Vascular endothelial growth factor biology for regenerative angiogenesis
DOI: https://doi.org/10.4414/smw.2019.20011
Andrea
Uccelliab, Thomas
Wolffb, Paolo
Valenteab, Nunzia
Di Maggioab, Matteo
Pellegrinoab, Lorenz
Gürkeb, Andrea
Banfiab, Roberto
Gianni-Barreraab
aCell and Gene Therapy, Department of Biomedicine, University of Basel, Switzerland
bVascular Surgery, Department of Surgery, Basel University Hospital, Switzerland
Summary
Despite major advances in medical, catheter-based or surgical treatment, cardiovascular diseases such as peripheral artery disease and coronary artery disease still cause significant morbidity and mortality. Furthermore, many patients do not qualify for catheter-based treatment or bypass surgery because of advanced disease or surgical risk. There is therefore an urgent need for novel treatment strategies. Therapeutic angiogenesis aims to restore blood flow to ischaemic tissue by stimulating the growth of new blood vessels through the local delivery of angiogenic factors, and may thus be an attractive treatment alternative for these patients. Angiogenesis is a complex process and the growth of normal, stable and functional vasculature depends on the coordinated interplay of different cell types and growth factors. Vascular endothelial growth factor-A (VEGF) is the fundamental regulator of vascular growth and the key target of therapeutic angiogenesis approaches. However, first-generation clinical trials of VEGF gene therapy have been disappointing, and a clear clinical benefit has yet to be established. In particular, VEGF delivery (a) appears to have a very limited therapeutic window in vivo: low doses are safe but mostly inefficient, whereas higher doses become rapidly unsafe; and (b) requires a sustained expression in vivo of at least about four weeks to achieve stable vessels that persist after cessation of the angiogenic stimulus. Here we will review the current understanding of how VEGF induces the growth of normal or pathological blood vessels, what limitations for the controlled induction of safe and efficient angiogenesis are intrinsically linked to the biological properties of VEGF, and how this knowledge can guide the design of more effective strategies for therapeutic angiogenesis.
Ischaemic cardiovascular disease and clinical trials of therapeutic angiogenesis
Atherosclerotic cardiovascular disease is the most frequent cause of death in Western societies. Despite advances in medical and surgical therapy, the morbidity and mortality of coronary heart disease (CAD) and peripheral artery disease (PAD) remain very high [1]. Slowly growing atherosclerotic plaques cause progressive obstruction of feeding arteries and chronic clinical symptoms in the lower limbs, the heart or the brain, but their sudden rupture with subsequent thrombosis leads to acute events like myocardial infarction or stroke. The clinical picture of progressive CAD and PAD is extremely debilitating for the patient, with chest pain (angina) and severe dyspnoea or muscle pain in the lower limbs while walking short distances (claudication) progressing to pain at rest and to ischaemic ulcers and gangrene, requiring major amputations (PAD). As the major risk factors are obesity, diabetes and smoking, the prevalence of PAD and CAD is predicted to further increase in Western-style societies. Furthermore, many patients are not candidates for surgical treatment, because of unfavourable anatomy of the obstructions, diffuse disease or significant comorbidities [1]. Therapeutic angiogenesis aims to restore sufficient blood flow to ischaemic tissues by the generation of new vascular networks through the delivery of specific growth factors. It would provide a much-needed additional approach for the treatment of ischaemic conditions.
The master regulator of vascular growth is vascular endothelial growth factor-A (VEGF). Already, only a few years after its isolation and molecular cloning [2, 3], the first uncontrolled clinical attempts to induce therapeutic angiogenesis with its gene delivery were reported in PAD or CAD patients [4, 5], generating great excitement. However, despite success in preclinical models and early clinical studies, subsequent double-blind, randomised and placebo-controlled studies failed to show clear clinical efficacy for protein or gene delivery of either VEGF or other angiogenic factors (see comprehensive reviews by Losordo [6] and Rubanyi [7]).
All accumulated biological evidence supports the notion that VEGF is the key factor capable of kick-starting the complex cascade of events leading to the assembly and maturation of new vascular networks [8], suggesting that the choice of target does not explain these clinical failures. On the other hand, retrospective analyses identified several factors for the unsatisfactory results of VEGF gene therapy in clinical trials. Among these, the difficulty of delivering sufficient VEGF into the target tissues at safe vector doses is a key issue to overcome in order to achieve a therapeutic effect and correct ischaemia [9]. Here, we will review key aspects of VEGF biology that have significant implications for its therapeutic exploitation.
Angiogenesis and arteriogenesis
Angiogenesis is the growth or expansion of micro-vascular capillary networks from pre-existing ones and is driven by ischaemia and hypoxia. However, hypoxia-induced angiogenesis alone is not sufficient to restore physiological blood flow after an ischaemic event. In fact, capillaries are responsible for the metabolic exchanges between blood and tissue, but feeding arteries are required to supply sufficient blood flow to satisfy the tissue needs. In contrast, arteriogenesis is the formation of mature, functional arteries through the functional remodelling of pre-existing and little-perfused interconnecting arterioles after an occlusion of large conductance arteries, thereby bypassing the block and restoring blood flow to ischaemic tissue downstream [10]. Unlike angiogenesis, arteriogenesis is driven mostly by haemodynamic factors, such as an increase in stretch or fluid shear stress on endothelium, rather than hypoxia [11]. However, angiogenesis induced by the delivery of growth factors such as VEGF has been shown to drive the enlargement of upstream collateral arteries through increased shear stress and gap junction-mediated retrograde signalling along vessel walls, thereby effectively producing a biological bypass and restoring downstream perfusion [12–14]. The ability of micro-vascular expansion by growth factor delivery to drive upstream arteriogenesis has been clearly proven in animal models. Evidence for the same process in human patients undergoing pro-angiogenic therapies has also been reported [15–17], but thorough investigations have been hampered by the general lack of efficacy of first-generation clinical trials of therapeutic angiogenesis.
It must be remembered that preclinical studies in small, young rodents poorly recapitulate the clinical situation of aging human patients with several comorbidities, such as diabetes and hyperlipidaemia [18]. Better models have been introduced, including larger size (rabbit, pig) and accompanying disease conditions (diabetes and obesity in db/db mice or hyperlipidaemia in LDL-receptor knock-out mice). However, despite these improvements, the usefulness of preclinical animal models should probably be limited to establishing proof-of-principle, while therapeutic efficacy should be more appropriately determined in controlled clinical trials.
In conclusion, therapeutic angiogenesis can lead to arteriogenesis and so restore tissue perfusion, but a more comprehensive understanding of the complex underlying biology is required to significantly improve the efficacy of the treatment strategies and to exploit this concept in the clinical arena.
VEGF ligands and receptors
The mammalian VEGF family consists of five different polypeptides: VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF). Besides these canonical VEGFs, two other related factors have been isolated, one from the genome of the Orf virus (VEGF-E) [19] and one from the venom of the Eastern Cottonmouth snake (VEGF-F, or vammin) [20]. These do not play a role in mammalian angiogenesis. Despite this small number of members, the complexity of the group is high, due to alternative splicing (VEGF-A, VEGF-B and PlGF) and post-translational proteolytic processing (VEGF-A, VEGF-C and VEGF-D) [21]. VEGF-A, which was originally identified as vascular permeability factor (VPF), was the first member of this family to be characterised [22]. It is the most important family member, controlling blood vessel growth in both physiological and pathological angiogenesis. VEGF-B and PlGF play more accessory or tissue-specific roles, while VEGF-C and -D mostly regulate lymphatic vessels [8]. VEGF molecules bind to three distinct tyrosine kinase receptors in mammals: VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1), which can also exist as soluble forms, named sVEGFR-1 and sVEGFR-2 respectively [23], and VEGFR-3 (Flt-3). Ligand binding leads to receptor dimerisation, which is followed by autophosphorylation of the intracellular kinase domain and subsequent initiation of the signal transduction cascade. Depending on the ligand, both homodimeric and heterodimeric receptor complexes can be formed [23]. Furthermore, the three VEGF-Receptors are also able to interact with co-receptors of the neuropilin family (neuropilin-1, NRP1 and neuropilin-2, NRP2), macromolecules like heparan sulphate and proteoglycans (syndecan and glypican), non-VEGF-binding auxiliary proteins like vascular endothelial cadherin (VE-cadherin), integrins, ephrin-B2, and protein tyrosine phosphatase [24]. VEGFR-1, which can also bind to VEGF-B and PlGF [25], is the receptor with the highest affinity for VEGF-A, but it displays only weak tyrosine phosphorylation activity. As a consequence, the interaction of VEGFR-1 with the ligand does not start the signal transduction cascade efficiently [25], and VEGFR-1 may rather act as both a decoy receptor by sequestering VEGF-A [26] and as a modulator of the activity of VEGFR-2 homodimers [27]. VEGFR-2 is able to bind to VEGF-A with an affinity 10-fold lower than that of VEGFR-1, but it is the main mediator of VEGF-A activity in endothelial cell differentiation, proliferation, migration, angiogenesis and vessel permeabilisation [23]. VEGFR-2 is also able to bind to the processed forms of VEGF-C and VEGF-D [28]. VEGFR-3 is activated by VEGF-C and VEGF-D and is mainly implicated in the regulation of lymphatic endothelial cells [29].
VEGF and the extracellular matrix
Physiological vascular growth is finely regulated by the interaction of VEGF-A (from here on referred to simply as VEGF) with the extracellular matrix. The human Vegf-a gene is organised into eight exons, which generate multiple isoforms by alternative splicing of exons 6, 7 and 8, but all share the same N-terminal amino acid sequence, which contains the receptor-interacting site [30]. Furthermore, exon 8 contains a second (distal) splice site, which can generate an alternate sequence for the last six residues of all VEGF isoforms. This is important because the canonical isoforms, termed VEGFxxx (where “xxx” indicates the amino acid number of the mature protein), are pro-angiogenic, whereas the alternate versions, termed VEGFxxxb, are anti-angiogenic. Both the VEGFxxx and VEGFxxxb families of isoforms bind to their cognate receptors, but only the former are able to activate them [31]. While the anti-angiogenic VEGFxxxb isoforms certainly play important regulatory roles, here we will focus on the canonical, pro-angiogenic VEGF isoforms for their role in therapeutic angiogenesis.
The three major pro-angiogenic VEGF isoforms each differ in length between rodents and humans by one amino acid, with VEGF188/189 (rodent/human) encoded by exons 1–8, VEGF164/165 missing exon 6, and VEGF120/121 missing exons 6 and 7 [31]. As exons 6 and 7 encode heparin-binding domains, the key functional difference between these isoforms is their increasing affinity for extracellular matrix, from the shorter to the longer molecules [32]. Elegant studies in transgenic mice that selectively express only one of the principal VEGF isoforms without changes to the total expression levels showed that their matrix affinity is a key factor controlling physiological vascular morphogenesis. VEGF120 does not bind matrix and induces vascular networks with poor branching and abnormally enlarged diameters, whereas VEGF188 binds strongly and also causes defective vascular networks, but with opposite features, namely excessive branching and very small capillaries. In contrast, VEGF164 has intermediate affinity for extracellular matrix and is the only isoform capable of inducing physiologically patterned vasculature in the absence of the others [33]. Remarkably, the combination of VEGF120 and VEGF188 also leads to normal angiogenesis in the absence of VEGF164, showing that a balanced matrix-binding affinity, rather than a specific isoform, is required for proper vascular morphogenesis [33].
Cellular mechanisms of vascular growth by VEGF delivery: sprouting vs intussusception
The best understood mode of vascular growth is sprouting, by which new vessels invade avascular areas of tissue guided by gradients of factors, e.g. during embryonic development. However, sprouting is not the only way vessels can grow. It has become increasingly clear in the last decade that the alternative mechanism of intussusception (or splitting angiogenesis) plays an important role in the expansion and remodelling of vessel networks in already vascularised tissues, both in development and in post-natal life [34, 35].
Sprouting angiogenesis relies on the functional specification of endothelial cells into either tip or stalk cells. Tip cells do not have a lumen, extend numerous thin filopodia through which they can sense VEGF gradients, and migrate towards the VEGF source, thereby guiding the nascent sprout. Stalk cells, which instead proliferate just behind the tip, form the body of the sprout and start the process of lumen formation (fig. 1A) [36]. Physiological patterning of sprouting vessels requires a balanced formation of tip and stalk cells, which is finely regulated by Notch signalling. In fact, tip cells increase expression of the Notch ligand Delta-like-4 (Dll4) in response to VEGF signalling. This activates Notch1 in adjacent cells, instructing them in turn to downregulate VEGF-R2 and Dll4 and acquire the stalk phenotype instead [37].
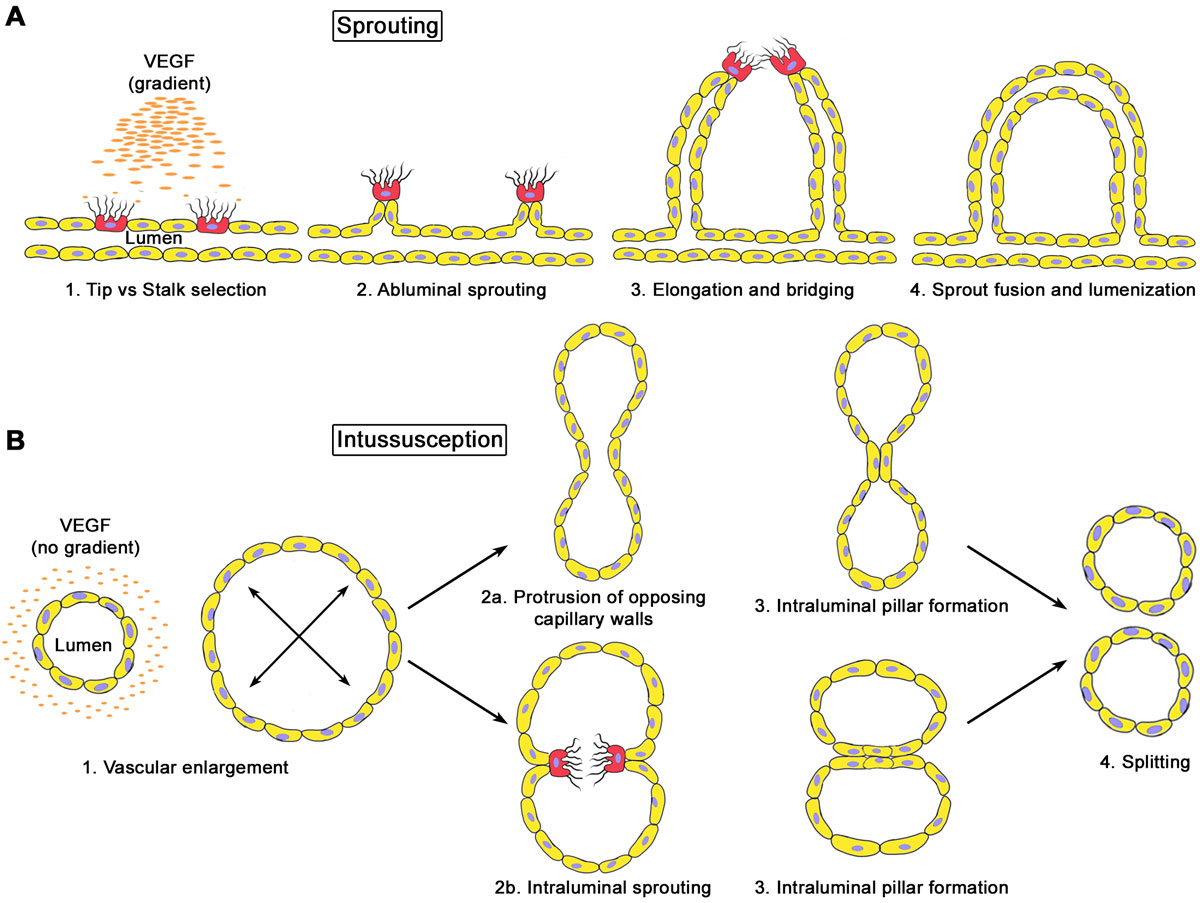
Figure 1
Mechanisms of angiogenesis: sprouting (A) vs intussusception, or splitting (B). A. VEGF gradients in the extracellular matrix induce the functional specification of endothelial tip cells (in red), which migrate towards the gradient source, and stalk cells (in yellow), which proliferate behind the tip, forming ab-luminal sprouts that fuse together and generate new vessels. B. In the absence of a gradient, all endothelial cells respond to VEGF by assuming a stalk phenotype without tip cells. The subsequent proliferation without migration leads to circumferential enlargement of vessels without sprouting followed by formation of intra-luminal endothelial pillars which fuse together and cause longitudinal splitting into two new vessels.
In contrast, intussusception (literally meaning “growth within itself”) can be considered the opposite of sprouting, with morphogenic events taking place inside the lumen rather than towards the ab-luminal side of vessels. Its hallmark is the formation of trans-luminal tissue pillars, which can occur either through a zone of contact between the endothelial cells of opposite capillary walls, with subsequent reorganisation of the endothelial junctions and invasion of the pillar core by myofibroblasts, or through the extension and fusion of intraluminal protrusions made exclusively of endothelial cells (intraluminal sprouting). Subsequently, transluminal tissue pillars align along the length of the pre-existing vessel, progressively fuse together and divide the affected vascular segment longitudinally into new, individual vascular structures (fig. 1B) [38, 39].
Recent evidence has shown that VEGF over-expression at the doses required to induce functional benefit and to restore tissue perfusion after ischaemia in the therapeutic target tissue of skeletal muscle induces angiogenesis essentially without sprouting, but rather through a first step of circumferential vascular enlargement followed by intussusception and longitudinal splitting [40]. It is not completely clear what determines whether VEGF induces sprouting or splitting angiogenesis. However, unpublished results, based on testable scenarios predicted by a computational model of Notch signalling [41], suggest that therapeutic doses of VEGF may saturate the very limited amount of extracellular matrix between muscle fibres, and that the lack of a concentration gradient may cause endothelium to acquire an all-stalk phenotype, leading to proliferation without migration and therefore circumferential enlargement rather than sprouting (Gianni-Barrera, Bentley, Gerhardt and Banfi, unpublished results). The molecular regulation of intussusception remains poorly understood due to a paucity of appropriate models, and its elucidation will be key to providing a rational basis for the design of therapeutic angiogenesis strategies.
Therapeutic considerations for VEGF delivery
VEGF dose: the importance of the microenvironment
VEGF dose is tightly regulated during development. In fact, it has been shown that relatively minor changes in its expression levels, such as a 50% reduction [42, 43] or a 2- to 3-fold increase [44], are lethal for the embryo at an early stage. The exquisite potency of VEGF is also confirmed in postnatal tissues, as robust over-expression of VEGF by different gene therapy vectors has been reported to cause the growth of aberrant angioma-like vascular structures in several tissues, including skeletal muscle [45, 46] and the myocardium [45, 47]. Preclinical data also showed a surprisingly narrow therapeutic window for VEGF gene delivery, such that low vector doses are safe but not sufficiently effective to yield a therapeutic benefit, whereas only slightly higher doses rapidly become unsafe [45]. Subsequent studies have discovered that the spatial distribution of VEGF in the tissue is key to determining its therapeutic window. In fact, work with monoclonal populations of transduced muscle progenitors, in which every cell produces the same level of factor, has shown that the switch between normal and aberrant angiogenesis by VEGF, as well as its therapeutic potential, depend strictly on the amount of factor localised in the microenvironment around each producing cell, and not simply on the total dose [48, 49]. Indeed, when the VEGF dose is homogeneously distributed in tissue, a wide range of microenvironmental VEGF doses are capable of inducing exclusively physiological microvascular networks, until a threshold level is reached, above which aberrant angiogenesis is initiated [48]. This is due to the fact that VEGF binds tightly to extracellular matrix [32], so that heterogeneous levels do not average out with each other in tissue. Therefore, “hotspots” of excessive expression can cause toxic effects even in the case of rather low total doses of factor.
This concept is schematically depicted in figure 2. When VEGF delivery leads to broadly heterogeneous levels of expression, as is typically the case with gene therapy approaches, a robust vector dose will produce effective levels in some areas but also toxic amounts in others, depending on the variable copy numbers transducing different cells in vivo (red curve in panel A). Reducing the total vector dose delivered, however, will not significantly change the degree of heterogeneity in the level distribution in the microenvironment (i.e. the breadth of the curve), but only shift it globally towards lower expression (blue and black curves in panel A). In such a scenario, total vector doses that are sufficiently low to completely avoid all “hotspots” of excessive microenvironmental expression (red dots in the upper boxes above the curves) will actually lead to mostly ineffective levels (grey dots above the black curve), and therefore a significant loss of therapeutic efficacy. The consequence of this is the rapid change from toxic (red and blue curves) to ineffective total vector doses (black curve), and an apparently very narrow or absent therapeutic window. However, if the microenvironmental distribution of expression levels can be controlled, e.g. by the delivery of monoclonal populations of transduced progenitors in which every cell produces the same amount of VEGF [48], or by the controlled release of VEGF protein from fibrin hydrogels [50], the microenvironmental levels achieved in tissue can be rather homogenous (e.g. only the safe and effective levels represented by the blue dots at the top of panel B), with a tight distribution around a desired mean dose (represented by the tight curves in panel B). This enables the totality of the delivered dose to be placed within the therapeutic window, so that both safety and efficacy can be achieved, as shown in murine models of hind limb ischaemia and wound-healing [49, 50].
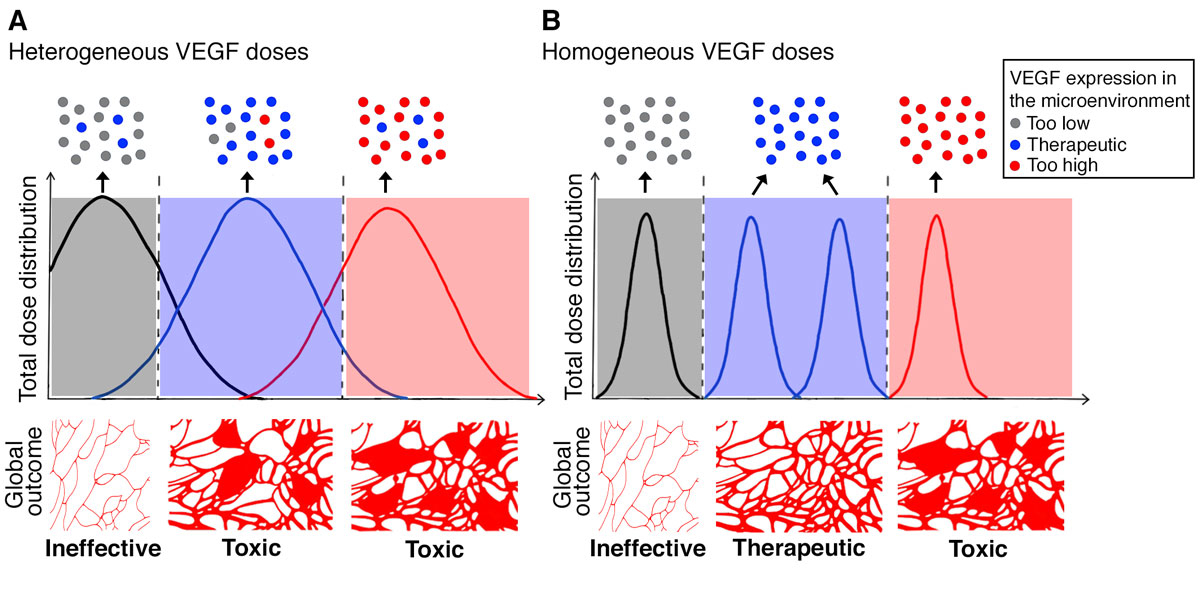
Figure 2
Functional outcomes of VEGF dose distribution in tissue. A. Heterogeneous dose distributions (e.g. by gene therapy vectors) lead to some hotspots of excessive expression that remain localised in the microenvironment around producing cells (red spots on upper panels) and lead to toxic effects. Reducing the total dose does not completely avoid toxic hotspots, even if therapeutic levels are achieved in some areas (blue spots), until the total dose is so low that mostly inefficient levels are achieved (grey spots), leading to a global outcome that goes from toxic to inefficient, apparently without a significant therapeutic window (lower panels). B. Homogeneous distribution of the total dose in the tissue allows consistent therapeutic levels to be achieved in the microenvironment around each expressing cell and unveils the therapeutic window of VEGF delivery.
Duration of the VEGF stimulus
In order to be therapeutically useful, newly induced vessels need to persist indefinitely after the end of the angiogenic stimulus. Experiments with transgenic animals [51] or viral vectors [52] affording inducible control of VEGF expression, or VEGF blockade by treatment with the receptor-body aflibercept [48] showed that VEGF stimulation shorter than about four weeks is insufficient to stabilise normal newly induced vessels, leading to their regression. Therefore, while transient delivery of a powerful factor like VEGF is desirable for safety reasons, sustained expression is necessary to ensure efficacy: an understanding of the mechanisms of vascular stabilisation is crucial for the development of treatments to ensure new vessel persistence despite transient and safe VEGF delivery.
Vascular stabilisation requires the maturation of newly induced vessels, i.e. the recruitment of mural cells (pericytes and smooth muscle cells) and the establishment of cell-to-cell contact between them and the endothelium. This process is mediated by Platelet-Derived Growth Factor-BB (PDGF-BB), produced by activated endothelial cells. In fact, association with pericytes is necessary to render vessels independent of continued VEGF expression [53, 54]. Interference with PDGF-BB/PDGF-Rβ signalling in transgenic mice and the subsequent defects in pericyte coverage lead to vascular abnormalities, unabated endothelial proliferation, the growth of microaneurysms, bleeding and embryonic lethality [55, 56]. Pericytes regulate several aspects of endothelium biology by establishing a complex molecular cross-talk [57]. TGF-β1 and angiopoietin/Tie2 signalling in particular have well-established roles in inducing vessel stabilisation. TGF-β1 has pleiotropic functions and can regulate both endothelial proliferation and quiescence through its receptors Alk1 and Alk5 respectively, possibly as a function of its dose [58]. Activation of TGF-β1-Alk5-SMAD2/3 specifically promotes vessel stabilisation by inhibiting endothelial migration and proliferation, as well as by stimulating mural cell differentiation and the deposition of extracellular matrix and the vascular basal lamina [59]. Angiopoietins (Ang), on the other hand, regulate vascular activation through the endothelial-specific receptor Tie2. Ang2, which is a context-dependent partial agonist, predominantly acts as a negative regulator of Tie2 signalling. It is released by endothelial cells upon their stimulation with VEGF and is crucial for their activation and the kick-starting of the angiogenic process. Ang1, in contrast, is produced by pericytes, activates Tie2 and plays a crucial role in vascular stabilisation by: (1) providing a survival signal to endothelial cells; (2) inhibiting vascular permeability; and (3) promoting further pericyte recruitment [57].
Although pericytes have long been considered the only regulators of vascular maturation and stabilisation, it has become increasingly clear that circulating bone marrow-derived cells also play an important role. In particular, a population of CD11b+ monocytes has been found to express the VEGF co-receptor neuropilin-1 (Nrp-1), and are therefore named Nrp1-expressing monocytes (NEM). These cells infiltrate the site of VEGF-induced angiogenesis and were originally described as promoting artery formation through the release of paracrine factors that stimulate smooth muscle cell recruitment, such as Ang1, TGF-β and PBGF-BB [60]. However, we recently found that NEM also play a key role in the stabilisation of newly induced microvascular networks, allowing them to achieve VEGF-independence and indefinite persistence [61]. Interestingly, although Nrp1 can be a co-receptor for VEGF, NEM are recruited to areas of VEGF-induced angiogenesis by the other Nrp1 ligand, Semaphorin3A (Sema3A), which is produced by activated endothelium in a VEGF dose-dependent fashion. In conditions with moderate VEGF expression, Sema3A expression by endothelium leads to robust recruitment of NEM, which induce endothelial quiescence and vascular stabilisation by activating TGF-β1 signalling through SMAD2/3. TGF-β1, in turn, potently upregulates Sema3A expression by endothelium, generating a powerful pro-stabilisation feedback loop. However, high VEGF doses directly inhibit Sema3A expression by endothelium, disrupting these molecular events and resulting in a dose-dependent delay of vascular stabilisation until its complete inhibition. From a therapeutic perspective, local treatment with recombinant Sema3A protein restored NEM recruitment and ensured vessel stabilisation despite uncontrolled VEGF expression. It also ensured vessel persistence despite a transient VEGF delivery, by a clinically relevant adenoviral vector, of less than two weeks [61], providing a novel molecular target to accelerate stabilisation of VEGF-induced angiogenesis.
Outlook
VEGF is the master regulator of angiogenesis, both in health and disease, and remains the key target for the therapeutic induction of vascular growth. However, exploiting VEGF’s formidable potency in a controlled and safe manner is a significant challenge that requires a thorough understanding of its complex biology, as outlined above.
One class of approaches aim to control VEGF-R2 stimulation in order to avoid excessive activation and aberrant angiogenesis. To achieve this, VEGF needs to be delivered at precisely controlled microenvironmental doses in a clinically applicable way, e.g. by providing recombinant VEGF protein engineered to be crosslinked in a fibrin matrix, so that the factor is released only through cell-mediated degradation of the matrix, and the dose can be precisely controlled and homogeneously distributed [50]. An alternative ligand may activate VEGF-R2 more gently and therefore require less stringent dose control. For example, VEGF-B does not bind VEGF-R2, but activates it indirectly by displacing endogenous VEGF-A from docking sites and making it available for signalling through VEGF-R2. As receptor activation is limited to the amount of endogenously available VEGF-A, VEGF-B overexpression does not easily cause excessive VEGF-R2 signalling. However, VEGF-B actions are tissue-specific and appear restricted to myocardium [62, 63]. Also, the proteolytically processed mature form of VEGF-D (VEGF-DΔCΔN) has been shown to acquire a strong blood angiogenic potential compared to the native, unprocessed VEGF-D, comparable to that of VEGF-A. However, as it is has significantly less matrix affinity than VEGF-A, it caused more diffuse effects and more physiological vascular growth [64]. Intramyocardial adenoviral delivery of VEGF-DΔCΔN is currently being evaluated in refractory angina patients in a phase I/IIa clinical trial [65].
A second class of approaches focus on correcting and preventing the side effects of robust and effective but uncontrolled VEGF delivery through the co-delivery of a second factor. As pericytes play a key role in regulating endothelial activation and proliferation by VEGF, increasing pericyte recruitment through PDGF-BB co-delivery has been shown to prevent aberrant angiogenesis and yield physiological and mature microvascular networks regardless of VEGF dose [66, 67], and to be effective in models of limb and cardiac ischaemia [66, 68]. On the other hand, ephrinB2/EphB4 signalling in the pericyte-endothelial crosstalk has recently been identified as the pathway responsible for switching aberrant angiogenesis to normal regardless of VEGF dose [69], providing a more specific, druggable molecular target for correcting VEGF side-effects. In fact, EphB4 stimulation finely tunes ERK1/2 signalling downstream of the activated VEGF-R2, effectively acting as a partial inhibitor, and therefore ensures moderate endothelial proliferation despite robust and uncontrolled VEGF expression [69].
Based on recent advances in the understanding of VEGF biology and the cascade of events downstream of VEGF-R2 activation that control endothelial activation, vascular morphogenesis and subsequent maturation and stabilisation, it can be envisioned that complementary targeting of specific molecular targets with systemic drug treatments could ensure a safe and effective outcome of VEGF gene delivery. For example, a combination of adenoviral VEGF delivery to skeletal muscle, leading to robust but uncontrolled and transient gene expression, can be combined with pharmacological stimulation of specific pathways that will prevent aberrant vascular growth (e.g. EphB4 [69]) and accelerate stabilisation (e.g. Sema3A [61]) to yield only homogeneously normal and stable microvascular networks.
References
1
Benjamin
EJ
,
Virani
SS
,
Callaway
CW
,
Chamberlain
AM
,
Chang
AR
,
Cheng
S
, et al.; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137(12):e67–492. doi:.https://doi.org/10.1161/CIR.0000000000000558
2
Keck
PJ
,
Hauser
SD
,
Krivi
G
,
Sanzo
K
,
Warren
T
,
Feder
J
, et al.
Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246(4935):1309–12. doi:.https://doi.org/10.1126/science.2479987
3
Leung
DW
,
Cachianes
G
,
Kuang
WJ
,
Goeddel
DV
,
Ferrara
N
. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–9. doi:.https://doi.org/10.1126/science.2479986
4
Isner
JM
,
Pieczek
A
,
Schainfeld
R
,
Blair
R
,
Haley
L
,
Asahara
T
, et al.
Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet. 1996;348(9024):370–4. doi:.https://doi.org/10.1016/S0140-6736(96)03361-2
5
Losordo
DW
,
Vale
PR
,
Symes
JF
,
Dunnington
CH
,
Esakof
DD
,
Maysky
M
, et al.
Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98(25):2800–4. doi:.https://doi.org/10.1161/01.CIR.98.25.2800
6
Gupta
R
,
Tongers
J
,
Losordo
DW
. Human studies of angiogenic gene therapy. Circ Res. 2009;105(8):724–36. doi:.https://doi.org/10.1161/CIRCRESAHA.109.200386
7
Rubanyi
GM
. Mechanistic, technical, and clinical perspectives in therapeutic stimulation of coronary collateral development by angiogenic growth factors. Mol Ther. 2013;21(4):725–38. doi:.https://doi.org/10.1038/mt.2013.13
8
Potente
M
,
Gerhardt
H
,
Carmeliet
P
. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–87. doi:.https://doi.org/10.1016/j.cell.2011.08.039
9
Ylä-Herttuala
S
,
Bridges
C
,
Katz
MG
,
Korpisalo
P
. Angiogenic gene therapy in cardiovascular diseases: dream or vision?
Eur Heart J. 2017;38(18):1365–71. doi:.https://doi.org/10.1093/eurheartj/ehw547
10
Cai
W
,
Schaper
W
. Mechanisms of arteriogenesis. Acta Biochim Biophys Sin (Shanghai). 2008;40(8):681–92. doi:.https://doi.org/10.1093/abbs/40.8.681
11
Persson
AB
,
Buschmann
IR
. Vascular growth in health and disease. Front Mol Neurosci. 2011;4:14. doi:.https://doi.org/10.3389/fnmol.2011.00014
12
Annex
BH
. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. 2013;10(7):387–96. doi:.https://doi.org/10.1038/nrcardio.2013.70
13
Pries
AR
,
Höpfner
M
,
le Noble
F
,
Dewhirst
MW
,
Secomb
TW
. The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer. 2010;10(8):587–93. doi:.https://doi.org/10.1038/nrc2895
14
Rissanen
TT
,
Korpisalo
P
,
Markkanen
JE
,
Liimatainen
T
,
Ordén
MR
,
Kholová
I
, et al.
Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation. 2005;112(25):3937–46. doi:.https://doi.org/10.1161/CIRCULATIONAHA.105.543124
15
Baumgartner
I
,
Pieczek
A
,
Manor
O
,
Blair
R
,
Kearney
M
,
Walsh
K
, et al.
Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation. 1998;97(12):1114–23. doi:.https://doi.org/10.1161/01.CIR.97.12.1114
16
Mäkinen
K
,
Manninen
H
,
Hedman
M
,
Matsi
P
,
Mussalo
H
,
Alhava
E
, et al.
Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol Ther. 2002;6(1):127–33. doi:.https://doi.org/10.1006/mthe.2002.0638
17
Tsurumi
Y
,
Takeshita
S
,
Chen
D
,
Kearney
M
,
Rossow
ST
,
Passeri
J
, et al.
Direct intramuscular gene transfer of naked DNA encoding vascular endothelial growth factor augments collateral development and tissue perfusion. Circulation. 1996;94(12):3281–90. doi:.https://doi.org/10.1161/01.CIR.94.12.3281
18
Lähteenvuo
J
,
Ylä-Herttuala
S
. Advances and Challenges in Cardiovascular Gene Therapy. Hum Gene Ther. 2017;28(11):1024–32. doi:.https://doi.org/10.1089/hum.2017.129
19
Ogawa
S
,
Oku
A
,
Sawano
A
,
Yamaguchi
S
,
Yazaki
Y
,
Shibuya
M
. A novel type of vascular endothelial growth factor, VEGF-E (NZ-7 VEGF), preferentially utilizes KDR/Flk-1 receptor and carries a potent mitotic activity without heparin-binding domain. J Biol Chem. 1998;273(47):31273–82. doi:.https://doi.org/10.1074/jbc.273.47.31273
20
Yamazaki
Y
,
Tokunaga
Y
,
Takani
K
,
Morita
T
. C-terminal heparin-binding peptide of snake venom VEGF specifically blocks VEGF-stimulated endothelial cell proliferation. Pathophysiol Haemost Thromb. 2005;34(4-5):197–9. doi:.https://doi.org/10.1159/000092423
21
Ferrara
N
. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell. 2010;21(5):687–90. doi:.https://doi.org/10.1091/mbc.e09-07-0590
22
Senger
DR
,
Galli
SJ
,
Dvorak
AM
,
Perruzzi
CA
,
Harvey
VS
,
Dvorak
HF
. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219(4587):983–5. doi:.https://doi.org/10.1126/science.6823562
23
Koch
S
,
Claesson-Welsh
L
. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2(7):a006502. doi:.https://doi.org/10.1101/cshperspect.a006502
24
Grünewald
FS
,
Prota
AE
,
Giese
A
,
Ballmer-Hofer
K
. Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim Biophys Acta. 2010;1804(3):567–80. doi:.https://doi.org/10.1016/j.bbapap.2009.09.002
25
Ito
N
,
Wernstedt
C
,
Engström
U
,
Claesson-Welsh
L
. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J Biol Chem. 1998;273(36):23410–8. doi:.https://doi.org/10.1074/jbc.273.36.23410
26
Ferrara
N
,
Gerber
HP
,
LeCouter
J
. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76. doi:.https://doi.org/10.1038/nm0603-669
27
Cudmore
MJ
,
Hewett
PW
,
Ahmad
S
,
Wang
KQ
,
Cai
M
,
Al-Ani
B
, et al.
The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat Commun. 2012;3(1):972. doi:.https://doi.org/10.1038/ncomms1977
28
Haiko
P
,
Makinen
T
,
Keskitalo
S
,
Taipale
J
,
Karkkainen
MJ
,
Baldwin
ME
, et al.
Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28(15):4843–50. doi:.https://doi.org/10.1128/MCB.02214-07
29
Takahashi
H
,
Shibuya
M
. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond). 2005;109(3):227–41. doi:.https://doi.org/10.1042/CS20040370
30
Krilleke
D
,
Ng
YS
,
Shima
DT
. The heparin-binding domain confers diverse functions of VEGF-A in development and disease: a structure-function study. Biochem Soc Trans. 2009;37(6):1201–6. doi:.https://doi.org/10.1042/BST0371201
31
Harper
SJ
,
Bates
DO
. VEGF-A splicing: the key to anti-angiogenic therapeutics?
Nat Rev Cancer. 2008;8(11):880–7. doi:.https://doi.org/10.1038/nrc2505
32
Park
JE
,
Keller
GA
,
Ferrara
N
. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4(12):1317–26. doi:.https://doi.org/10.1091/mbc.4.12.1317
33
Ruhrberg
C
,
Gerhardt
H
,
Golding
M
,
Watson
R
,
Ioannidou
S
,
Fujisawa
H
, et al.
Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16(20):2684–98. doi:.https://doi.org/10.1101/gad.242002
34
De Spiegelaere
W
,
Casteleyn
C
,
Van den Broeck
W
,
Plendl
J
,
Bahramsoltani
M
,
Simoens
P
, et al.
Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J Vasc Res. 2012;49(5):390–404. doi:.https://doi.org/10.1159/000338278
35
Gianni-Barrera
R
,
Bartolomeo
M
,
Vollmar
B
,
Djonov
V
,
Banfi
A
. Split for the cure: VEGF, PDGF-BB and intussusception in therapeutic angiogenesis. Biochem Soc Trans. 2014;42(6):1637–42. doi:.https://doi.org/10.1042/BST20140234
36
Blanco
R
,
Gerhardt
H
. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 2013;3(1):a006569. doi:.https://doi.org/10.1101/cshperspect.a006569
37
Hellström
M
,
Phng
LK
,
Hofmann
JJ
,
Wallgard
E
,
Coultas
L
,
Lindblom
P
, et al.
Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–80. doi:.https://doi.org/10.1038/nature05571
38
Egginton
S
. Invited review: activity-induced angiogenesis. Pflugers Arch. 2009;457(5):963–77. doi:.https://doi.org/10.1007/s00424-008-0563-9
39
Makanya
AN
,
Hlushchuk
R
,
Djonov
VG
. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. 2009;12(2):113–23. doi:.https://doi.org/10.1007/s10456-009-9129-5
40
Gianni-Barrera
R
,
Trani
M
,
Fontanellaz
C
,
Heberer
M
,
Djonov
V
,
Hlushchuk
R
, et al.
VEGF over-expression in skeletal muscle induces angiogenesis by intussusception rather than sprouting. Angiogenesis. 2013;16(1):123–36. doi:.https://doi.org/10.1007/s10456-012-9304-y
41
Bentley
K
,
Gerhardt
H
,
Bates
PA
. Agent-based simulation of notch-mediated tip cell selection in angiogenic sprout initialisation. J Theor Biol. 2008;250(1):25–36. doi:.https://doi.org/10.1016/j.jtbi.2007.09.015
42
Carmeliet
P
,
Ferreira
V
,
Breier
G
,
Pollefeyt
S
,
Kieckens
L
,
Gertsenstein
M
, et al.
Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–9. doi:.https://doi.org/10.1038/380435a0
43
Ferrara
N
,
Carver-Moore
K
,
Chen
H
,
Dowd
M
,
Lu
L
,
O’Shea
KS
, et al.
Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439–42. doi:.https://doi.org/10.1038/380439a0
44
Miquerol
L
,
Langille
BL
,
Nagy
A
. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127(18):3941–6.
45
Schwarz
ER
,
Speakman
MT
,
Patterson
M
,
Hale
SS
,
Isner
JM
,
Kedes
LH
, et al.
Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat--angiogenesis and angioma formation. J Am Coll Cardiol. 2000;35(5):1323–30. doi:.https://doi.org/10.1016/S0735-1097(00)00522-2
46
Springer
ML
,
Chen
AS
,
Kraft
PE
,
Bednarski
M
,
Blau
HM
. VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell. 1998;2(5):549–58. doi:.https://doi.org/10.1016/S1097-2765(00)80154-9
47
Lee
RJ
,
Springer
ML
,
Blanco-Bose
WE
,
Shaw
R
,
Ursell
PC
,
Blau
HM
. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000;102(8):898–901. doi:.https://doi.org/10.1161/01.CIR.102.8.898
48
Ozawa
CR
,
Banfi
A
,
Glazer
NL
,
Thurston
G
,
Springer
ML
,
Kraft
PE
, et al.
Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Invest. 2004;113(4):516–27. doi:.https://doi.org/10.1172/JCI18420
49
von Degenfeld
G
,
Banfi
A
,
Springer
ML
,
Wagner
RA
,
Jacobi
J
,
Ozawa
CR
, et al.
Microenvironmental VEGF distribution is critical for stable and functional vessel growth in ischemia. FASEB J. 2006;20(14):2657–9. doi:.https://doi.org/10.1096/fj.06-6568fje
50
Sacchi
V
,
Mittermayr
R
,
Hartinger
J
,
Martino
MM
,
Lorentz
KM
,
Wolbank
S
, et al.
Long-lasting fibrin matrices ensure stable and functional angiogenesis by highly tunable, sustained delivery of recombinant VEGF164. Proc Natl Acad Sci USA. 2014;111(19):6952–7. doi:.https://doi.org/10.1073/pnas.1404605111
51
Dor
Y
,
Djonov
V
,
Abramovitch
R
,
Itin
A
,
Fishman
GI
,
Carmeliet
P
, et al.
Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21(8):1939–47. doi:.https://doi.org/10.1093/emboj/21.8.1939
52
Tafuro
S
,
Ayuso
E
,
Zacchigna
S
,
Zentilin
L
,
Moimas
S
,
Dore
F
, et al.
Inducible adeno-associated virus vectors promote functional angiogenesis in adult organisms via regulated vascular endothelial growth factor expression. Cardiovasc Res. 2009;83(4):663–71. doi:.https://doi.org/10.1093/cvr/cvp152
53
Benjamin
LE
,
Golijanin
D
,
Itin
A
,
Pode
D
,
Keshet
E
. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103(2):159–65. doi:.https://doi.org/10.1172/JCI5028
54
Benjamin
LE
,
Hemo
I
,
Keshet
E
. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125(9):1591–8.
55
Hellström
M
,
Kalén
M
,
Lindahl
P
,
Abramsson
A
,
Betsholtz
C
. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–55.
56
Lindahl
P
,
Johansson
BR
,
Levéen
P
,
Betsholtz
C
. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–5. doi:.https://doi.org/10.1126/science.277.5323.242
57
Gaengel
K
,
Genové
G
,
Armulik
A
,
Betsholtz
C
. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29(5):630–8. doi:.https://doi.org/10.1161/ATVBAHA.107.161521
58
Goumans
MJ
,
Valdimarsdottir
G
,
Itoh
S
,
Rosendahl
A
,
Sideras
P
,
ten Dijke
P
. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21(7):1743–53. doi:.https://doi.org/10.1093/emboj/21.7.1743
59
van Meeteren
LA
,
ten Dijke
P
. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347(1):177–86. doi:.https://doi.org/10.1007/s00441-011-1222-6
60
Zacchigna
S
,
Pattarini
L
,
Zentilin
L
,
Moimas
S
,
Carrer
A
,
Sinigaglia
M
, et al.
Bone marrow cells recruited through the neuropilin-1 receptor promote arterial formation at the sites of adult neoangiogenesis in mice. J Clin Invest. 2008;118(6):2062–75. doi:.https://doi.org/10.1172/JCI32832
61
Groppa
E
,
Brkic
S
,
Bovo
E
,
Reginato
S
,
Sacchi
V
,
Di Maggio
N
, et al.
VEGF dose regulates vascular stabilization through Semaphorin3A and the Neuropilin-1+ monocyte/TGF-β1 paracrine axis. EMBO Mol Med. 2015;7(10):1366–84. doi:.https://doi.org/10.15252/emmm.201405003
62
Kivelä
R
,
Bry
M
,
Robciuc
MR
,
Räsänen
M
,
Taavitsainen
M
,
Silvola
JM
, et al.
VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6(3):307–21. doi:.https://doi.org/10.1002/emmm.201303147
63
Lähteenvuo
JE
,
Lähteenvuo
MT
,
Kivelä
A
,
Rosenlew
C
,
Falkevall
A
,
Klar
J
, et al.
Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation. 2009;119(6):845–56. doi:.https://doi.org/10.1161/CIRCULATIONAHA.108.816454
64
Rissanen
TT
,
Markkanen
JE
,
Gruchala
M
,
Heikura
T
,
Puranen
A
,
Kettunen
MI
, et al.
VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ Res. 2003;92(10):1098–106. doi:.https://doi.org/10.1161/01.RES.0000073584.46059.E3
65
Hartikainen
J
,
Hassinen
I
,
Hedman
A
,
Kivelä
A
,
Saraste
A
,
Knuuti
J
, et al.
Adenoviral intramyocardial VEGF-DΔNΔC gene transfer increases myocardial perfusion reserve in refractory angina patients: a phase I/IIa study with 1-year follow-up. Eur Heart J. 2017;38(33):2547–55. doi:.https://doi.org/10.1093/eurheartj/ehx352
66
Banfi
A
,
von Degenfeld
G
,
Gianni-Barrera
R
,
Reginato
S
,
Merchant
MJ
,
McDonald
DM
, et al.
Therapeutic angiogenesis due to balanced single-vector delivery of VEGF and PDGF-BB. FASEB J. 2012;26(6):2486–97. doi:.https://doi.org/10.1096/fj.11-197400
67
Gianni-Barrera
R
,
Butschkau
A
,
Uccelli
A
,
Certelli
A
,
Valente
P
,
Bartolomeo
M
, et al.
PDGF-BB regulates splitting angiogenesis in skeletal muscle by limiting VEGF-induced endothelial proliferation. Angiogenesis. 2018;21(4):883–900. doi:.https://doi.org/10.1007/s10456-018-9634-5
68
Kupatt
C
,
Hinkel
R
,
Pfosser
A
,
El-Aouni
C
,
Wuchrer
A
,
Fritz
A
, et al.
Cotransfection of vascular endothelial growth factor-A and platelet-derived growth factor-B via recombinant adeno-associated virus resolves chronic ischemic malperfusion role of vessel maturation. J Am Coll Cardiol. 2010;56(5):414–22. doi:.https://doi.org/10.1016/j.jacc.2010.03.050
69
Groppa
E
,
Brkic
S
,
Uccelli
A
,
Wirth
G
,
Korpisalo-Pirinen
P
,
Filippova
M
, et al.
EphrinB2/EphB4 signaling regulates non-sprouting angiogenesis by VEGF. EMBO Rep. 2018;19(5):e45054. doi:.https://doi.org/10.15252/embr.201745054