Design and evaluation of a multi-epitope assembly peptide vaccine against Acinetobacter baumannii infection in mice
DOI: https://doi.org/10.4414/smw.2019.20052
Shan
Ren, Lina
Guan, Yao
Dong, Chaoli
Wang, Li
Feng, Yongen
Xie
Research Institute of Immunology and Molecular Biology, North Sichuan Medical College, Nanchong City, Sichuan Province, China
Summary
AIM
To design a multi-epitope assembly peptide (MEP) of Acinetobacter baumannii and evaluate its immunogenicity and protective immunity in Balb/c mice.
METHODS
The T- and B-cell epitopes of outer membrane proteins FilF and NucAb from A. baumannii were predicted and identified by using bioinformatics software and immunological tests. Peptides with predicted high adhesin probability from A. baumannii Ata protein was used as the backbone, two B-cell epitopes and one CD4+ T-cell epitope from FilF were linked to the N-terminal of the backbone, and two B-cell epitopes and one CD4+ T-cell epitope from NucAb were linked to the C-terminal of the backbone to construct the MEP. The gene of the MEP was expressed in E. coli BL21, and its immunogenicity and protective efficacy were evaluated in Balb/c mice.
RESULTS
A recombinant protein with a molecular weight of about 37 kDa was successfully purified, and was identified as the recombinant multi-epitope assembly peptide (rMEP) by Western blot analysis. The animal tests showed that the rMEP was highly immunogenic and could induce high levels of IgG antibody and provide potent protection (88.9%) against lethal doses of A. baumannii.
CONCLUSIONS
This is the first report of the design and study of a rMEP vaccine against A. baumannii. The results indicate that the rMEP is a promising vaccine candidate for the control of infections caused by A. baumannii.
Introduction
Acinetobacter baumannii is a common conditional pathogenic bacterium which mainly causes hospital infections, such as intubation-associated pneumonia, traumatic infections, urogenital infections and catheter related sepsis [1, 2]. Commonly used antibacterial drugs cannot effectively control A. baumannii related infectious disease because drug resistance of A. baumannii is becoming a very serious issue, especially for nosocomial infections [3, 4]. Vaccination may be an alternative strategy for the prevention of A. baumannii infections. However, there is still no licensed vaccine against A. baumannii.
During the last decade, the development of vaccines against A. baumannii has primarily focused on various forms of recombinant antigens [5–7]. Animal studies showed that some single recombinant protein based vaccines provided only partial protection against A. baumannii challenge, especially for heterogeneous bacterial strains, therefore the vaccine design needs to be optimised further [8, 9]. Recently, many new approaches to vaccine design have emerged, including the multi-epitope assembly peptide (MEP), which contains B-cell epitopes, T-cell epitopes or other specific residues from several different proteins. This kind of vaccine has been proved effective for the control of several other pathogens in animal models [10, 11].
Protein FilF is a putative pilus assembly protein located in the outer membrane of A. baumannii, and it was found to be highly conserved among the A. baumannii strains. Animal vaccination with recombinant FilF generated high antibody titres and provided partial protection against a lethal dose of A. baumannii [12]. Another protein, named NucAb, was predicted to possess many attributes of a promising vaccine candidate, such as outer membrane localisation, high adhesin probability, non-homology to human proteins and presence of B-cell and T-cell epitopes with high affinity to HLA alleles prevalent in a North Indian population [13]. Ata is a trimeric autotransporter protein, which is important for biofilm formation, binding to extracellular matrix/basal membrane proteins, and the adhesion of A. baumannii cells to collagen type IV. In a pneumonia model of infection in both immunocompetent and immunocompromised mice, antiserum to Ata significantly reduced the levels of A. baumannii ATCC 17978 and two multidrug-resistant strains in the lungs of infected mice [14]. All these studies indicate that FilF, NucAb and Ata are ideal targets for vaccine design.
In this study, a MEP based on the above described three antigens of A. baumannii was designed, expressed recombinantly and purified, and its immunogenicity and protective immunity were evaluated in Balb/c mice. The results showed that the recombinant multi-epitope assembly peptide (rMEP) had strong and specific antigenicity and induced potent protective immunity in Blab/c mice, providing a basis for the development of novel vaccines against A. baumannii.
Materials and methods
Materials
Experimental animals
Seventy-two pathogen-free Balb/c female mice were obtained from the Laboratory Animal Centre of Chongqing Medical University (Chongqing, China) and held in a specific-pathogen-free laboratory. The mice were 6 to 8 weeks old at the time of vaccination. Mice were housed six to a cage and fed commercial mouse chow. Animal experiments were approved by the Committee on Ethics and Experimentation at the Institute of Medical Biology, Chinese Academy of Medical Sciences (project ID: SCXKCHUAN2018-18).
Bacterial strains and plasmid vector
Acinetobacter baumannii ATCC19606 strain was obtained from China General Microbiological Culture Collection Centre (CGMCC, China). Prokaryotic expressing vector pColdIand Escherichia coli BL21 strain were purchased from Takara Company (Takara Biotechnology Co., Ltd).
Methods
Design of the MEP
The B-cell epitopes of FilF (protein ID: WP_001034582.1) and NucAb (protein ID: WP_000847219.1) were predicted with BepiPred 1.0 (threshold value 0.35) and ABCpred (threshold value 0.51) software [15, 16]. The mouse H-2-IAb and H-2-IAd T-cell epitopes of FilF and NucAb were predicted with NetMHCII2.3 software (http://www.cbs.dtu.dk/services/NetMHCII/) and Rankpep software (http://imed.med.ucm.es/Tools/rankpep.html) [17, 18]. Common B-cell (or T-cell) epitopes predicted by two different softwares were picked out for the following analysis. The predicted B-cell epitopes were chemically synthesised and identified by serological experiments, and the predicted T-cell epitopes were chemically synthesised and identified by splenocytes stimulation test (splenocytes were isolated from A. baumannii ATCC19606 whole cell protein immunised mice). The probable adhesin active peptide of Ata (Protein ID: WP_001045602.1) was predicted with Vaxign software (http://www.violinet.org/vaxign/) [19]. The B- and T-cell epitopes of antigen Ata were also analysed with the above described software. Based on the above analysis, four B-cell epitopes and two T-cell epitopes from FilF and NucAb, and a 202 amino acid peptide of Ata were selected to design the MEP, and peptide GGGGSGGGG was used as the flexible linker between the peptides.
Molecular cloning, expression and purification of the rMEP
The gene of the designed MEP was chemically synthesised (by Sangon Biotechnology Company, Shanghai, China), and cloned into the sites between BamHIand SalIof plasmid pColdI to construct the recombinant plasmid pColdI-MEP, and thereafter transferred into E. coli BL21. Protein expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 15°C for 24 hours and analysed with sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). The 6×His-tagged recombinant protein was purified through nickel affinity chromatography. Briefly, the bacteria were harvested from 200 ml of bacteria culture by centrifugation and washed with 1 × phosphate buffered saline (PBS) (pH 7.4); the bacterial pellet was resuspended with 30 ml lysis buffer (20 mM tris-HCl, 8 M Urea, 500 mM NaCl, pH 8.0) and lysed by ultrasound on ice for 20 minutes. The lysis supernatant was collected by centrifugation (10,000×g, 20 min, 4°C). The collected supernatant was loaded on an Ni-NTA column previously equilibrated with equilibration buffer (20 mM tris-HCl, 8 M Urea, 500 mM NaCl, 10 mM imidazole, pH 8.0), the flow-through discarded, and the nonspecific proteins were removed by washing with five column volumes of washing buffer (20 mM tris-HCl, 8 M urea, 500 mM NaCl, 10 mM imidazole, pH 8.0). The bound protein was eluted with elution buffer (20 mM tris-HCl, 8 M urea, 500 mM NaCl, 500 mM imidazole, pH 8.0), eluted fractions were collected and analysed by 12% SDS-PAGE, and the recombinant protein was identified by Western-blot. The purified protein was dialysed in PBS buffer to remove urea and imidazole. Protein concentration was measured by using Bradford Protein Assay kit (Thermo Fisher Scientific Co., Ltd China).
Immunization of Balb/c mice
Sixty-three Balb/c female mice were randomly divided into three groups: an rMEP-immunised group, an adjuvant-treated group and a PBS-treated group. Each mouse in the rMEP group was immunised subcutaneously with 30 μg purified rMEP in 100 μl PBS formulated with an equal volume of Freund’s complete adjuvant (Sigma), and boosted with the same dose of rMEP in 100 μl PBS formulated with 100 μl Freund’s incomplete adjuvant (Sigma) on days 14 and 21. In the adjuvant-treated group, each mouse was injected subcutaneously with 100 μl Freund’s complete adjuvant on the first day and 100 μl Freund’s incomplete adjuvant on days 14 and 21. In the PBS-treated group, each mouse was injected subcutaneously with 100 μl PBS on day 0, 14 and 21. Six mice were randomly selected from each group for immunological assay. The other animals in each group were used for challenge testing.
Antibody titre measurement with ELISA
Blood and serum samples were taken from the inner canthus of six mice in each group on days 7 and 21after the third immunisation. Sera were tested through indirect enzyme-linked immunosorbent assay (ELISA) to assess antigen-specific antibody titres. Briefly, ELISA plate was coated with 0.2 μg per well of rMEP overnight, and blocked with PBST (0.01 M PBS, 0.05% Tween 20) containing 5% bovine serum albumin (BSA) (w/v) at room temperature for 3 hours. After three plate washes with PBST, 100 μl of 2-fold serially diluted serum samples (beginning at a 1/50 dilution) were added to the corresponding wells and incubated at 37 °C for 1.5 hours. The plate was washed three times with PBST, then 100 μl horseradish peroxidase (HRP)-conjugated donkey anti-mouse IgG (BBI Life Science) diluted in PBST (containing 1% BSA, 1/5000 dilution) was added to each well and incubated at 37 °C for 1 hour. After washing three times with PBST, 100 μl of 3,3´5,5´-tetramethylbenzidine (TMB) substrate solution was added to each well, mixed and incubated at room temperature in the dark for 20 minutes, The reaction was stopped with the addition of 50 μl of 2 M H2SO4, optical density (OD) values were determined at 450 nm on a microplate reader (Bio-Rad iMark). The endpoint titre was defined as the highest dilution at which the OD at 450 nm was at least 0.1 above that of the background well; in the background wells serum was replaced with PBS [20, 21].
Antiadhesive capacity of antiserum to rMEP
Cell culture plates (96-well) were coated with 5 μg collagen type IV (Sigma Aldrich) per well overnight, blocked with PBS containing 5% BSA for 3 hours at room temperature, washed with PBS three times. To each well were added 5×105 colony-forming units (CFU) of A. baumannii cells which had been pre-incubated with either anti-rMEP antibodies or the control mouse serum in a total volume of 100 μl (diluted 1/500 in PBS) for 30 minutes at 37 °C and incubated for 1 hour at 37 °C. The wells were washed with PBS four times and rinsed with 100 μl 0.25% trypsin solution to release the bound bacteria. The rinsed solution was serially diluted, plated on LB agar plates, and incubated at 37 °C overnight. The adherent A. baumannii cells were numbered and calculated.
Antigen-specific proliferation and cytokine secretion of splenocytes
Six mice in each group were sacrificed after the second blood sampling taken from the inner canthus. Spleens were taken aseptically and cut into pieces with surgical scissors, and then transferred to a 200 mesh stainless steel net which was put into a 16 cm2 culture dish containing 6 ml PBS. The spleen pieces were squeezed with syringe needle cores to release the tissue cells and the splenocytes were isolated with commercial mouse lymphocyte separation medium (Sangon Biotch. Shanghai, China). The splenocytes were washed and resuspended in complete RPMI 1640 medium supplemented with 10% fetal calf serum. Splenocytes were seeded in 96-well flat-bottomed plates (100 μl per well of 5×105 cells), and rMEP was added at a final concentration of 20 μg/ml (splenocytes from three untreated normal Balb/c mice were used as negative control), After incubation for 46 hours, CCK-8 reagent (10 μl) was added to each well and incubate continued for 2 hours at 37 °C. OD values were measured at 450 nm with a microplate reader. The proliferation degree was expressed as the stimulation index (SI) calculated as A450 nm experimental group / A450 nm negative control group. For detection of cytokine secretion, splenocytes were seeded in 96-well cell culture plates (100 μl per well of 5×106 cells), and rMEP was added at a final concentration of 20 μg/ml. After incubation for 72 hours, the culture supernatants were collected for cytokine detection. Interleukin-4 (IL-4) and interferon-gamma (IFN-γ) were examined by use of mouse cytokine EIA kits according to the manufacturer’s instructions.
Challenge of mice with A. baumannii ATCC19606 strain
A. baumannii ATCC19606 strain was grown in LB broth to the late-logarithmic phase at 37 °C. Cells were harvested by centrifugation at 4000×g for 10 minutes, washed and resuspended in PBS. Three weeks after the third immunisation, 15 mice in each group were injected intraperitoneally with 2×108 CFU/per mouse of A. baumannii cells. Nine mice in each group were randomly selected for the observation of survival rate everyday post-infection. The other six mice were used to detect the bacterial load in mouse blood 12 hours post-infection and to evaluate pathological changes in lung tissue 24 hours post-infection.
Detection of the bacterial loads in mice blood
Blood samples were aseptically taken from the caudal vein of five mice in each group 12 hours post-challenge, 10 μl of each blood sample was serially diluted with PBS and spread plated on Luria-Bertani (LB) agar plates followed by incubation at 37 °C overnight. The colony forming units (CFU) were counted and the total bacterial CFU in 1 ml blood calculated. The results were expressed as the mean of log10 CFU in 1 ml blood of the infected mice.
Histopathological analysis
Three mice challenged with A. baumannii in each group and three untreated normal mice were sacrificed 24 hours post-challenge, and lung specimens were fixed in 10% neutrally buffered formalin and paraffin embedded. Deparaffinised sections were stained with haematoxylin-eosin and observed under microscope at 100× magnification. The pathological changes were recorded by a pathologist with no prior knowledge of the treatment groups.
Statistical analysis
All the statistical analysis were processed with SPSS software version 18.0 (SPSS Inc, Chicago, IL, USA). Data were analysed with one-way analysis of variance (ANOVA) followed by Dunnett’s test. The data were considered significantly different at p <0.05.
Results
The epitope prediction, the design of the MEP, the expression and purification of the rMEP, the immunisation and the challenge test were independently completed in our laboratory. The pathological changes in the mouse lung specimens were assessed in a teaching hospital of our college. The total number of Balb/c mice used in this study was 72. Nine of these mice were left as the untreated normal mice. Each of the three test groups included 21 mice. Six mice in each test group were randomly picked out for serum IgG, splenocyte proliferation and cytokine secretion assays at days 7 and 21 after the third immunisation. Fifteen mice in each test group were used in the challenge test at day 21 after the third immunisation. Nine mice challenged with A. baumannii in each group were randomly picked out for the observation of survival rate. The other six mice challenged with A. baumannii in each group were used for detection of blood bacterial loads and lung tissue pathology. Because one of the six mice challenged with A. baumannii in the PBS-treated group and one of the six mice challenged with A. baumannii in the adjuvant-treated group died at 12 hours post-challenge, bacterial load data were collected from only five mice in each group. Because three of the six mice challenged with A. baumannii in the PBS-treated group and two of the six mice challenged with A. baumannii in the adjuvant-treated group died at 24 hours post-challenge, only three mice in each group were used for the detection of pathological changes in lung tissue.
Epitope prediction and design of MEP
Nine peptides from the B-cell prediction results (four from FilF, five from NucAb) were chemically synthesised. The serological testing showed that two of the four peptides from FilF and four of the five peptides from NucAb were recognised by serum from mice immunised with A. baumannii whole cell protein (table 1). Four B-cell epitopes (FilF469–484, FilF162–177, NucAb130–141 and NucAb613–628) were picked out for the design of the MEP (because NucAb130–141 and NucAb613–628 showed much higher OD values than NucAb185–197 and NucAb293–304, only NucAb130–141 and NucAb613–628 were selected for the design of the MEP). Six peptides from the T-cell prediction results (three from FilF, three from NucAb) were chemically synthesised. Splenocytes from the A. baumannii whole cell protein immunised mice could be stimulated by four of the six synthetic peptides (table 2). Two T-cell epitopes (FilF116–125 and NucAb405–420) with relatively high stimulation index (SI) were picked out for the design of the MEP. A 202 amino acid peptide showed high adhesin probability (0.734) was predicted by Vaxign software and selected for the backbone of the MEP. Bioinformatics analysis showed that the 202 amino acid peptide also contained several B- and T-cell epitopes of the antigen Ata. Because the 202 amino acid peptide was inevitably to be used as the backbone of the MEP in this study, its B- and T-cell epitopes were not identified. The designed MEP for construction by genetic engineering is shown in figure 1.
Table 1 Serological detection and analysis of the synthetic peptides from B-cell epitope prediction.
|
Antigen
|
Peptides synthesised according to the B-cell epitope prediction (amino acid loci at the protein)
|
Diluted antiserum obtained from A. baumannii whole cell protein immunised mice (produced by our laboratory)
|
|
1/50
|
1/100
|
1/500
|
| FilF |
FQSIGLERGDNVVGDL(FilF162–177) |
(+) |
(+) |
(+) |
| VLTKAKPTQMIANAQT (FilF319–334) |
(–) |
(–) |
(–) |
| YGNIRTDIKPNATATD (FilF469–484) |
(+) |
(+) |
(+) |
| ASGANLTTYDNKTVNW(FilF577–592) |
(–) |
(–) |
(–) |
| NucAb |
RIGERPEAGWGT (NucAb130–141) |
(+) |
(+) |
(+) |
| NLSGTTTQPPIETVSC(NucAb185–197) |
(+) |
(+) |
(+) |
| QQDIQTCNSNMA (NucAb293–304) |
(+) |
(+) |
(+) |
| SKIIFDDGYNNQ (NucAb386–397) |
(–) |
(–) |
(–) |
| VDANSSDADQNDGQGC (NucAb613–628) |
(+) |
(+) |
(+) |
Table 2 Detection of the synthetic peptides induced proliferation of splenocytes from A.baumannii whole cell protein immunised mice.
|
Antigen
|
Peptides synthesized according to the T-cell epitope prediction (amino acid loci at the protein)
|
The stimulation index (SI) of the splenocytes from A. baumannii whole cell protein immunized mice (mean ± SD)
|
| FilF |
LWPFALTTIAL (FilF 6–16) |
2.51 ± 0.33 |
| TFYLQGAKNPRKVELG (FilF 96–111) |
0.96 ± 11 |
| DSVSKIQMTV (FilF 116–125) |
3.71 ± 0. 48 |
| NucAb |
YVQTPDTKARANVSNA (NucAb 239–254) |
1.21 ± 0.30 |
| TNFSAANTLRSGYQLK (NucAb 405-420) |
3.41 ± 0.16 |
| KIVSALKSIDA (NucAb 499–509) |
2.75 ± 0.42 |
Expression and purification of the rMEP
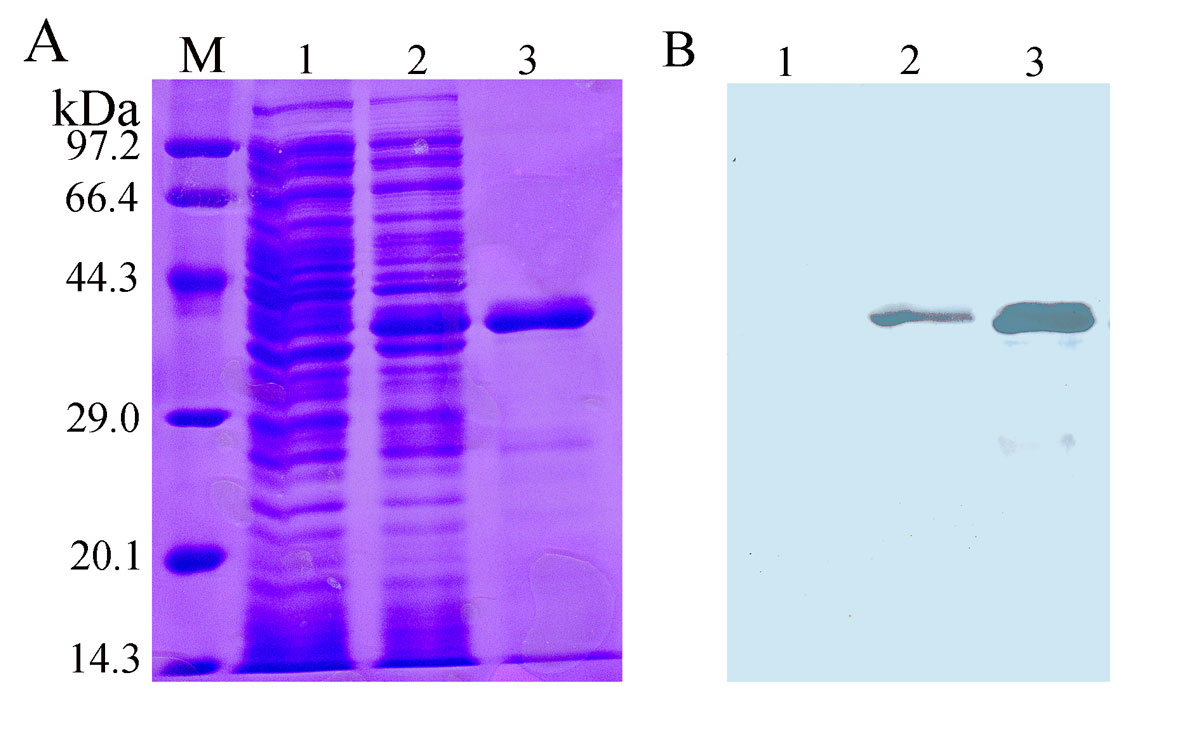
The corresponding gene of the designed MEP was chemically synthesised and cloned into plasmid pColdI. After DNA sequencing, the recombinant plasmid pColdI-MEP was transformed into E.coli BL21 and induced with 1 mM IPTG. Protein expression analysis showed that the recombinant protein was mainly expressed in soluble form (data not shown). As shown in figure 2, E.coli BL21 containing pColdI-MEP expressed a recombinant protein with a molecular weight of about 37 kDa, and the recombinant protein could be purified with nickel affinity chromatography column and recognised by anti-6×His antibody. These results demonstrated that the rMEP was successfully expressed and purified. Protein concentration analysis showed that about 3.5 mg rMEP with high purity can be purified from 200 ml bacterial culture.
Antigen-specific antibody in the serum of the immunised mice
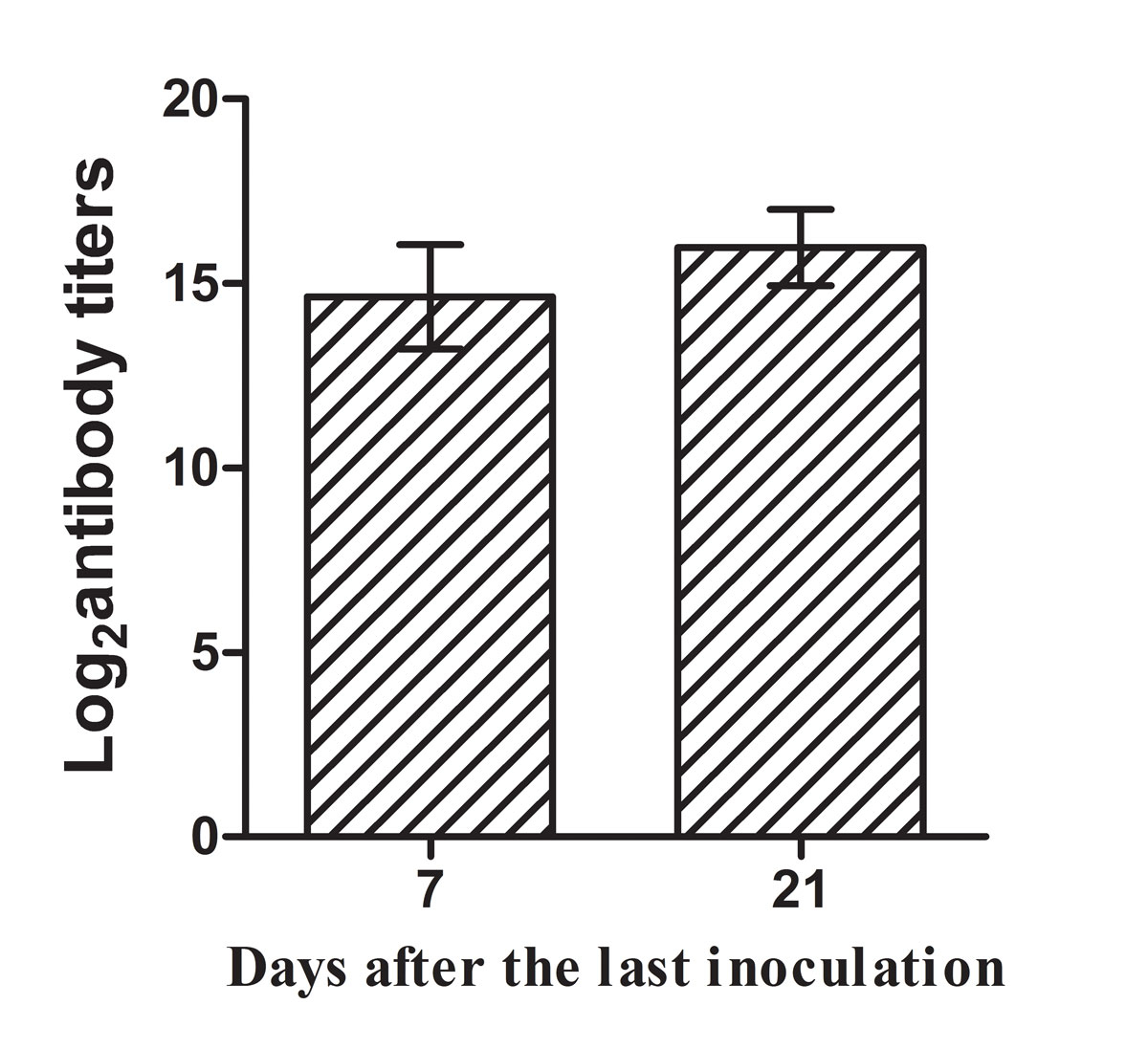
Blood and serum samples were taken from the inner canthus of six mice in each group at day 7 and 21 after the third immunisation. Sera were tested with indirect ELISA to assess antigen-specific antibody titres. The endpoint titre was defined as the highest dilution at which the OD at 450 nm was at least 0.1 above that of the background well; in the background wells serum was replaced with PBS. The log2 antibody titres were calculated and defined as the antigen-specific antibody level. Mice immunised with rMEP protein produced high level of IgG antibody at day 7 and 21 after the third immunisation (fig. 3). No antigen-specific antibody was detected in the serum of the mice from the PBS control group or the adjuvant-treated group (data not shown).
Antibody to rMEP blocks the binding of A. baumannii to collagen type IV
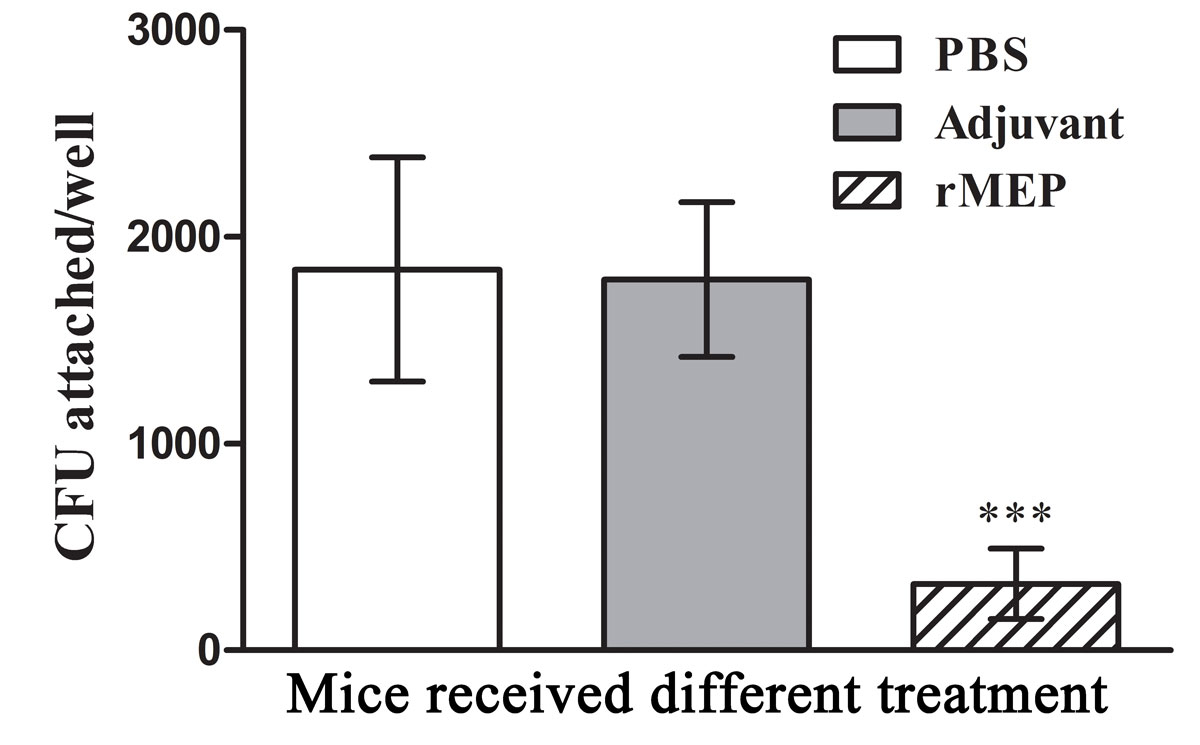
Cell culture plates (96-well) were coated with 5 μg collagen type IV per well overnight. After blocking and plate washing three times, A. baumannii ATCC strain 19606 cells were incubated for 30 minutes with sera from six mice in each group (1/500 dilution) and added to the corresponding well. Each well tested a serum sample from one mouse. Plates were incubated for 1 hour at 37 °C and washed with PBS. The attached bacteria were rinsed with trypsin solution and plated on LB agar followed by incubation at 37 °C overnight; bacterial CFU were counted and compared. The results showed that incubation of A. baumannii 19606 strain with antibodies to rMEP resulted in a dramatic decrease in the binding of bacterial cells to collagen type IV compared with the antiserum from the adjuvant- or PBS-treated mice (p <0.001; fig. 4).
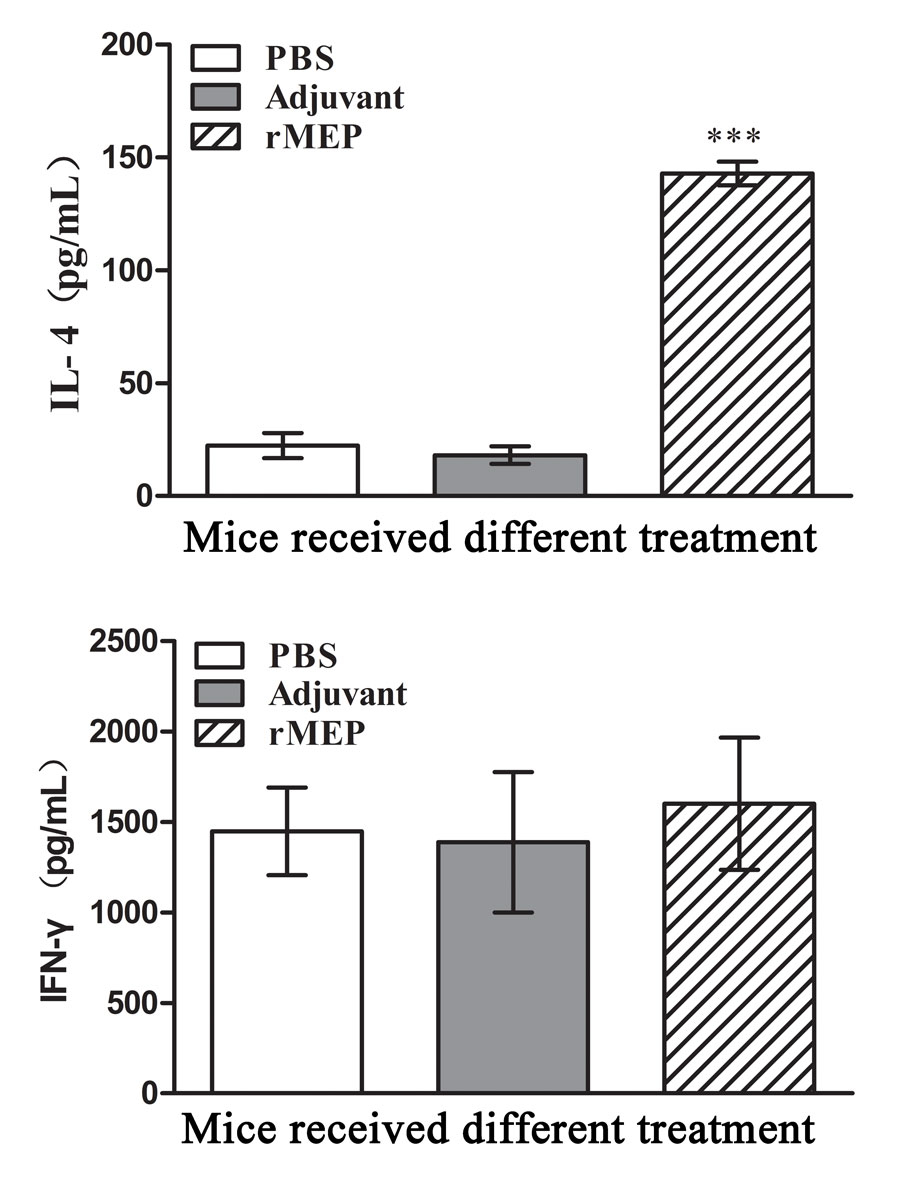
Splenocyte proliferation and cytokine secretion
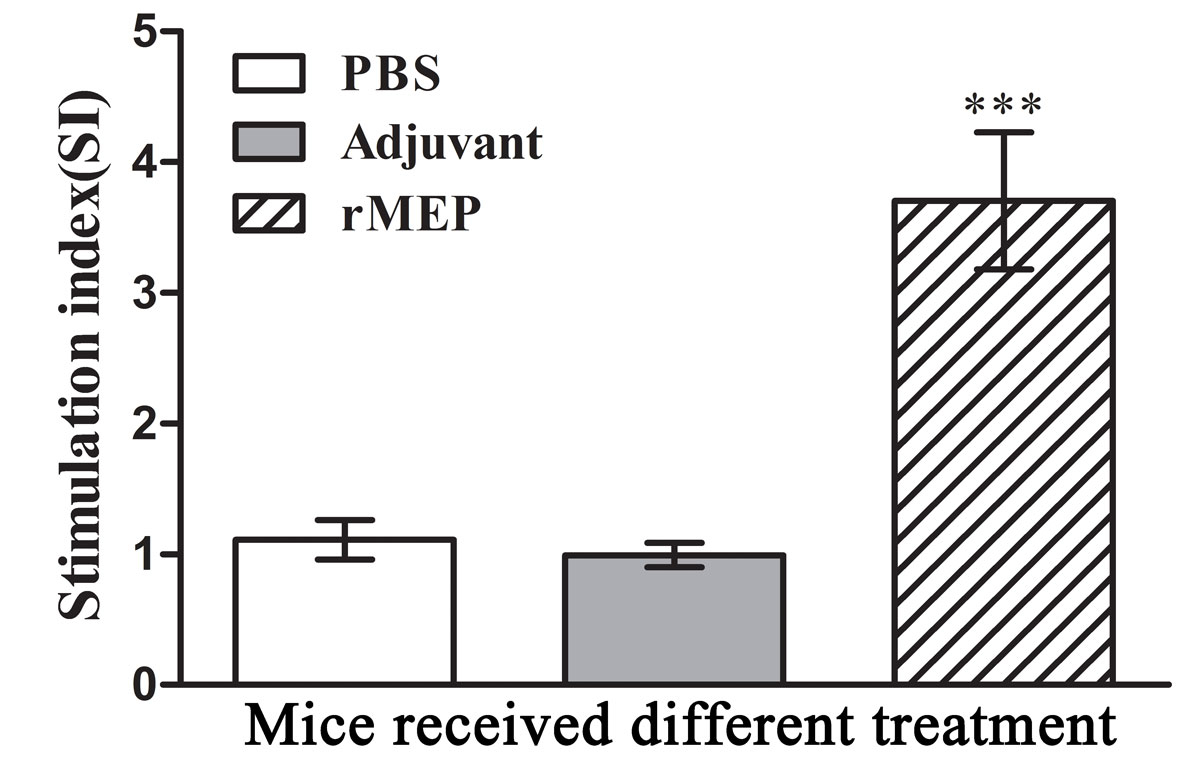
Splenocytes were isolated from six mice in each group at day 21 after the third immunisation and cultivated with the stimulation of rMEP. The antigen-specific proliferation of the splenocytes was detected using CCK-8 reagents. IL-4 and IFN-γ secretion in the cell culture supernatants were detected by means of a commercial EIA kit. Splenocytes from the rMEP-immunised mice showed much a higher antigen-specific proliferation rate than splenocytes from either adjuvant- or PBS-treated mice (p <0.001; fig. 5). Splenocytes from the rMEP-immunised mice produced higher levels of IL-4 than splenocytes from the other two groups (p <0.001), whereas IFN-γ secretion showed no difference between the three groups (fig. 6).
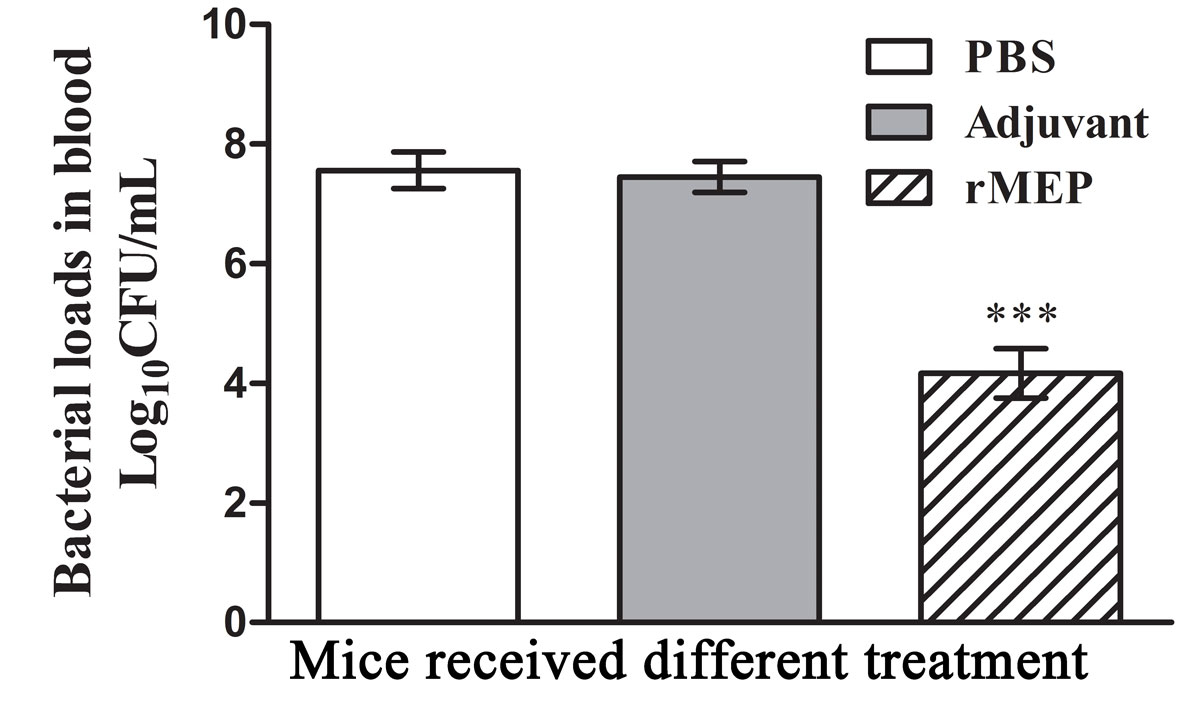
Bacterial loads in mice blood
Blood samples taken from five mice in each group at 12 hours post-challenge were serially diluted and spread plated on LB agar plates followed by incubation at 37 °C overnight. The number CFUs was counted, and the log
10
CFU/ml blood was calculated and compared. As shown in figure 7, mice immunised with rMEP had lower bacterial loads in the blood than mice from the adjuvant- or PBS-treated groups (p <0.001).
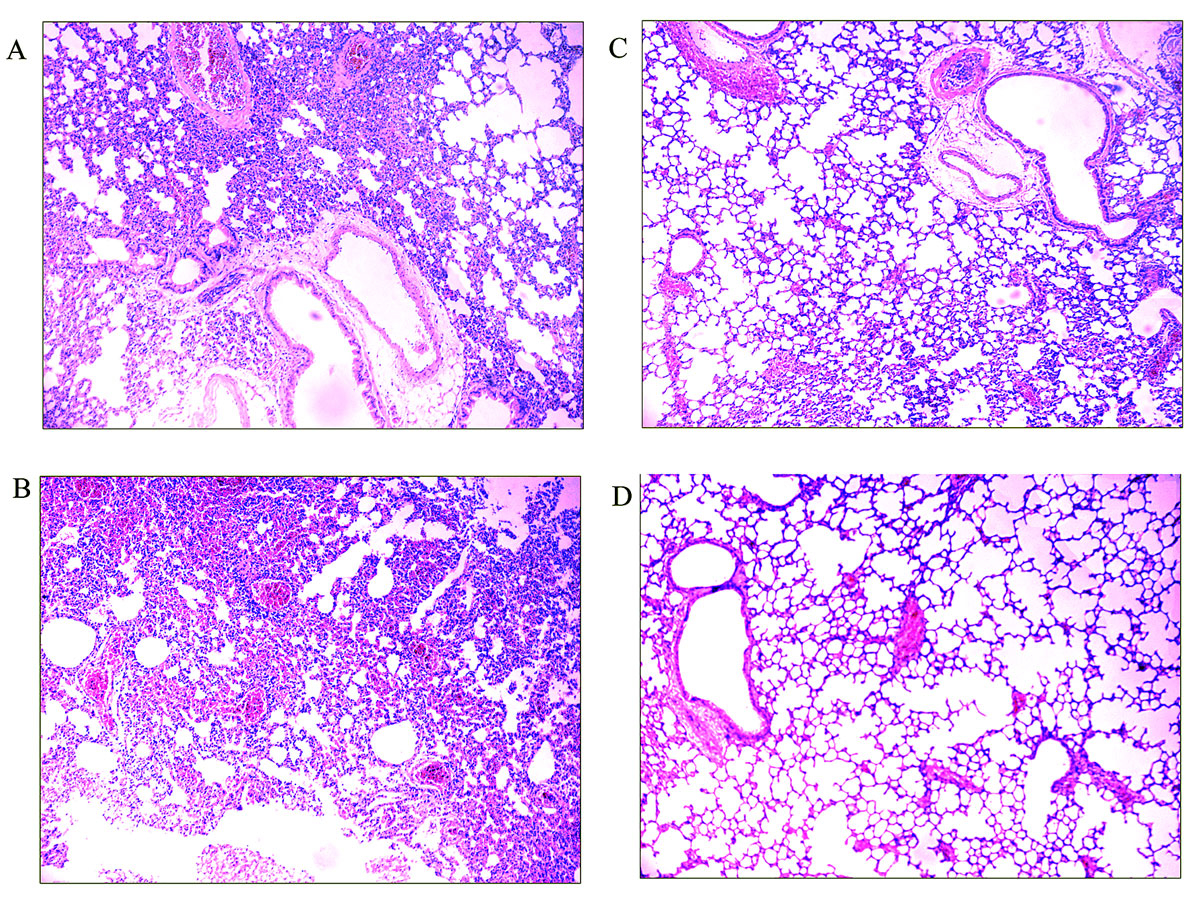
Pathological changes in mouse lung tissue
Lung specimens were taken from three mice challenged with A. baumannii in each group and three untreated normal mice at 24 hours post-challenge. Lung tissue sections were stained with haematoxylin-eosin and observed under a microscope. As shown in figure 8, lung tissue from either the PBS-treated mice or the adjuvant treated-mice showed widespread and severe interstitial pneumonia with intensive infiltration of inflammatory cells, especially in the perivascular areas. Lung tissue from rMEP-immunised mice appeared almost normal, with only very small number of inflammatory cells in the perivascular areas.
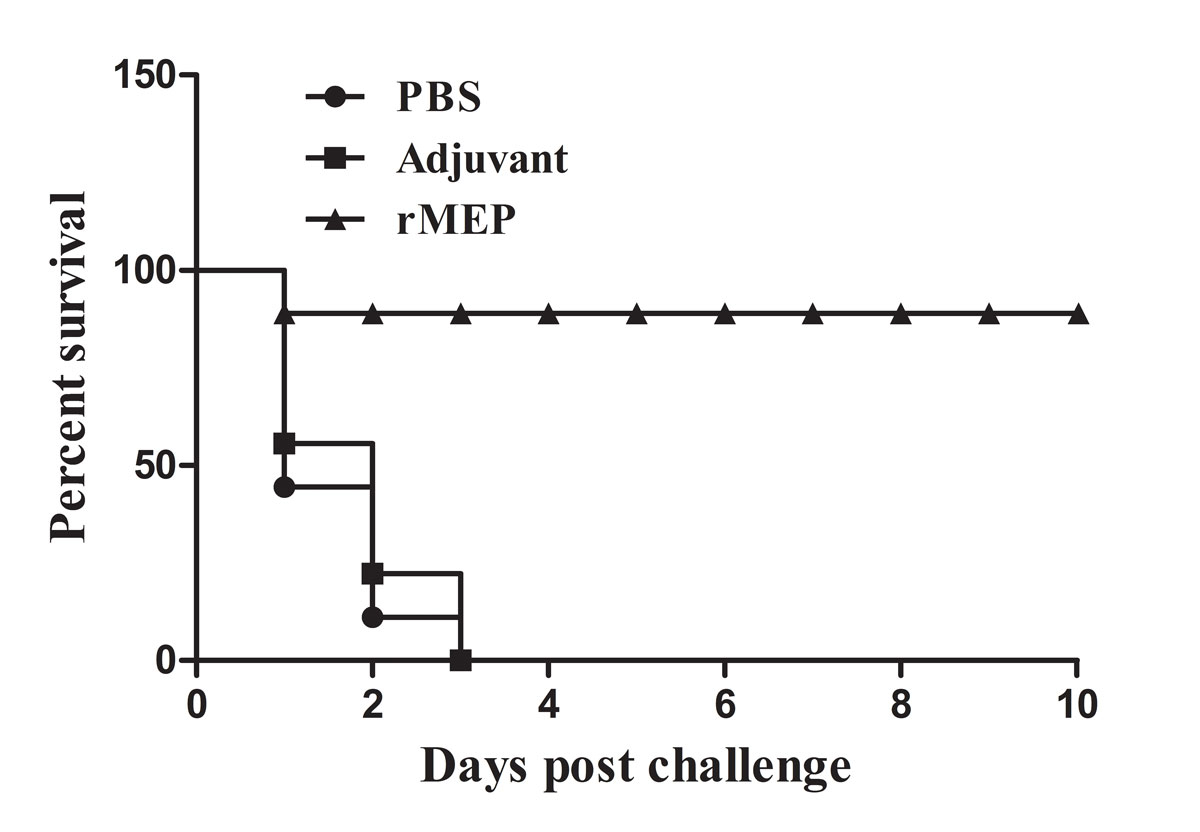
Mice immunised with rMEP had a high survival rate post-challenge
The protective efficacy of the rMEP was also determined by observing the survival rate after challenge with a lethal dose of A. baumannii. Nine mice challenged with A. baumannii were randomly selected for recording of survival every day until 10 days after the challenge. As shown in figure 9, all the mice in the PBS-treated and adjuvant-treated groups died within 72 hours post-challenge. Mice immunised with rMEP showed potent protection (88.9% survival at 10 days post-challenge).
Discussion
Several kinds of vaccine candidates against A. baumannii have been studied, such as inactivated whole cell vaccines (IWCs), outer membrane vesicles (OMVs), outer membrane complexes (OMCs) and subunit vaccines [22–25]. Active and passive immunisation with IWCs, OMVs and OMCs protected mice against bacterial challenge. However, owing to their complex composition and potential safety concerns, none is the ideal vaccine candidate [26–28]. Subunit vaccines based on a single recombinant protein or chimeric proteins have been evaluated for their antigenicity and protective efficacy in the past few years, but to date no A. baumannii vaccine has entered phase I clinical trials [29, 30]. More effort needs to be put into A. baumannii vaccine design and development. So, for the first time we designed a multi-epitope assembly peptide based on three proteins from A. baumannii, and its immunogenicity and protective efficacy were evaluated in Balb/c mice.
Antigen and epitope selection is the first critical step in constructing an effective multi-epitope vaccine. The advances in comparative genomics and bioinformatics enable researchers to capture more information for vaccine development [31, 32]. In the present study, we used multiple bioinformatics software for the prediction of B- and T-cell epitopes, which increased the reliability of the predicted peptides. In the subsequent serological and splenocyte proliferation tests, immuodominant epitopes were successfully picked out. The method described in this study may provide reference for future studies. The adhesin active peptide of Ata was predicted by using Vaxigen software. Although a preliminary experiment to identify its adhesin activity was not conducted, the anti-adhesive capacity of antiserum to rMEP confirmed the prediction.
To construct an ideal multi-epitope vaccine, the conformation of the recombinant protein is very important. If the recombinant protein is mainly expressed as an inclusion body, the natural conformation is hard to ensure even through refolding experiments, and epitope exposure will be severely disturbed. In this study, a prokaryotic expressing plasmid pColdI was used, and the cspA (cold shock protein A) promoter and related elements have been incorporated into this vector to up-regulate target protein expression at a relatively low temperature (12–15 °C). This vector allows expression of target proteins at high yield and increased solubility as compared with conventional E. coli expression systems. The rMEP is mainly expressed in soluble form. This soluble expression ensured the correct conformation of the rMEP, and provided convenience for the subsequent purification and immunisation testing.
High titres of antigen-specific antibody were detected in the serum of mice immunised with rMEP after boosting twice. This suggested that rMEP immunisation was capable of inducing a significant humoral response in mice. The rMEP vaccination induced much higher levels of antigen-specific IL-4 production from splenocytes of immunised mice, whereas the IFN-γ secretion showed no difference compared with the control group, which indicated that rMEP induced a predominant Th2-type immune response. The currently available research data indicate that A. baumannii is mainly an extracellular bacterial pathogen, and there is no evidence for the requirement of the CD8+ T-cell mediated immune response in the control of the infection [33–35], so we did not involve CD8+ T cell epitopes in the MEP design, and the cytotoxic T-cell response was not detected in this study.
Challenge experiments showed that the rMEP-immunised mice acquired potent protection (88.9%) against A. baumannii infection. The bacterial load in the blood of rMEP-inoculated mice after infection was much lower than that of the control group. Almost no pathological change was observed in the lung tissue of the rMEP-immunised mice post-challenge. These results indicate that the high titre of antigen-specific antibody contributed to the potent protection in the rMEP-immunised mice. Previous published data showed that protection in mice immunised with either rNucAb or rFilF was not so satisfactory (20~40% survival with rNucAb immunisation and 50% survival with rFilF) [12, 13]. It can be concluded that the rMEP induced more potent protective immunity than its individual antigens. As we did not compare the immunogenicity and protective immunity of the rMEP with any one of the previously published candidate vaccines, further study will be needed to make this comparison.
In summary, a recombinant multi-epitope assembly peptide (rMEP) from A. baumannii was designed, expressed recombinantly and purified, and its immunogenicity and protective efficacy were evaluated in Balb/c mice. The results showed that rMEP was capable of inducing a remarkable humoral immune response and potent protection against lethal dose of A. baumannii challenge. It can be concluded that rMEP is a promising subunit vaccine candidate for the prevention of A. baumannii infections.
References
1
Garnacho
J
,
Sole-Violan
J
,
Sa-Borges
M
,
Diaz
E
,
Rello
J
. Clinical impact of pneumonia caused by Acinetobacter baumannii in intubated patients: a matched cohort study. Crit Care Med. 2003;31(10):2478–82. doi:.https://doi.org/10.1097/01.CCM.0000089936.09573.F3
2
Kim
SW
,
Choi
CH
,
Moon
DC
,
Jin
JS
,
Lee
JH
,
Shin
JH
, et al.
Serum resistance of Acinetobacter baumannii through the binding of factor H to outer membrane proteins. FEMS Microbiol Lett. 2009;301(2):224–31. doi:.https://doi.org/10.1111/j.1574-6968.2009.01820.x
3
Dijkshoorn
L
,
Nemec
A
,
Seifert
H
. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii
. Nat Rev Microbiol. 2007;5(12):939–51. doi:.https://doi.org/10.1038/nrmicro1789
4
McConnell
MJ
,
Rumbo
C
,
Bou
G
,
Pachón
J
. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii
. Vaccine. 2011;29(34):5705–10. doi:.https://doi.org/10.1016/j.vaccine.2011.06.001
5
Lin
L
,
Tan
B
,
Pantapalangkoor
P
,
Ho
T
,
Hujer
AM
,
Taracila
MA
, et al.
Acinetobacter baumannii rOmpA vaccine dose alters immune polarization and immunodominant epitopes. Vaccine. 2013;31(2):313–8. doi:.https://doi.org/10.1016/j.vaccine.2012.11.008
6
Sefidi
MD
,
Rasooli
I
,
Owlia
P
,
Talei
D
,
Astaneh
SD
,
Nazarian
S
. Adjuvant role of Pseudomonas flagellin for Acinetobacter baumannii biofilm associated protein. World J Methodol. 2016;6(3):190–9. doi:.https://doi.org/10.5662/wjm.v6.i3.190
7
Huang
W
,
Wang
S
,
Yao
Y
,
Xia
Y
,
Yang
X
,
Long
Q
, et al.
OmpW is a potential target for eliciting protective immunity against Acinetobacter baumannii infections. Vaccine. 2015;33(36):4479–85. doi:.https://doi.org/10.1016/j.vaccine.2015.07.031
8
Chiang
MH
,
Sung
WC
,
Lien
SP
,
Chen
YZ
,
Lo
AF
,
Huang
JH
, et al.
Identification of novel vaccine candidates against Acinetobacter baumannii using reverse vaccinology. Hum Vaccin Immunother. 2015;11(4):1065–73. doi:.https://doi.org/10.1080/21645515.2015.1010910
9
Perez
F
,
Bonomo
RA
. Vaccines for Acinetobacter baumannii: thinking “out of the box”. Vaccine. 2014;32(22):2537–9. doi:.https://doi.org/10.1016/j.vaccine.2014.03.031
10
Yang
W
,
Jackson
DC
,
Zeng
Q
,
McManus
DP
. Multi-epitope schistosome vaccine candidates tested for protective immunogenicity in mice. Vaccine. 2000;19(1):103–13. doi:.https://doi.org/10.1016/S0264-410X(00)00165-1
11
Xu
Q
,
Ma
X
,
Wang
F
,
Li
H
,
Zhao
X
. Evaluation of a multi-epitope subunit vaccine against avian leukosis virus subgroup J in chickens. Virus Res. 2015;210:62–8. doi:.https://doi.org/10.1016/j.virusres.2015.06.024
12
Singh
R
,
Garg
N
,
Shukla
G
,
Capalash
N
,
Sharma
P
. Immunoprotective efficacy of Acinetobacter baumannii outer membrane protein, FilF, predicted in silico as a potential vaccine candidate. Front Microbiol. 2016;7:158. doi:.https://doi.org/10.3389/fmicb.2016.00158
13
Garg
N
,
Singh
R
,
Shukla
G
,
Capalash
N
,
Sharma
P
. Immunoprotective potential of in silico predicted Acinetobacter baumannii outer membrane nuclease, NucAb. Int J Med Microbiol. 2016;306(1):1–9. doi:.https://doi.org/10.1016/j.ijmm.2015.10.005
14
Bentancor
LV
,
Routray
A
,
Bozkurt-Guzel
C
,
Camacho-Peiro
A
,
Pier
GB
,
Maira-Litrán
T
. Evaluation of the trimeric autotransporter Ata as a vaccine candidate against Acinetobacter baumannii infections. Infect Immun. 2012;80(10):3381–8. doi:.https://doi.org/10.1128/IAI.06096-11
15
Larsen
JE
,
Lund
O
,
Nielsen
M
. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2(1):2. doi:.https://doi.org/10.1186/1745-7580-2-2
16
Saha
S
,
Raghava
GP
. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40–8. doi:.https://doi.org/10.1002/prot.21078
17
Nielsen
M
,
Lund
O
,
Buus
S
,
Lundegaard
C
. MHC class II epitope predictive algorithms. Immunology. 2010;130(3):319–28. doi:.https://doi.org/10.1111/j.1365-2567.2010.03268.x
18
Reche
PA
,
Glutting
JP
,
Zhang
H
,
Reinherz
EL
. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56(6):405–19. doi:.https://doi.org/10.1007/s00251-004-0709-7
19
He
Y
,
Xiang
Z
,
Mobley
HL
. Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol. 2010;2010:297505. doi:.https://doi.org/10.1155/2010/297505
20
García-Quintanilla
M
,
Pulido
MR
,
Pachón
J
,
McConnell
MJ
. Immunization with lipopolysaccharide-deficient whole cells provides protective immunity in an experimental mouse model of Acinetobacter baumannii infection. PLoS One. 2014;9(12):e114410. doi:.https://doi.org/10.1371/journal.pone.0114410
21
Huang
W
,
Wang
S
,
Yao
Y
,
Xia
Y
,
Yang
X
,
Li
K
, et al.
Employing Escherichia coli-derived outer membrane vesicles as an antigen delivery platform elicits protective immunity against Acinetobacter baumannii infection. Sci Rep. 2016;6(1):37242. doi:.https://doi.org/10.1038/srep37242
22
McConnell
MJ
,
Domínguez-Herrera
J
,
Smani
Y
,
López-Rojas
R
,
Docobo-Pérez
F
,
Pachón
J
. Vaccination with outer membrane complexes elicits rapid protective immunity to multidrug-resistant Acinetobacter baumannii. Infect Immun. 2011;79(1):518–26. doi:.https://doi.org/10.1128/IAI.00741-10
23
Lin
L
,
Tan
B
,
Pantapalangkoor
P
,
Ho
T
,
Hujer
AM
,
Taracila
MA
, et al.
Acinetobacter baumannii rOmpA vaccine dose alters immune polarization and immunodominant epitopes. Vaccine. 2013;31(2):313–8. doi:.https://doi.org/10.1016/j.vaccine.2012.11.008
24
McConnell
MJ
,
Pachón
J
. Active and passive immunization against Acinetobacter baumannii using an inactivated whole cell vaccine. Vaccine. 2010;29(1):1–5. doi:.https://doi.org/10.1016/j.vaccine.2010.10.052
25
McConnell
MJ
,
Rumbo
C
,
Bou
G
,
Pachón
J
. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii. Vaccine. 2011;29(34):5705–10. doi:.https://doi.org/10.1016/j.vaccine.2011.06.001
26
Moriel
DG
,
Beatson
SA
,
Wurpel
DJ
,
Lipman
J
,
Nimmo
GR
,
Paterson
DL
, et al.
Identification of novel vaccine candidates against multidrug-resistant Acinetobacter baumannii
. PLoS One. 2013;8(10):e77631. doi:.https://doi.org/10.1371/journal.pone.0077631
27
Ahmad
TA
,
Tawfik
DM
,
Sheweita
SA
,
Haroun
M
,
El-Sayed
LH
. Development of immunization trials against Acinetobacter baumannii
. Trials Vaccinol. 2016;5:53–60. doi:.https://doi.org/10.1016/j.trivac.2016.03.001
28
Garcia-Quintanilla
M
,
Pulido
MR
,
McConnell
MJ
. First steps towards a vaccine against Acinetobacter baumannii
. Curr Pharm Biotechnol. 2013;14(10):897–902. doi:.https://doi.org/10.2174/1389201014666131226123511
29
Guo
SJ
,
Ren
S
,
Xie
YE
. Evaluation of the protective efficacy of a fused OmpK/Omp22 protein vaccine candidate against Acinetobacter baumannii infection in mice. Biomed Environ Sci. 2018;31(2):155–8.
30
Chen
W
. Current advances and challenges in the development of Acinetobacter vaccines. Hum Vaccin Immunother. 2015;11(10):2495–500. doi:.https://doi.org/10.1080/21645515.2015.1052354
31
Sette
A
,
Rappuoli
R
. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33(4):530–41. doi:.https://doi.org/10.1016/j.immuni.2010.09.017
32
Hosseingholi
EZ
,
Rasooli
I
,
Gargari
SL
. In silico analysis of Acinetobacter baumannii phospholipase D as a subunit vaccine candidate. Acta Biotheor. 2014;62(4):455–78. doi:.https://doi.org/10.1007/s10441-014-9226-8
33
Russo
TA
,
Beanan
JM
,
Olson
R
,
MacDonald
U
,
Luke
NR
,
Gill
SR
, et al.
Rat pneumonia and soft-tissue infection models for the study of Acinetobacter baumannii biology. Infect Immun. 2008;76(8):3577–86. doi:.https://doi.org/10.1128/IAI.00269-08
34
Bist
P
,
Dikshit
N
,
Koh
TH
,
Mortellaro
A
,
Tan
TT
,
Sukumaran
B
. The Nod1, Nod2, and Rip2 axis contributes to host immune defense against intracellular Acinetobacter baumannii infection. Infect Immun. 2014;82(3):1112–22. doi:.https://doi.org/10.1128/IAI.01459-13
35
García-Patiño
MG
,
García-Contreras
R
,
Licona-Limón
P
. The immune response against Acinetobacter baumannii, an emerging pathogen in nosocomial infections. Front Immunol. 2017;8:441. doi:.https://doi.org/10.3389/fimmu.2017.00441