Development of a novel class of interleukin-2 immunotherapies for metastatic cancer
DOI: https://doi.org/10.4414/smw.2019.14697
Onur
Boymanab, Natalia
Arenas-Ramireza
aDepartment of Immunology, University Hospital Zurich, Switzerland
bFaculty of Medicine, University of Zurich, Switzerland
Summary
Tumour immunotherapy, and particularly immue checkpoint inhibitors, have resulted in considerable response rates in patients with metastatic cancer. However, most of these approaches are limited to immunogenic tumours. Based on its ability to stimulate cytotoxic T cells, interleukin-2 (IL-2) has been used to treat patients with metastatic melanoma and metastatic kidney cancer. Clinical efficacy achieved through high doses is countered by severe adverse effects on vascular endothelial cells and various organs, a short in vivo half-life, and the stimulation of regulatory T cells that counteract antitumour immune responses. Accumulating evidence suggests that IL-2 receptor β (CD122)-biased IL-2 formulations address the shortcomings of IL-2 cancer immunotherapy. This knowledge stems from studies using CD122-biased IL-2/anti-IL-2 antibody complexes (IL-2 complexes), which preferentially stimulate CD8+ T cells, while interaction with regulatory T cells and vascular endothelial cells is disfavoured by the anti-IL-2 antibody used. CD122-biased IL-2 complexes, when assessed in different mouse cancer models, cause stronger antitumour effects and significantly less adverse effects than high-dose IL-2. A recently developed and characterised anti-human IL-2 antibody, termed NARA1, forms human CD122-biased IL-2 complexes. Alternative strategies based on this concept, such as site-directed pegylation and mutation of IL-2, have also been pursued. Moreover, recent data have shown that a combination of CD122-biased IL-2 formulations with immune checkpoint inhibitors, antigen-specific immunotherapy and epigenetic modifying drugs results in synergistic anti-cancer effects in various tumour models. Thus, CD122-biased IL-2 approaches constitute a novel class of immunotherapy for metastatic cancer that has the potential to complement and increase the efficacy of other antitumour strategies.
Introduction
Cancer is a major health problem of our time. With an estimated 8.8 million deaths globally in 2015, cancer is the second leading cause of death worldwide [1]. Moreover, the economic impact of cancer in 2010 was estimated at about 1.16 trillion US dollars, further emphasising the urgency and importance of fighting this disease. Whereas localised cancer can often be cured using surgery and radiation, metastatic disease, where cancer cells spread to distant sites of the body, is often incurable. The current treatment options for metastatic cancer include, in addition to the aforementioned approaches, chemotherapy, agents targeting cancer-associated pathways, angiogenesis inhibitors and tumour immunotherapy. In fact, the recent impact of immune checkpoint inhibitors on advanced cancer has firmly established the importance of tumour immunotherapy in the treatment of metastatic disease [2].
However, stimulation of the immune system to fight cancer is not a new concept. In the late 19th century, the New York surgeon William Coley injected cancer patients intratumourally with bacterial extracts, with the aim of eliciting an antitumour immune response [3]. Yet almost a century separates Coley's initial clinical trial from the discovery of tumour antigens and the use of the first molecularly defined immunotherapies, including interleukin-2 (IL-2), for metastatic disease [4, 5]. In 2010, the use of a monoclonal antibody (mAb) blocking the immune checkpoint cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) was reported to show a significant improvement in the overall survival of patients with metastatic melanoma [6], leading the field towards the research and development of immune checkpoint inhibitors. While the clinical use of immune checkpoint inhibitors and the science behind them are transforming tumour immunotherapy and immunology [2], IL-2-based immunotherapy does provide some advantages that justify renewed interest in this approach against advanced cancer.
IL-2 immunotherapy
IL-2 was discovered, purified and molecularly characterised between 1976 and 1983 [7–9]. A small cytokine of 15.5–16 kDa consisting of four α-helices, IL-2 is a member of the common cytokine receptor γ chain (γc) that also includes IL-4, IL-7, IL-9, IL-15 and IL-21 [10]. In the early 1990s, IL-2 was the first immunotherapy used and approved in metastatic melanoma and metastatic renal cell carcinoma. In these indications, high-dose IL-2 therapy results in an overall response rate of 15-17% and in about 5-10% durable and complete disease remission [5]. A course of high-dose IL-2 therapy consists of two cycles of 600,000–720,000 international units/kg, given every 8 hours for up to 12-15 doses per cycle, and further courses of treatment are repeated every 2 months if tolerated by the patient [11]. Therapeutic response rates can increase to 30-70% when IL-2 is combined with adoptive T cell transfer together with nonmyeloablative conditioning or with cancer vaccines.
Despite these promising outcomes, IL-2-based approaches have not been widely adopted. The reasons for this include the short half-life of IL-2, which makes its administration to patients inefficient, its dose-dependent, toxic adverse effects on the vasculature and other organs, and its stimulation of regulatory T (Treg) cells that dampen antitumour immune responses [12, 13]. A previously held conviction paralleled the adverse effects of IL-2 with its antitumour activities. It was thought that IL-2 activated cytotoxic T cells and natural killer (NK) cells to attack cancerous cells but, simultaneously, the same cells released vaso-active substances, including tumour necrosis factor (TNF), that resulted in vascular (or capillary) leak syndrome (VLS) [14–19]. However, in 2010 we were able to demonstrate that IL-2-mediated toxicity and VLS was the result of a two-step process, whereby, firstly IL-2 binds to and damages endothelial cells, and in a second phase, IL-2-activated T and NK cells release pro-inflammatory cytokines including TNF [20]. The association of IL-2 with endothelial cells is facilitated by the expression of IL-2 receptor α (IL-2Rα, also called CD25) on endothelial cells, the levels of which further increase upon IL-2 immunotherapy in vivo, whereas the antitumour activity of IL-2 is independent of CD25 but requires cells expressing IL-2Rβ (also termed CD122) and γc [20]. These findings opened up the possibility of using modified IL-2 approaches, such as CD122-biased IL-2/anti-IL-2 monoclonal antibody complexes (IL-2 complexes; see below) [13, 21–23] and IL-2 muteins [24]. Our 2010 Krieg et al. report also motivated Charych and colleagues [25] to PEGylate IL-2 with the aim of biasing IL-2 to preferentially bind to CD122 [26]. These approaches are based on the differential expression of IL-2R subunits, as discussed in the next chapter.
IL-2 approaches biased to different IL-2 receptors
IL-2Rs include dimeric IL-2Rs consisting of CD122 and γc, or trimeric IL-2Rs made of CD122, γc and CD25 [12, 13]. The CD122–γc heterodimer mediates signal transduction. Conversely, CD25 by itself can serve as a non-signalling, low-affinity IL-2R, with a dissociation constant (Kd) of about 10–8 M. Upon association with CD25, thus forming the trimeric IL-2R, the binding property of the dimeric CD122–γc IL-2R increases from intermediate (Kd about 10–9 M) to high affinity (Kd about 10–10 M to 10–11 M) [13]. Thus, the principal function of CD25 appears to be to increase the affinity of the IL-2R. However, CD25 can also exert other functions. CD25 is prominently expressed on Treg cells, where the excess of CD25 could serve as a sink, thereby taking IL-2 away from effector T cells [27, 28]. Moreover, as mentioned above, endothelial cells express CD25, maybe to sense IL-2 signals during inflammation, thereby locally increasing the leakiness of the vasculature to allow influx of leukocytes and to concentrate IL-2 molecules on their surface for transiting T cells [20, 28].
In the context of cancer immunotherapy, IL-2 formulations that allow IL-2 binding to the dimeric CD122–γc IL-2R but disfavour the association of IL-2 with CD25 (thus termed CD122-biased) could be advantageous. Strategies to obtain CD122-biased IL-2 formulations that have been pursued include, to name but a few, (i) CD122-directed IL-2 complexes, in which the anti-IL-2 mAb covers the CD25-binding site (fig. 1A–C), (ii) IL-2 muteins with a mutation at the CD25-binding site, (iii) IL-2 carrying polyethylene glycol (PEG) groups at the CD25-binding site and (iv) a fusion protein of IL-2 and CD25. PEGylation requires exclusive targeting of the IL-2 residues involved in CD25 interaction, which so far has not been achieved. Thus, PEGylation of accessible lysine (K) residues of human IL-2, as in NKTR-214 [26], will also target epitopes that are involved in CD122–γc binding [13]. A CD25–IL-2 fusion protein consists of two functional parts that interact with each other with low affinity (10–8 M), unlike the CD25 mimobody NARA1 (see below). IL-2 muteins have the advantage of consisting of only one molecule, but they are considerably immunogenic and have a short half-life [13]. Conversely, CD122-directed IL-2 complexes are made of natural (i.e. non-immunogenic) IL-2 bound by a high-affinity mAb. While natural IL-2 has a short in vivo half-life, measured in minutes, due to rapid renal clearance, CD122-directed IL-2 complexes have an in vivo half-life of days [21, 23]. The increased in vivo half-life is the result of the association of the small (15.5-16 kDa) IL-2 with an antibody of 150 kDa [23], and this principle also applies to other cytokines, including IL-1β, IL-3, IL-4, IL-6, IL-7, IL-15, interferon-γ, and granulocyte colony-stimulating factor [22, 30–38]. IL-2 complexes, in comparison to complexes made of most other cytokines, are particular because, depending on the anti-IL-2 mAb used, they preferentially stimulate either CD122high cells expressing CD122–γc dimeric IL-2Rs or CD25high cells carrying trimeric IL-2Rs. This principle, first described in 2006 [22], states that CD122-biased IL-2 complexes (e.g. mouse IL-2/S4B6 complexes or human IL-2/NARA1 complexes) preferentially stimulate CD122high cells, including antigen-experienced (memory) CD8+ T cells and NK cells. Conversely, CD25-biased IL-2 complexes (e.g. mouse IL-2/JES6-1 complexes or human IL-2/5344 complexes) selectively activate CD25high cells such as thymus-derived CD25high Treg cells [12, 13, 19]. Mechanistically, mAbs that form CD122-biased IL-2 complexes bind to and cover IL-2 epitopes that usually interact with CD25, whereas mAbs that form CD25-biased IL-2 complexes associate with and temporally obstruct IL-2 epitopes that interact with CD122 and γc [13].
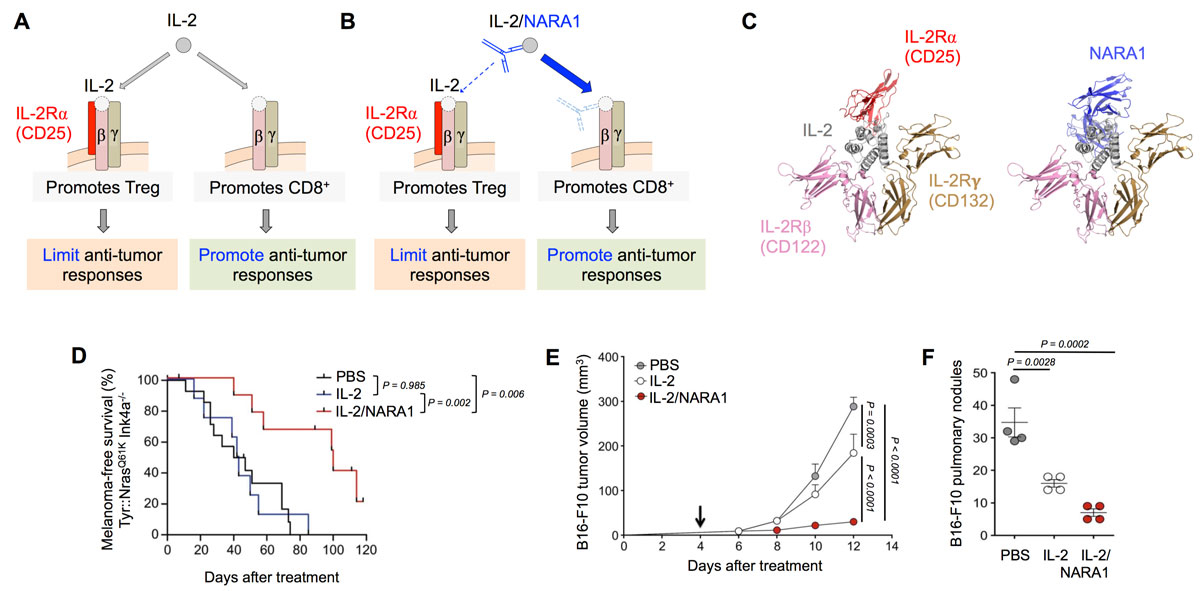
Figure 1
IL-2 complexes stimulate potent antitumour CD8+ T cell responses. (A–B) IL-2 activates CD25high CD4+ Treg and CD122high CD8+ T cells, whereas CD122-biased IL-2 complexes (such as IL-2/NARA1 complexes) preferentially stimulate CD122high CD8+ T cells. (C) NARA1 binds with 10-fold higher affinity than CD25 to the CD25 epitope of IL-2. (D–F) IL-2/NARA1 complexes elicit potent antitumour responses in Tyr::N-RasQ61K Ink4a–/–
mice (that spontaneously develop melanoma; ref [29]) (D) as well as in the cutaneous (E) and pulmonary B16F10 mouse melanoma models (F). Adapted from refs [12, 21].
CD122-directed IL-2 complexes have been shown in several preclinical cancer models to exert superior antitumour responses compared to IL-2, either as a monotherapy or in combination with other immune stimulating agents, cancer vaccines and epigenetic modifying agents [20, 21, 24, 39–42]. Cancer types where CD122-biased IL-2 formulations have shown efficacy include melanoma (B16F10 and Tyr::N-RasQ61K Ink4a–/–
mice), lung carcinoma (Lewis lung carcinoma), colon carcinoma (MC38), prostate carcinoma (TRAMP-C1), sarcoma (MCA-205) and leukaemia (BCL1) [20, 21, 24, 39–43] (fig. 1D–F). As for epigenetic modification, a combination of IL-2/NARA1 complexes with an inhibitor of enhancer of zeste homologue 2 (EZH2) was particularly advantageous. Thus, systemic or tumour-intrinsic EZH2 inhibition in different mouse models of melanoma was able to reverse the features of adaptive resistance to tumour immunotherapy such as silencing of immunogenicity and antigen presentation, as well as upregulation of programmed death ligand 1 (PD-L1, also known as CD274) on melanoma cells [42]. Recently, a monoclonal anti-human IL-2 antibody, termed NARA1, has been generated and characterised. It forms CD122-directed human IL-2 complexes and is suitable for clinical development [21]. Because it mimics CD25's binding to the CD25 epitope of human IL-2, NARA1 was also dubbed a CD25 mimobody. However, unlike CD25, NARA1 binds with high affinity (10–9 M) to IL-2. Similar to their mouse analogues, IL-2/NARA1 complexes preferentially stimulate CD8+ T cells and NK cells while sparing Treg cells and endothelial cells, and show strong efficacy in several mouse tumour models [21] (fig. 1D–F).
CD122-biased IL-2 therapies currently in development are listed in table 1.
Table 1 CD122-biased IL-2 therapies in development.
Human IL-2 (hIL-2)-based formulations that have been reported to show an IL-2 receptor β (CD122) bias, i.e. preference of CD122 over IL-2 receptor α (CD25) binding, are listed according to their state of development.
Product
(Company)
|
CD122 bias
|
Description
|
Status
|
Ref.
|
NKTR-214
(Nektar)
|
+ |
hIL-2 PEGylated on lysine (K) residues, resulting in up to 11 PEGs per hIL-2 (6 on average), of which 6 are in or adjacent to the CD25 epitope (thus resulting in CD122 bias), and 3-4 are in or adjacent to the CD122 epitope (thus favouring CD25 bias) |
6 Phase I to III studies |
CT, [26] |
RG7461 (RO6874281)
(Roche)
|
++ |
hIL-2 carrying the mutations F42A, Y45A and L72G, located in the CD25-binding epitope of hIL-2 and fused to a mAb targeting fibroblast activation protein-alpha (FAP) |
4 Phase I to II studies |
CT, [44] |
ALKS 4230
(Alkermes)
|
++ |
hIL-2–CD25 fusion protein consisting of complementary mutated hIL-2 and CD25 |
Phase I recruiting |
CT |
ANV329 & ANV629
= humanised NARA1
(Anaveon)
|
++ |
Humanised NARA1 mAb binding with high affinity and affinity-matured to the CD25 epitope of hIL-2 |
Pre-clinical |
Anaveon, [21] |
Concluding remarks
As CD122-biased IL-2 formulations are being tested in patients, the first clinical data are becoming available, including potential advantages and disadvantages [13, 44, 45]. Due to its different but proven mode of action, IL-2 immunotherapy could complement many anti-cancer approaches, including immune checkpoint inhibitors, adoptive cell transfer regimens and vaccination, as well as strategies that are not considered typical immunotherapies (such as epigenetic modifying agents), to name a few. In adoptive cell transfer approaches, CD122-biased IL-2 complexes could help to rescue T cells that have been strongly activated in vitro and to keep these cells alive, as suggested by a number of preclinical studies [10, 39–41, 46]. Notably, IL-2 immunotherapy might even contribute to turning a “cold” (i.e. poorly immunogenic) tumour into a “hot” (i.e. immunogenic) one, e.g. by the upregulation of PD-L1 on cancer cells [45]. However, to date, we do not know how these different processes will best benefit from CD122-biased IL-2 complexes, or whether IL-2 signals will be required in the tumour and the tumour microenvironment, and/or in secondary lymphoid organs. Future preclinical and clinical studies will shed light on these open questions on the biology and use of IL-2 and biased IL-2 formulations.
Acknowledgments
We thank Catherine Crowley-Kühn for reading and editing the manuscript.
References
1WHO. Cancer. World Health Organization, Media Centre, Fact Sheet, Updated February 2017. http://www.who.int/mediacentre/factsheets/fs297/en/
2
Sharma
P
,
Allison
JP
. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi:.https://doi.org/10.1126/science.aaa8172
3
Coley
WB
. The treatment of malignant tumours by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;105(5):487–510.
4
Coulie
PG
,
Van den Eynde
BJ
,
van der Bruggen
P
,
Boon
T
. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–46. doi:.https://doi.org/10.1038/nrc3670
5
Rosenberg
SA
. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–8. doi:.https://doi.org/10.4049/jimmunol.1490019
6
Hodi
FS
,
O’Day
SJ
,
McDermott
DF
,
Weber
RW
,
Sosman
JA
,
Haanen
JB
, et al.
Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:.https://doi.org/10.1056/NEJMoa1003466
7
Morgan
DA
,
Ruscetti
FW
,
Gallo
R
. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193(4257):1007–8. doi:.https://doi.org/10.1126/science.181845
8
Robb
RJ
,
Smith
KA
. Heterogeneity of human T-cell growth factor(s) due to variable glycosylation. Mol Immunol. 1981;18(12):1087–94. doi:.https://doi.org/10.1016/0161-5890(81)90024-9
9
Taniguchi
T
,
Matsui
H
,
Fujita
T
,
Takaoka
C
,
Kashima
N
,
Yoshimoto
R
, et al.
Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302(5906):305–10. doi:.https://doi.org/10.1038/302305a0
10
Raeber
ME
,
Zurbuchen
Y
,
Impellizzieri
D
,
Boyman
O
. The role of cytokines in T-cell memory in health and disease. Immunol Rev. 2018;283(1):176–93. doi:.https://doi.org/10.1111/imr.12644
11
Klapper
JA
,
Downey
SG
,
Smith
FO
,
Yang
JC
,
Hughes
MS
,
Kammula
US
, et al.
High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113(2):293–301. doi:.https://doi.org/10.1002/cncr.23552
12
Boyman
O
,
Sprent
J
. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–90. doi:.https://doi.org/10.1038/nri3156
13
Arenas-Ramirez
N
,
Woytschak
J
,
Boyman
O
. Interleukin-2: Biology, Design and Application. Trends Immunol. 2015;36(12):763–77. doi:.https://doi.org/10.1016/j.it.2015.10.003
14
Anderson
TD
,
Hayes
TJ
,
Gately
MK
,
Bontempo
JM
,
Stern
LL
,
Truitt
GA
. Toxicity of human recombinant interleukin-2 in the mouse is mediated by interleukin-activated lymphocytes. Separation of efficacy and toxicity by selective lymphocyte subset depletion. Lab Invest. 1988;59(5):598–612.
15
Gately
MK
,
Anderson
TD
,
Hayes
TJ
. Role of asialo-GM1-positive lymphoid cells in mediating the toxic effects of recombinant IL-2 in mice. J Immunol. 1988;141(1):189–200.
16
Peace
DJ
,
Cheever
MA
. Toxicity and therapeutic efficacy of high-dose interleukin 2. In vivo infusion of antibody to NK-1.1 attenuates toxicity without compromising efficacy against murine leukemia. J Exp Med. 1989;169(1):161–73. doi:.https://doi.org/10.1084/jem.169.1.161
17
Assier
E
,
Jullien
V
,
Lefort
J
,
Moreau
JL
,
Di Santo
JP
,
Vargaftig
BB
, et al.
NK cells and polymorphonuclear neutrophils are both critical for IL-2-induced pulmonary vascular leak syndrome. J Immunol. 2004;172(12):7661–8. doi:.https://doi.org/10.4049/jimmunol.172.12.7661
18
Fraker
DL
,
Langstein
HN
,
Norton
JA
. Passive immunization against tumor necrosis factor partially abrogates interleukin 2 toxicity. J Exp Med. 1989;170(3):1015–20. doi:.https://doi.org/10.1084/jem.170.3.1015
19
Boyman
O
,
Surh
CD
,
Sprent
J
. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther. 2006;6(12):1323–31. doi:.https://doi.org/10.1517/14712598.6.12.1323
20
Krieg
C
,
Létourneau
S
,
Pantaleo
G
,
Boyman
O
. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci USA. 2010;107(26):11906–11. doi:. Correction in: Proc Natl Acad Sci USA. 2012;109(1):345. doi:https://doi.org/10.1073/pnas.1119897109https://doi.org/10.1073/pnas.1002569107
21
Arenas-Ramirez
N
,
Zou
C
,
Popp
S
,
Zingg
D
,
Brannetti
B
,
Wirth
E
, et al.
Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci Transl Med. 2016;8(367):367ra166. doi:.https://doi.org/10.1126/scitranslmed.aag3187
22
Boyman
O
,
Kovar
M
,
Rubinstein
MP
,
Surh
CD
,
Sprent
J
. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311(5769):1924–7. doi:.https://doi.org/10.1126/science.1122927
23
Létourneau
S
,
van Leeuwen
EM
,
Krieg
C
,
Martin
C
,
Pantaleo
G
,
Sprent
J
, et al.
IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proc Natl Acad Sci USA. 2010;107(5):2171–6. doi:.https://doi.org/10.1073/pnas.0909384107
24
Levin
AM
,
Bates
DL
,
Ring
AM
,
Krieg
C
,
Lin
JT
,
Su
L
, et al.
Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–33. doi:.https://doi.org/10.1038/nature10975
25
Garber
K
. Cytokine resurrection: engineered IL-2 ramps up immuno-oncology responses. Nat Biotechnol. 2018;36(5):378–9. doi:.https://doi.org/10.1038/nbt0518-378
26
Charych
DH
,
Hoch
U
,
Langowski
JL
,
Lee
SR
,
Addepalli
MK
,
Kirk
PB
, et al.
NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin Cancer Res. 2016;22(3):680–90. doi:.https://doi.org/10.1158/1078-0432.CCR-15-1631
27
Pandiyan
P
,
Zheng
L
,
Ishihara
S
,
Reed
J
,
Lenardo
MJ
. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–62. doi:.https://doi.org/10.1038/ni1536
28
Létourneau
S
,
Krieg
C
,
Pantaleo
G
,
Boyman
O
. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. 2009;123(4):758–62. doi:.https://doi.org/10.1016/j.jaci.2009.02.011
29
Zingg
D
,
Debbache
J
,
Schaefer
SM
,
Tuncer
E
,
Frommel
SC
,
Cheng
P
, et al.
The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6(1):6051. doi:.https://doi.org/10.1038/ncomms7051
30
Spohn
G
,
Arenas-Ramirez
N
,
Bouchaud
G
,
Boyman
O
. Endogenous polyclonal anti-IL-1 antibody responses potentiate IL-1 activity during pathogenic inflammation. J Allergy Clin Immunol. 2017;139(6):1957–1965.e3. doi:.https://doi.org/10.1016/j.jaci.2016.09.033
31
Jones
AT
,
Ziltener
HJ
. Enhancement of the biologic effects of interleukin-3 in vivo by anti-interleukin-3 antibodies. Blood. 1993;82(4):1133–41.
32
Finkelman
FD
,
Madden
KB
,
Morris
SC
,
Holmes
JM
,
Boiani
N
,
Katona
IM
, et al.
Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151(3):1235–44.
33
Klein
B
,
Brailly
H
. Cytokine-binding proteins: stimulating antagonists. Immunol Today. 1995;16(5):216–20. doi:.https://doi.org/10.1016/0167-5699(95)80161-8
34
Montero-Julian
FA
,
Klein
B
,
Gautherot
E
,
Brailly
H
. Pharmacokinetic study of anti-interleukin-6 (IL-6) therapy with monoclonal antibodies: enhancement of IL-6 clearance by cocktails of anti-IL-6 antibodies. Blood. 1995;85(4):917–24.
35
Boyman
O
,
Ramsey
C
,
Kim
DM
,
Sprent
J
,
Surh
CD
. IL-7/anti-IL-7 mAb complexes restore T cell development and induce homeostatic T Cell expansion without lymphopenia. J Immunol. 2008;180(11):7265–75. doi:.https://doi.org/10.4049/jimmunol.180.11.7265
36
Schmutz
S
,
Bosco
N
,
Chappaz
S
,
Boyman
O
,
Acha-Orbea
H
,
Ceredig
R
, et al.
Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol. 2009;183(4):2217–21. doi:.https://doi.org/10.4049/jimmunol.0802911
37
Rosalia
RA
,
Arenas-Ramirez
N
,
Bouchaud
G
,
Raeber
ME
,
Boyman
O
. Use of enhanced interleukin-2 formulations for improved immunotherapy against cancer. Curr Opin Chem Biol. 2014;23:39–46. doi:.https://doi.org/10.1016/j.cbpa.2014.09.006
38
Woytschak
J
,
Keller
N
,
Krieg
C
,
Impellizzieri
D
,
Thompson
RW
,
Wynn
TA
, et al.
Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration during Infection and Inflammation. Immunity. 2016;45(1):172–84. doi:.https://doi.org/10.1016/j.immuni.2016.06.025
39
Verdeil
G
,
Marquardt
K
,
Surh
CD
,
Sherman
LA
. Adjuvants targeting innate and adaptive immunity synergize to enhance tumor immunotherapy. Proc Natl Acad Sci USA. 2008;105(43):16683–8. doi:.https://doi.org/10.1073/pnas.0805054105
40
Cho
HI
,
Reyes-Vargas
E
,
Delgado
JC
,
Celis
E
. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res. 2012;72(8):1986–95. doi:.https://doi.org/10.1158/0008-5472.CAN-11-3246
41
Redmond
WL
,
Triplett
T
,
Floyd
K
,
Weinberg
AD
. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PLoS One. 2012;7(4):e34467. doi:.https://doi.org/10.1371/journal.pone.0034467
42
Zingg
D
,
Arenas-Ramirez
N
,
Sahin
D
,
Rosalia
RA
,
Antunes
AT
,
Haeusel
J
, et al.
The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017;20(4):854–67. doi:.https://doi.org/10.1016/j.celrep.2017.07.007
43
Tomala
J
,
Chmelova
H
,
Mrkvan
T
,
Rihova
B
,
Kovar
M
. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. 2009;183(8):4904–12. doi:.https://doi.org/10.4049/jimmunol.0900284
44
Klein
C
,
Waldhauer
I
,
Nicolini
VG
,
Freimoser-Grundschober
A
,
Nayak
T
,
Vugts
DJ
, et al.
Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. OncoImmunology. 2017;6(3):e1277306. doi:.https://doi.org/10.1080/2162402X.2016.1277306
45Press release by Bristol-Myers Squibb & Nektar Therapeutics: Preliminary data for NKTR-214 in combination with Opdivo (nivolumab) for patients with stage IV metastatic melanoma, renal cell carcinoma, and urothelial cancers presented at ASCO 2018.
46
Klevorn
LE
,
Berrien-Elliott
MM
,
Yuan
J
,
Kuehm
LM
,
Felock
GD
,
Crowe
SA
, et al.
Rescue of Tolerant CD8+ T Cells during Cancer Immunotherapy with IL2:Antibody Complexes. Cancer Immunol Res. 2016;4(12):1016–26. doi:.https://doi.org/10.1158/2326-6066.CIR-16-0159