DC-SIGN, L-SIGN |
[64, 65] |
| Influenza A H3N2, PR8 |
MMR, MGL |
[66] |
| Influenza A H5N1 |
DC-SIGN |
[65] |
| Influenza A H7N7 |
EGFR c-Met receptor |
[67, 68] |
| Influenza B |
Receptor-binding specificity of HA |
|
[69] |
| Influenza C |
Receptor-binding specificity of HEF |
|
[70] |
| Influenza D |
Open receptor-binding cavity of HEF |
|
[71] |
|
Bacterial aggressors
|
| Gram-positive bacteria |
TLR-2 |
Receptor for LP, LTA, PG |
[72, 73] |
| Gram-negative bacteria |
TLR-4 |
Receptor for LPS |
[72] |
|
Pseudomonas aeruginosa
|
TLR-5 |
Receptor for flagellin |
[73, 74] |
| Gram positive and negative |
asialoGM1 |
Receptor for flagellin and pilli |
[75, 76] |
|
Staphylococcus aureus
|
TNF-α R1, EGFR |
Receptor for staphylococcal protein A |
[77, 78] |
| General bacterial responses |
NOD1 and NOD2 |
Intracellular receptor for PG |
[79, 80] |
| IL-2R |
Expressed in mucosal lymphocytes, macrophages, NK |
|
| MHC class I |
Antigen-presenting cells (nasal epithelial, dendritic, macrophages, CD4 T) |
[81] |
| MHC class II |
Antigen-presenting cells (nasal epithelial, dendritic, phagocytes, CD8 T) |
[82] |
Table 3 Defence reaction of respiratory cells to viral and bacterial aggressors
|
Response
|
Molecule(s)
|
Function/Mechanism
|
References
|
|
Viral aggressors
|
| Interferons |
IFN-α |
Transcription of type I and type III IFNs, IL-6, IL-8, CXCL5 |
[83–85] |
| IFN-β |
| IFN-λ |
Transcription of type III IFNs |
[86] |
| Interleukins |
IL-4, IL-5, IL-13, IL-33 |
Induce Th2 immune responses only asthmatics subjects |
[87] |
IL-1A, IL-1B, IL-6, IL-17C, IL-23A, IL-28A,
IL-28B, IL-29, IL-32 |
Induced by influenza A viruses but not RSV, type III interferons |
[68] |
| IL-25, IL-33, TSLP |
Regulate Th2 immune responses |
[88, 89] |
| Chemokines |
TGF-β |
Enhances innate lymphoid cell function, mediates IFN suppression |
[90, 91] |
| CCL2, CCL8 and CXCL5 |
Induced by influenza A viruses but not RSV |
[68] |
|
Bacterial aggressors
|
Pro-inflammatory cytokines/
chemokines |
IL-6, IL-8, mucins |
Upon activation of transcription factor NF-κB by IL-1 and TNF-α |
[92] |
| IL-1β |
Stimulates production of pro-inflammatory molecules by parenchymal airway cells |
[93] |
| GM-CSF, G-CSF |
Induces recruitment, activation and survival of PMNs |
[94] |
| TGF-α, TGF-β |
Promote airway remodelling and fibrosis |
[95] |
| MCP-1 |
Activation of airway host defence |
[96] |
| TNF-α |
Activation of neutrophils |
[97] |
| Pro- and anti- inflammatory molecules |
RANTES |
Migration and homing of T cells during infection |
[98] |
| IFN-α/β |
Activate type I IFN signalling in the airway |
[99] |
| GRO-α, ENA-78, GCP-2 |
Involved in neutrophilic activation/inflammation, chemotactic effects |
[100] |
| Antimicrobial proteins/peptides, molecules |
SP-A, SP-D |
Bind to bacterial adhesins, induce agglutination, macrophage recruitment |
[101–103] |
| Cathelicidin |
Binds and neutralises bacterial LPS, protects against endotoxic shock |
[104] |
| α-, β- and θ-defensins |
Roles in inflammation, airway repair and immune responses |
[105] |
| Mucins |
Defensive molecules, form barrier against pathogens, cross talk with TLRs |
[106] |
| Lactoferrin, lysozyme |
|
[107] |
However, in some cases, pathogens such as HRV can invade the respiratory immune system and produce cytotoxic molecules that disrupt the paracellular permeability of the respiratory epithelium leading to long-term damage (table 4), metaplasia of the mucus producing goblet cells, increased extracellular matrix deposition, airway thickening, and remodelling (fig. 1, panel C). Tissue remodelling takes place not only on the sinus tissues but also on the bronchial tissues, and this can promote the chronic and persistent inflammatory phenotype of the UAD. Unveiling the molecular mechanism of HRV binding in the nasal mucosa holds the potential to provide opportunities for therapeutic interventions in the onset of the low respiratory disease.
Table 4 Pathological responses of the airways leading to chronic asthma and long-term damage
|
Aggressors
|
Long-term effect
|
References
|
| Viral and bacterial aggressors |
Metaplasia of goblet cells |
[112] |
| Increased extracellular matrix deposition |
[113] |
| Hyperplasia of respiratory epithelium |
[114, 115] |
| Thickness of the reticular basement membrane and submucosal collagen |
[116] |
| Ineffective function of respiratory epithelium |
[117] |
| Increased eosinophilia |
[118] |
| Increased neutrophils, damage of resident respiratory cells |
[119] |
| ILC2 and ILC3 |
[120] |
Surfactant proteins A and D as mediators of the sinonasal immunity
Other major players of the upper and lower airway innate immunity system are the SP-A and SP-D, which are secreted by the airway epithelium and are recognised as pattern recognition receptors that play a role in microbial phagocytosis. These calcium-dependent proteins are soluble pattern recognition receptors that bind microbial pathogens and target them for elimination mediated by other phagocytotic cells such as the macrophages [121]. Surfactant proteins also are known to modulate inflammatory response of the airway immune system [122, 123]. Innate immune molecules SP-A and SP-D can either eliminate the microbial pathogens directly by binding to them or indirectly by modulating phagocytic cells such as lymphocytes and AM [124]. Additionally, SP-A and SP-D have been shown to identify, bind, and aggregate upper and lower airway viruses such as RSV and influenza A [125, 126]. HRV infections, albeit critical for sinusitis and shown to exacerbate asthma, have not been yet tested whether they are inactivated by SP-A and SP-D.
Innate molecules SP-A and SP-D are immunolocalised in sinonasal tissues of CRS patients and healthy controls [127], and SP-A mRNA was found to be increased in CRS patients’ submucosal glands [101, 128]. In our studies, we have also shown that protein and mRNA levels of SP-A are increased significantly in CRS patients’ biopsies compared to healthy controls. Additionally, we have established that although SP-A is primarily synthesised by the type II cells in the lung, it is also expressed in the sinonasal epithelium and plays an important role during bacterial infections [101]. SP-D has also been detected in the submucosal glands at protein and mRNA level by means of tissue histology, enzyme-linked immunosorbent assay, and real-time quantitative polymerase reaction [129]. The same group of researchers showed that SP-D is expressed abundantly in CRS nasal tissues but is not detected in tissues of patients suffering from allergic fungal sinusitis. Interestingly, CRS explant models stimulated by fungal antigens were associated with lower levels of SP-D, and authors brought forward the notion that low SP-D levels probably result in ineffective clearance of pathogens in the sinus mucosa and the latter confers to chronic sinusitis [130]. Other animal studies with fungal allergens in the lower airways have shown that both SP-A and SP-D enhance phagocytosis and binding of the glycosylated antigens of Aspergillus fumigatus, and when SP-A is administered exogenously, it blocks the histamine release from the sensitised basophils, reduces eosinophilic infiltration, polarises a marked shift from Th2 cytokine responses to Th1, and thus protects from fungal-induced hypersensitivity and asthma [131].
Aside from their role in their innate immunity, SP-A and SP-D also exert adaptive immunity responses though their interaction with dendritic cells (DCs) [132, 133]. While SP-D has been shown to augment DCs maturation and increase their ability to present antigens to the lymphocytes, SP-A inhibits the maturation of DCs. Thus, surfactant proteins are considered as one of the most important regulators of the respiratory DCs-mediated adaptive immunity [22]. Another example of their adaptive immunoregulatory functions is found in an in vitro model where SP-A and SP-D have been found to inhibit T lymphocyte proliferation after allergen challenge. All the above reinforce the notion that surfactant proteins ameliorate the inflammatory responses [134, 135]. Of the two innate immunity surfactant molecules, SP-A has been shown to have regulatory effects either directly on AM function and the proteomic expression profile of AM or indirectly via the production of cytokines and chemokines by AM [124, 136, 137].
Based on the above, we have put together a model (fig. 1) to depict succinctly the mechanisms employed by the viral (fig. 1, Panel A) and the bacterial (fig. 1, Panel B) aggressors to invade the upper airway respiratory epithelium (table 2), the defence reaction of the cells (table 3), and the pathological responses of the airway that lead to tissue remodelling and long-term damage (fig. 1, Panel C; table 4). We conclude that SP-A and SP-D bridge the innate and adaptive immunity mechanisms of the nasal epithelium to bring together a well-orchestrated mechanism that can fight effectively pathogens such as bacteria, viruses, allergens and other environmental insults.
Acknowledgements
We would like to thank the Arizona State University and Kaiser Permanente Medical Center for granting us access to the scientific literature listed in the present review.
Author contributions
GTN reviewed the relevant literature, designed the structure of the review article, integrated and synthesised published data, contributed to manuscript writing and prepared figures. SS reviewed the relevant literature and integrated and synthesised published data.
References
1
Bhattacharyya
N
. The economic burden and symptom manifestations of chronic rhinosinusitis. Am J Rhinol. 2003;17(1):27–32. doi:.https://doi.org/10.1177/194589240301700106
2
Barnett
SB
,
Nurmagambetov
TA
. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145–52. doi:.https://doi.org/10.1016/j.jaci.2010.10.020
3
Corren
J
. Allergic rhinitis and asthma: how important is the link?
J Allergy Clin Immunol. 1997;99(2):S781–6. doi:.https://doi.org/10.1016/S0091-6749(97)70127-1
4
Guerra
S
,
Sherrill
DL
,
Baldacci
S
,
Carrozzi
L
,
Pistelli
F
,
Di Pede
F
, et al.
Rhinitis is an independent risk factor for developing cough apart from colds among adults. Allergy. 2005;60(3):343–9. doi:.https://doi.org/10.1111/j.1398-9995.2005.00717.x
5
Krouse
JH
,
Krouse
HJ
. Asthma, rhinitis, and the unified airway. ORL Head Neck Nurs. 2013;31(4):6–10.
6
Krouse
JH
. The unified airway. Facial Plast Surg Clin North Am. 2012;20(1):55–60. doi:.https://doi.org/10.1016/j.fsc.2011.10.006
7
Kocevar
VS
,
Bisgaard
H
,
Jönsson
L
,
Valovirta
E
,
Kristensen
F
,
Yin
DD
, et al.
Variations in pediatric asthma hospitalization rates and costs between and within Nordic countries. Chest. 2004;125(5):1680–4. doi:.https://doi.org/10.1378/chest.125.5.1680
8
Stachler
RJ
. Comorbidities of asthma and the unified airway. Int Forum Allergy Rhinol. 2015;5(1, Suppl 1):S17–22. doi:.https://doi.org/10.1002/alr.21615
9
Guerra
S
,
Sherrill
DL
,
Martinez
FD
,
Barbee
RA
. Rhinitis as an independent risk factor for adult-onset asthma. J Allergy Clin Immunol. 2002;109(3):419–25. doi:.https://doi.org/10.1067/mai.2002.121701
10
Jacobs
SE
,
Lamson
DM
,
St George
K
,
Walsh
TJ
. Human rhinoviruses. Clin Microbiol Rev. 2013;26(1):135–62. doi:.https://doi.org/10.1128/CMR.00077-12
11
Gavala
ML
,
Bertics
PJ
,
Gern
JE
. Rhinoviruses, allergic inflammation, and asthma. Immunol Rev. 2011;242(1):69–90. doi:.https://doi.org/10.1111/j.1600-065X.2011.01031.x
12
Foxman
EF
,
Iwasaki
A
. Genome-virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol. 2011;9(4):254–64. doi:.https://doi.org/10.1038/nrmicro2541
13
Doctor
TH
,
Trivedi
SS
,
Chudasama
RK
. Pulmonary function test in healthy school children of 8 to 14 years age in south Gujarat region, India. Lung India. 2010;27(3):145–8. doi:.https://doi.org/10.4103/0970-2113.68317
14
Gupta
S
,
Siddiqui
S
,
Haldar
P
,
Raj
JV
,
Entwisle
JJ
,
Wardlaw
AJ
, et al.
Qualitative analysis of high-resolution CT scans in severe asthma. Chest. 2009;136(6):1521–8. doi:.https://doi.org/10.1378/chest.09-0174
15
Jensen
SP
,
Lynch
DA
,
Brown
KK
,
Wenzel
SE
,
Newell
JD
. High-resolution CT features of severe asthma and bronchiolitis obliterans. Clin Radiol. 2002;57(12):1078–85. doi:.https://doi.org/10.1053/crad.2002.1104
16
Van Zele
T
,
Claeys
S
,
Gevaert
P
,
Van Maele
G
,
Holtappels
G
,
Van Cauwenberge
P
, et al.
Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61(11):1280–9. doi:.https://doi.org/10.1111/j.1398-9995.2006.01225.x
17
Akdis
CA
,
Bachert
C
,
Cingi
C
,
Dykewicz
MS
,
Hellings
PW
,
Naclerio
RM
, et al.
Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2013;131(6):1479–90. doi:.https://doi.org/10.1016/j.jaci.2013.02.036
18
Zhang
N
,
Van Zele
T
,
Perez-Novo
C
,
Van Bruaene
N
,
Holtappels
G
,
DeRuyck
N
, et al.
Different types of T-effector cells orchestrate mucosal inflammation in chronic sinus disease. J Allergy Clin Immunol. 2008;122(5):961–8. doi:.https://doi.org/10.1016/j.jaci.2008.07.008
19
Smith
TL
,
Litvack
JR
,
Hwang
PH
,
Loehrl
TA
,
Mace
JC
,
Fong
KJ
, et al.
Determinants of outcomes of sinus surgery: a multi-institutional prospective cohort study. Otolaryngol Head Neck Surg. 2010;142(1):55–63. doi:.https://doi.org/10.1016/j.otohns.2009.10.009
20
Wenzel
SE
. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–25. doi:.https://doi.org/10.1038/nm.2678
21
Lötvall
J
,
Akdis
CA
,
Bacharier
LB
,
Bjermer
L
,
Casale
TB
,
Custovic
A
, et al.
Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355–60. doi:.https://doi.org/10.1016/j.jaci.2010.11.037
22
Noutsios
GT
,
Floros
J
. Childhood asthma: causes, risks, and protective factors; a role of innate immunity. Swiss Med Wkly. 2014;144:w14036. doi:.https://doi.org/10.4414/smw.2014.14036
23
Hinks
TS
,
Brown
T
,
Lau
LC
,
Rupani
H
,
Barber
C
,
Elliott
S
, et al.
Multidimensional endotyping in patients with severe asthma reveals inflammatory heterogeneity in matrix metalloproteinases and chitinase 3-like protein 1. J Allergy Clin Immunol. 2016;138(1):61–75. doi:.https://doi.org/10.1016/j.jaci.2015.11.020
24
Moore
WC
,
Meyers
DA
,
Wenzel
SE
,
Teague
WG
,
Li
H
,
Li
X
, et al.; National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–23. doi:.https://doi.org/10.1164/rccm.200906-0896OC
25
Haldar
P
,
Pavord
ID
,
Shaw
DE
,
Berry
MA
,
Thomas
M
,
Brightling
CE
, et al.
Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178(3):218–24. doi:.https://doi.org/10.1164/rccm.200711-1754OC
26
Tomassen
P
,
Vandeplas
G
,
Van Zele
T
,
Cardell
LO
,
Arebro
J
,
Olze
H
, et al.
Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137(5):1449–1456.e4. doi:.https://doi.org/10.1016/j.jaci.2015.12.1324
27
Nakayama
T
,
Yoshikawa
M
,
Asaka
D
,
Okushi
T
,
Matsuwaki
Y
,
Otori
N
, et al.
Mucosal eosinophilia and recurrence of nasal polyps - new classification of chronic rhinosinusitis. Rhinology. 2011;49(4):392–6.
28
Schlosser
RJ
,
London
SD
,
Gwaltney
JM, Jr
,
Gross
CW
. Microbiology of chronic frontal sinusitis. Laryngoscope. 2001;111(8):1330–2. doi:.https://doi.org/10.1097/00005537-200108000-00004
29
Soler
ZM
,
Hyer
JM
,
Ramakrishnan
V
,
Smith
TL
,
Mace
J
,
Rudmik
L
, et al.
Identification of chronic rhinosinusitis phenotypes using cluster analysis. Int Forum Allergy Rhinol. 2015;5(5):399–407. doi:.https://doi.org/10.1002/alr.21496
30
Ponikau
JU
,
Sherris
DA
,
Kephart
GM
,
Kern
EB
,
Gaffey
TA
,
Tarara
JE
, et al.
Features of airway remodeling and eosinophilic inflammation in chronic rhinosinusitis: is the histopathology similar to asthma?
J Allergy Clin Immunol. 2003;112(5):877–82. doi:.https://doi.org/10.1016/j.jaci.2003.08.009
31
Tagaya
E
,
Tamaoki
J
. Mechanisms of airway remodeling in asthma. Allergol Int. 2007;56(4):331–40. doi:.https://doi.org/10.2332/allergolint.R-07-152
32
Holgate
ST
. Pathogenesis of asthma. Clin Exp Allergy. 2008;38(6):872–97. doi:.https://doi.org/10.1111/j.1365-2222.2008.02971.x
33
Sly
PD
,
Kusel
M
,
Holt
PG
. Do early-life viral infections cause asthma?
J Allergy Clin Immunol. 2010;125(6):1202–5. doi:.https://doi.org/10.1016/j.jaci.2010.01.024
34
Gern
JE
. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84(15):7418–26. doi:.https://doi.org/10.1128/JVI.02290-09
35
Greve
JM
,
Davis
G
,
Meyer
AM
,
Forte
CP
,
Yost
SC
,
Marlor
CW
, et al.
The major human rhinovirus receptor is ICAM-1. Cell. 1989;56(5):839–47. doi:.https://doi.org/10.1016/0092-8674(89)90688-0
36
Hofer
F
,
Gruenberger
M
,
Kowalski
H
,
Machat
H
,
Huettinger
M
,
Kuechler
E
, et al.
Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci USA. 1994;91(5):1839–42. doi:.https://doi.org/10.1073/pnas.91.5.1839
37
Bønnelykke
K
,
Sleiman
P
,
Nielsen
K
,
Kreiner-Møller
E
,
Mercader
JM
,
Belgrave
D
, et al.
A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(1):51–5. doi:.https://doi.org/10.1038/ng.2830
38
Bochkov
YA
,
Watters
K
,
Ashraf
S
,
Griggs
TF
,
Devries
MK
,
Jackson
DJ
, et al.
Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci USA. 2015;112(17):5485–90. doi:.https://doi.org/10.1073/pnas.1421178112
39
Foxman
EF
,
Storer
JA
,
Fitzgerald
ME
,
Wasik
BR
,
Hou
L
,
Zhao
H
, et al.
Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proc Natl Acad Sci USA. 2015;112(3):827–32. doi:.https://doi.org/10.1073/pnas.1411030112
40
Mihaylova
VT
,
Kong
Y
,
Fedorova
O
,
Sharma
L
,
Dela Cruz
CS
,
Pyle
AM
, et al.
Regional differences in airway epithelial cells reveal tradeoff between defense against oxidative stress and defense against Rhinovirus. Cell Rep. 2018;24(11):3000–3007.e3. doi:.https://doi.org/10.1016/j.celrep.2018.08.033
41Warner JO, Boner A. Paediatric allergy and asthma. Vol. Chapter 18. 2012: Elsevier Ltd.
42
Bergeron
C
,
Tulic
MK
,
Hamid
Q
. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. 2010;17(4):e85–93. doi:.https://doi.org/10.1155/2010/318029
43
Jamieson
KC
,
Warner
SM
,
Leigh
R
,
Proud
D
. Rhinovirus in the Pathogenesis and Clinical Course of Asthma. Chest. 2015;148(6):1508–16. doi:.https://doi.org/10.1378/chest.15-1335
44
Ordoñez
CL
,
Khashayar
R
,
Wong
HH
,
Ferrando
R
,
Wu
R
,
Hyde
DM
, et al.
Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med. 2001;163(2):517–23. doi:.https://doi.org/10.1164/ajrccm.163.2.2004039
45
Van Bruaene
N
,
Bachert
C
. Tissue remodeling in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2011;11(1):8–11. doi:.https://doi.org/10.1097/ACI.0b013e32834233ef
46
Cho
GS
,
Moon
BJ
,
Lee
BJ
,
Gong
CH
,
Kim
NH
,
Kim
YS
, et al.
High rates of detection of respiratory viruses in the nasal washes and mucosae of patients with chronic rhinosinusitis. J Clin Microbiol. 2013;51(3):979–84. doi:.https://doi.org/10.1128/JCM.02806-12
47
Saitoh
T
,
Kusunoli
T
,
Yao
T
,
Kawano
K
,
Kojima
Y
,
Miyahara
K
, et al.
Relationship between epithelial damage or basement membrane thickness and eosinophilic infiltration in nasal polyps with chronic rhinosinusitis. Rhinology. 2009;47(3):275–9. doi:.https://doi.org/10.4193/Rhin08.109
48
Snidvongs
K
,
Chin
D
,
Sacks
R
,
Earls
P
,
Harvey
RJ
. Eosinophilic rhinosinusitis is not a disease of ostiomeatal occlusion. Laryngoscope. 2013;123(5):1070–4. doi:.https://doi.org/10.1002/lary.23721
49
Snidvongs
K
,
Lam
M
,
Sacks
R
,
Earls
P
,
Kalish
L
,
Phillips
PS
, et al.
Structured histopathology profiling of chronic rhinosinusitis in routine practice. Int Forum Allergy Rhinol. 2012;2(5):376–85. doi:.https://doi.org/10.1002/alr.21032
50
Murdoch
JR
,
Lloyd
CM
. Chronic inflammation and asthma. Mutat Res. 2010;690(1-2):24–39. doi:.https://doi.org/10.1016/j.mrfmmm.2009.09.005
51
Holtzman
MJ
. Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest. 2012;122(8):2741–8. doi:.https://doi.org/10.1172/JCI60325
52
Medzhitov
R
,
Janeway
C, Jr
. Innate immunity. N Engl J Med. 2000;343(5):338–44. doi:.https://doi.org/10.1056/NEJM200008033430506
53
McKenzie
EJ
,
Taylor
PR
,
Stillion
RJ
,
Lucas
AD
,
Harris
J
,
Gordon
S
, et al.
Mannose receptor expression and function define a new population of murine dendritic cells. J Immunol. 2007;178(8):4975–83. doi:.https://doi.org/10.4049/jimmunol.178.8.4975
54
Fraser
IP
,
Koziel
H
,
Ezekowitz
RA
. The serum mannose-binding protein and the macrophage mannose receptor are pattern recognition molecules that link innate and adaptive immunity. Semin Immunol. 1998;10(5):363–72. doi:.https://doi.org/10.1006/smim.1998.0141
55
Takizawa
R
,
Pawankar
R
,
Yamagishi
S
,
Takenaka
H
,
Yagi
T
. Increased expression of HLA-DR and CD86 in nasal epithelial cells in allergic rhinitics: antigen presentation to T cells and up-regulation by diesel exhaust particles. Clin Exp Allergy. 2007;37(3):420–33. doi:.https://doi.org/10.1111/j.1365-2222.2007.02672.x
56
Mukhopadhyay
S
,
Malik
P
,
Arora
SK
,
Mukherjee
TK
. Intercellular adhesion molecule-1 as a drug target in asthma and rhinitis. Respirology. 2014;19(4):508–13. doi:.https://doi.org/10.1111/resp.12285
57
Slater
L
,
Bartlett
NW
,
Haas
JJ
,
Zhu
J
,
Message
SD
,
Walton
RP
, et al.
Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6(11):e1001178. doi:.https://doi.org/10.1371/journal.ppat.1001178
58
Laza-Stanca
V
,
Stanciu
LA
,
Message
SD
,
Edwards
MR
,
Gern
JE
,
Johnston
SL
. Rhinovirus replication in human macrophages induces NF-kappaB-dependent tumor necrosis factor alpha production. J Virol. 2006;80(16):8248–58. doi:.https://doi.org/10.1128/JVI.00162-06
59
Chang
EH
,
Willis
AL
,
McCrary
HC
,
Noutsios
GT
,
Le
CH
,
Chiu
AG
, et al.
Association between the CDHR3 rs6967330 risk allele and chronic rhinosinusitis. J Allergy Clin Immunol. 2017;139(6):1990–1992.e2. doi:.https://doi.org/10.1016/j.jaci.2016.10.027
60
Kurt-Jones
EA
,
Popova
L
,
Kwinn
L
,
Haynes
LM
,
Jones
LP
,
Tripp
RA
, et al.
Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401. doi:.https://doi.org/10.1038/80833
61
Johnson
SM
,
McNally
BA
,
Ioannidis
I
,
Flano
E
,
Teng
MN
,
Oomens
AG
, et al.
Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog. 2015;11(12):e1005318. doi:.https://doi.org/10.1371/journal.ppat.1005318
62
Krusat
T
,
Streckert
HJ
. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch Virol. 1997;142(6):1247–54. doi:.https://doi.org/10.1007/s007050050156
63
Gonzalez
SF
,
Lukacs-Kornek
V
,
Kuligowski
MP
,
Pitcher
LA
,
Degn
SE
,
Kim
YA
, et al.
Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol. 2010;11(5):427–34. doi:.https://doi.org/10.1038/ni.1856
64
Londrigan
SL
,
Turville
SG
,
Tate
MD
,
Deng
YM
,
Brooks
AG
,
Reading
PC
. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol. 2011;85(6):2990–3000. doi:.https://doi.org/10.1128/JVI.01705-10
65
Wang
SF
,
Huang
JC
,
Lee
YM
,
Liu
SJ
,
Chan
YJ
,
Chau
YP
, et al.
DC-SIGN mediates avian H5N1 influenza virus infection in cis and in trans. Biochem Biophys Res Commun. 2008;373(4):561–6. doi:.https://doi.org/10.1016/j.bbrc.2008.06.078
66
Upham
JP
,
Pickett
D
,
Irimura
T
,
Anders
EM
,
Reading
PC
. Macrophage receptors for influenza A virus: role of the macrophage galactose-type lectin and mannose receptor in viral entry. J Virol. 2010;84(8):3730–7. doi:.https://doi.org/10.1128/JVI.02148-09
67
Eierhoff
T
,
Hrincius
ER
,
Rescher
U
,
Ludwig
S
,
Ehrhardt
C
. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010;6(9):e1001099. doi:.https://doi.org/10.1371/journal.ppat.1001099
68
Ioannidis
I
,
McNally
B
,
Willette
M
,
Peeples
ME
,
Chaussabel
D
,
Durbin
JE
, et al.
Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol. 2012;86(10):5422–36. doi:.https://doi.org/10.1128/JVI.06757-11
69
Wang
Q
,
Tian
X
,
Chen
X
,
Ma
J
. Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc Natl Acad Sci USA. 2007;104(43):16874–9. doi:.https://doi.org/10.1073/pnas.0708363104
70
Wang
M
,
Veit
M
. Hemagglutinin-esterase-fusion (HEF) protein of influenza C virus. Protein Cell. 2016;7(1):28–45. doi:.https://doi.org/10.1007/s13238-015-0193-x
71
Song
H
,
Qi
J
,
Khedri
Z
,
Diaz
S
,
Yu
H
,
Chen
X
, et al.
An open receptor-binding cavity of hemagglutinin-esterase-fusion glycoprotein from newly-identified influenza D virus: basis for its broad cell tropism. PLoS Pathog. 2016;12(1):e1005411. doi:.https://doi.org/10.1371/journal.ppat.1005411
72
Takeuchi
O
,
Hoshino
K
,
Kawai
T
,
Sanjo
H
,
Takada
H
,
Ogawa
T
, et al.
Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–51. doi:.https://doi.org/10.1016/S1074-7613(00)80119-3
73
Zhang
Z
,
Louboutin
JP
,
Weiner
DJ
,
Goldberg
JB
,
Wilson
JM
. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect Immun. 2005;73(11):7151–60. doi:.https://doi.org/10.1128/IAI.73.11.7151-7160.2005
74
DiMango
E
,
Ratner
AJ
,
Bryan
R
,
Tabibi
S
,
Prince
A
. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest. 1998;101(11):2598–605. doi:.https://doi.org/10.1172/JCI2865
75
Feldman
M
,
Bryan
R
,
Rajan
S
,
Scheffler
L
,
Brunnert
S
,
Tang
H
, et al.
Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66(1):43–51.
76
Krivan
HC
,
Roberts
DD
,
Ginsburg
V
. Many pulmonary pathogenic bacteria bind specifically to the carbohydrate sequence GalNAc beta 1-4Gal found in some glycolipids. Proc Natl Acad Sci USA. 1988;85(16):6157–61. doi:.https://doi.org/10.1073/pnas.85.16.6157
77
Gómez
MI
,
Lee
A
,
Reddy
B
,
Muir
A
,
Soong
G
,
Pitt
A
, et al.
Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10(8):842–8. doi:.https://doi.org/10.1038/nm1079
78
Gómez
MI
,
Seaghdha
MO
,
Prince
AS
. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 2007;26(3):701–9. doi:.https://doi.org/10.1038/sj.emboj.7601554
79
Girardin
SE
,
Boneca
IG
,
Carneiro
LA
,
Antignac
A
,
Jéhanno
M
,
Viala
J
, et al.
Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–7. doi:.https://doi.org/10.1126/science.1084677
80
Girardin
SE
,
Boneca
IG
,
Viala
J
,
Chamaillard
M
,
Labigne
A
,
Thomas
G
, et al.
Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–72. doi:.https://doi.org/10.1074/jbc.C200651200
81
Arebro
J
,
Tengroth
L
,
Razavi
R
,
Kumlien Georén
S
,
Winqvist
O
,
Cardell
LO
. Antigen-presenting epithelial cells can play a pivotal role in airway allergy. J Allergy Clin Immunol. 2016;137(3):957–60.e7. doi:.https://doi.org/10.1016/j.jaci.2015.08.053
82
Kalb
TH
,
Chuang
MT
,
Marom
Z
,
Mayer
L
. Evidence for accessory cell function by class II MHC antigen-expressing airway epithelial cells. Am J Respir Cell Mol Biol. 1991;4(4):320–9. doi:.https://doi.org/10.1165/ajrcmb/4.4.320
83
Zhu
Z
,
Tang
W
,
Gwaltney
JM, Jr
,
Wu
Y
,
Elias
JA
. Rhinovirus stimulation of interleukin-8 in vivo and in vitro: role of NF-kappaB. Am J Physiol. 1997;273(4):L814–24.
84
Bossios
A
,
Gourgiotis
D
,
Skevaki
CL
,
Saxoni-Papageorgiou
P
,
Lötvall
J
,
Psarras
S
, et al.
Rhinovirus infection and house dust mite exposure synergize in inducing bronchial epithelial cell interleukin-8 release. Clin Exp Allergy. 2008;38(10):1615–26. doi:.https://doi.org/10.1111/j.1365-2222.2008.03058.x
85
Donninger
H
,
Glashoff
R
,
Haitchi
HM
,
Syce
JA
,
Ghildyal
R
,
van Rensburg
E
, et al.
Rhinovirus induction of the CXC chemokine epithelial-neutrophil activating peptide-78 in bronchial epithelium. J Infect Dis. 2003;187(11):1809–17. doi:.https://doi.org/10.1086/375246
86
Contoli
M
,
Message
SD
,
Laza-Stanca
V
,
Edwards
MR
,
Wark
PA
,
Bartlett
NW
, et al.
Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12(9):1023–6. doi:.https://doi.org/10.1038/nm1462
87
Jackson
DJ
,
Makrinioti
H
,
Rana
BM
,
Shamji
BW
,
Trujillo-Torralbo
MB
,
Footitt
J
, et al.
IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–82. doi:.https://doi.org/10.1164/rccm.201406-1039OC
88
Gordon
ED
,
Locksley
RM
,
Fahy
JV
. Cross-talk between epithelial cells and type 2 immune signaling. The role of IL-25. Am J Respir Crit Care Med. 2016;193(9):935–6. doi:.https://doi.org/10.1164/rccm.201512-2534ED
89
Iyer
AS
,
Bhatt
SP
,
Garner
JJ
,
Wells
JM
,
Trevor
JL
,
Patel
NM
, et al.
Depression is associated with readmission for acute exacerbation of chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(2):197–203.
90
Bedke
N
,
Sammut
D
,
Green
B
,
Kehagia
V
,
Dennison
P
,
Jenkins
G
, et al.
Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS One. 2012;7(9):e44580. doi:.https://doi.org/10.1371/journal.pone.0044580
91
Denney
L
,
Byrne
AJ
,
Shea
TJ
,
Buckley
JS
,
Pease
JE
,
Herledan
GM
, et al.
Pulmonary epithelial cell-derived cytokine TGF-β1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity. 2015;43(5):945–58. doi:.https://doi.org/10.1016/j.immuni.2015.10.012
92
Strieter
RM
,
Belperio
JA
,
Keane
MP
. Cytokines in innate host defense in the lung. J Clin Invest. 2002;109(6):699–705. doi:.https://doi.org/10.1172/JCI0215277
93
Coulter
KR
,
Wewers
MD
,
Lowe
MP
,
Knoell
DL
. Extracellular regulation of interleukin (IL)-1beta through lung epithelial cells and defective IL-1 type II receptor expression. Am J Respir Cell Mol Biol. 1999;20(5):964–75. doi:.https://doi.org/10.1165/ajrcmb.20.5.3458
94
Saba
S
,
Soong
G
,
Greenberg
S
,
Prince
A
. Bacterial stimulation of epithelial G-CSF and GM-CSF expression promotes PMN survival in CF airways. Am J Respir Cell Mol Biol. 2002;27(5):561–7. doi:.https://doi.org/10.1165/rcmb.2002-0019OC
95
Kumar
RK
,
Herbert
C
,
Foster
PS
. Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin Exp Allergy. 2004;34(4):567–75. doi:.https://doi.org/10.1111/j.1365-2222.2004.1917.x
96
Rao
SP
,
Hayashi
T
,
Catanzaro
A
. Release of monocyte chemoattractant protein (MCP)-1 by a human alveolar epithelial cell line in response to mycobacterium avium. FEMS Immunol Med Microbiol. 2000;29(1):1–7. doi:.https://doi.org/10.1111/j.1574-695X.2000.tb01497.x
97
Gómez
MI
,
Sokol
SH
,
Muir
AB
,
Soong
G
,
Bastien
J
,
Prince
AS
. Bacterial induction of TNF-alpha converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. J Immunol. 2005;175(3):1930–6. doi:.https://doi.org/10.4049/jimmunol.175.3.1930
98
Leidal
KG
,
Munson
KL
,
Denning
GM
. Small molecular weight secretory factors from Pseudomonas aeruginosa have opposite effects on IL-8 and RANTES expression by human airway epithelial cells. Am J Respir Cell Mol Biol. 2001;25(2):186–95. doi:.https://doi.org/10.1165/ajrcmb.25.2.4273
99
Parker
D
,
Prince
A
. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol. 2011;32(12):582–8. doi:.https://doi.org/10.1016/j.it.2011.09.003
100
Sachse
F
,
von Eiff
C
,
Stoll
W
,
Becker
K
,
Rudack
C
. Induction of CXC chemokines in A549 airway epithelial cells by trypsin and staphylococcal proteases - a possible route for neutrophilic inflammation in chronic rhinosinusitis. Clin Exp Immunol. 2006;144(3):534–42. doi:.https://doi.org/10.1111/j.1365-2249.2006.03089.x
101
Noutsios
GT
,
Willis
AL
,
Ledford
JG
,
Chang
EH
. Novel role of surfactant protein A in bacterial sinusitis. Int Forum Allergy Rhinol. 2017;7(9):897–903. doi:.https://doi.org/10.1002/alr.21985
102
Ferguson
JS
,
Voelker
DR
,
Ufnar
JA
,
Dawson
AJ
,
Schlesinger
LS
. Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is independent of bacterial agglutination. J Immunol. 2002;168(3):1309–14. doi:.https://doi.org/10.4049/jimmunol.168.3.1309
103
Sever-Chroneos
Z
,
Krupa
A
,
Davis
J
,
Hasan
M
,
Yang
CH
,
Szeliga
J
, et al.
Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem. 2011;286(6):4854–70. doi:.https://doi.org/10.1074/jbc.M110.125567
104
Bals
R
,
Weiner
DJ
,
Moscioni
AD
,
Meegalla
RL
,
Wilson
JM
. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun. 1999;67(11):6084–9.
105
van Wetering
S
,
Sterk
PJ
,
Rabe
KF
,
Hiemstra
PS
. Defensins: key players or bystanders in infection, injury, and repair in the lung?
J Allergy Clin Immunol. 1999;104(6):1131–8. doi:.https://doi.org/10.1016/S0091-6749(99)70004-7
106
Kim
KC
. Role of epithelial mucins during airway infection. Pulm Pharmacol Ther. 2012;25(6):415–9. doi:.https://doi.org/10.1016/j.pupt.2011.12.003
107
Ganz
T
. Antimicrobial polypeptides in host defense of the respiratory tract. J Clin Invest. 2002;109(6):693–7. doi:.https://doi.org/10.1172/JCI0215218
108
van der Strate
BW
,
Beljaars
L
,
Molema
G
,
Harmsen
MC
,
Meijer
DK
. Antiviral activities of lactoferrin. Antiviral Res. 2001;52(3):225–39. doi:.https://doi.org/10.1016/S0166-3542(01)00195-4
109
Wilson
SS
,
Wiens
ME
,
Smith
JG
. Antiviral mechanisms of human defensins. J Mol Biol. 2013;425(24):4965–80. doi:.https://doi.org/10.1016/j.jmb.2013.09.038
110
Wilson
SS
,
Wiens
ME
,
Holly
MK
,
Smith
JG
. Defensins at the Mucosal Surface: Latest Insights into Defensin-Virus Interactions. J Virol. 2016;90(11):5216–8. doi:.https://doi.org/10.1128/JVI.00904-15
111
Grandvaux
N
,
tenOever
BR
,
Servant
MJ
,
Hiscott
J
. The interferon antiviral response: from viral invasion to evasion. Curr Opin Infect Dis. 2002;15(3):259–67. doi:.https://doi.org/10.1097/00001432-200206000-00008
112
Curran
DR
,
Cohn
L
. Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol. 2010;42(3):268–75. doi:.https://doi.org/10.1165/rcmb.2009-0151TR
113
Kuo
C
,
Lim
S
,
King
NJ
,
Johnston
SL
,
Burgess
JK
,
Black
JL
, et al.
Rhinovirus infection induces extracellular matrix protein deposition in asthmatic and nonasthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L951–7. doi:.https://doi.org/10.1152/ajplung.00411.2010
114
Ganesan
S
,
Comstock
AT
,
Sajjan
US
. Barrier function of airway tract epithelium. Tissue Barriers. 2013;1(4):e24997. doi:.https://doi.org/10.4161/tisb.24997
115
Hogan
BL
,
Barkauskas
CE
,
Chapman
HA
,
Epstein
JA
,
Jain
R
,
Hsia
CC
, et al.
Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–38. doi:.https://doi.org/10.1016/j.stem.2014.07.012
116
Wilson
JW
,
Li
X
. The measurement of reticular basement membrane and submucosal collagen in the asthmatic airway. Clin Exp Allergy. 1997;27(4):363–71. doi:.https://doi.org/10.1111/j.1365-2222.1997.tb00720.x
117
McGrath
JJC
,
Stampfli
MR
. The immune system as a victim and aggressor in chronic obstructive pulmonary disease. J Thorac Dis. 2018;10(S16, Suppl 17):S2011–7. doi:.https://doi.org/10.21037/jtd.2018.05.63
118
Drake
MG
,
Bivins-Smith
ER
,
Proskocil
BJ
,
Nie
Z
,
Scott
GD
,
Lee
JJ
, et al.
Human and mouse eosinophils have antiviral activity against parainfluenza virus. Am J Respir Cell Mol Biol. 2016;55(3):387–94. doi:.https://doi.org/10.1165/rcmb.2015-0405OC
119
Tang
FS
,
Van Ly
D
,
Spann
K
,
Reading
PC
,
Burgess
JK
,
Hartl
D
, et al.
Differential neutrophil activation in viral infections: Enhanced TLR-7/8-mediated CXCL8 release in asthma. Respirology. 2016;21(1):172–9. doi:.https://doi.org/10.1111/resp.12657
120
De Grove
KC
,
Provoost
S
,
Verhamme
FM
,
Bracke
KR
,
Joos
GF
,
Maes
T
, et al.
Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One. 2016;11(1):e0145961. doi:.https://doi.org/10.1371/journal.pone.0145961
121
Jäkel
A
,
Clark
H
,
Reid
KB
,
Sim
RB
. The human lung surfactant proteins A (SP-A) and D (SP-D) interact with apoptotic target cells by different binding mechanisms. Immunobiology. 2010;215(7):551–8. doi:.https://doi.org/10.1016/j.imbio.2009.09.005
122Floros J, Phelps DS. Pulmonary surfactant protein A; structure, expression, and its role in innate host defense, in Surfactant-Update of Intensive Care Medicine, G. Nakos and M.E. Lekka, Editors. 2002, University of Ioannina: Ioannina, Greece. p. 87-102.
123Floros J, Phelps DS. Pulmonary surfactant, in Anesthesia: Biologic Foundations, J. Biebuck, et al., Editors. 1997, Lippincott-Raven. p. 1259-1279.
124
Phelps
DS
. Surfactant regulation of host defense function in the lung: a question of balance. Pediatr Pathol Mol Med. 2001;20(4):269–92. doi:.https://doi.org/10.1080/15513810109168822
125
LeVine
AM
,
Gwozdz
J
,
Stark
J
,
Bruno
M
,
Whitsett
J
,
Korfhagen
T
. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103(7):1015–21. doi:.https://doi.org/10.1172/JCI5849
126
LeVine
AM
,
Whitsett
JA
,
Hartshorn
KL
,
Crouch
EC
,
Korfhagen
TR
. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol. 2001;167(10):5868–73. doi:.https://doi.org/10.4049/jimmunol.167.10.5868
127
Woodworth
BA
,
Lathers
D
,
Neal
JG
,
Skinner
M
,
Richardson
M
,
Young
MR
, et al.
Immunolocalization of surfactant protein A and D in sinonasal mucosa. Am J Rhinol. 2006;20(4):461–5. doi:.https://doi.org/10.2500/ajr.2006.20.2892
128
Lee
HM
,
Kang
HJ
,
Woo
JS
,
Chae
SW
,
Lee
SH
,
Hwang
SJ
. Upregulation of surfactant protein A in chronic rhinosinusitis. Laryngoscope. 2006;116(2):328–30. doi:.https://doi.org/10.1097/01.mlg.0000194223.22763.5f
129
Ooi
EH
,
Wormald
PJ
,
Carney
AS
,
James
CL
,
Tan
LW
. Surfactant protein d expression in chronic rhinosinusitis patients and immune responses in vitro to Aspergillus and alternaria in a nasal explant model. Laryngoscope. 2007;117(1):51–7. doi:.https://doi.org/10.1097/01.mlg.0000243196.75418.6f
130
Ooi
EH
,
Wormald
PJ
,
Tan
LW
. Innate immunity in the paranasal sinuses: a review of nasal host defenses. Am J Rhinol. 2008;22(1):13–9. doi:.https://doi.org/10.2500/ajr.2008.22.3127
131
Madan
T
,
Kishore
U
,
Singh
M
,
Strong
P
,
Clark
H
,
Hussain
EM
, et al.
Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest. 2001;107(4):467–75. doi:.https://doi.org/10.1172/JCI10124
132
Brinker
KG
,
Garner
H
,
Wright
JR
. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2003;284(1):L232–41. doi:.https://doi.org/10.1152/ajplung.00187.2002
133
Brinker
KG
,
Martin
E
,
Borron
P
,
Mostaghel
E
,
Doyle
C
,
Harding
CV
, et al.
Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1453–63. doi:.https://doi.org/10.1152/ajplung.2001.281.6.L1453
134
Wang
JY
,
Shieh
CC
,
You
PF
,
Lei
HY
,
Reid
KB
. Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am J Respir Crit Care Med. 1998;158(2):510–8. doi:.https://doi.org/10.1164/ajrccm.158.2.9709111
135
Borron
PJ
,
Mostaghel
EA
,
Doyle
C
,
Walsh
ES
,
McHeyzer-Williams
MG
,
Wright
JR
. Pulmonary surfactant proteins A and D directly suppress CD3+/CD4+ cell function: evidence for two shared mechanisms. J Immunol. 2002;169(10):5844–50. doi:.https://doi.org/10.4049/jimmunol.169.10.5844
136
Phelps
DS
,
Umstead
TM
,
Quintero
OA
,
Yengo
CM
,
Floros
J
. In vivo rescue of alveolar macrophages from SP-A knockout mice with exogenous SP-A nearly restores a wild type intracellular proteome; actin involvement. Proteome Sci. 2011;9(1):67. doi:.https://doi.org/10.1186/1477-5956-9-67
137
Crouch
E
,
Wright
JR
. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol. 2001;63(1):521–54. doi:.https://doi.org/10.1146/annurev.physiol.63.1.521