Hepatic manifestations of Wilson’s disease: 12-year experience in a Swiss tertiary referral centre
DOI: https://doi.org/10.4414/smw.2018.14699
Joana
Vieira Barbosaa, Montserrat
Fragaa, Joan
Saldarriagab, Philippe
Hiroza, Emiliano
Giostrac, Christine
Sempouxb, Peter
Ferencid, Darius
Moradpoura
aDivision of Gastroenterology and Hepatology, Centre Hospitalier Universitaire Vaudois, University of Lausanne, Switzerland
bInstitute of Pathology, Centre Hospitalier Universitaire Vaudois, University of Lausanne, Switzerland
cDivision of Gastroenterology and Hepatology, Geneva University Hospitals, Switzerland
dDivision of Gastroenterology and Hepatology, Department of Internal Medicine III, Medical University of Vienna, Austria
Summary
BACKGROUND AND AIM
Wilson’s disease is an inherited disorder of hepatic copper metabolism, leading to the accumulation of copper in the liver as well as the brain, cornea and other organs. Here, we describe the adult cases of hepatic Wilson’s disease diagnosed at the Division of Gastroenterology and Hepatology of the University Hospital Lausanne, Switzerland between September 2004 and August 2016.
METHODS
Clinical manifestations, results of diagnostic tests, management and outcomes of adult patients with hepatic Wilson’s disease were assessed based on standardised medical records. In addition, liver histology was reviewed and the lesional patterns were recorded.
RESULTS
Ten new adult cases of hepatic Wilson’s disease were diagnosed in our centre between September 2004 and August 2016. Male to female ratio was 1:1 and median age at diagnosis was 26 (range 18–56) years. Four patients presented with acute liver failure, four with persistently elevated liver function tests, and two with decompensated cirrhosis; none had neurological manifestations. Only one patient had a Kayser-Fleischer corneal ring. Median ceruloplasmin level at diagnosis was 0.13 (range <0.03–0.30) g/l, median 24-hour urinary copper excretion was 2.8 (range 0.3–77.3) μmol, and median hepatic copper concentration was 789 (range 284–1677) μg/g. At least one mutation in the ATP7B gene was identified in eight patients. Allelic frequency of the common H1069Q mutation was 19%. Leipzig score was ≥5 in all patients. Three patients presenting with acute liver failure and the two with decompensated cirrhosis underwent successful liver transplantation. One patient with acute liver failure recovered under chelation therapy, as predicted by a Dhawan score <11. D-penicillamine was used as first-line chelator treatment, with a subsequent switch to trientine due to adverse effects in three out of six patients.
CONCLUSIONS
The clinical presentation of hepatic Wilson’s disease is highly variable. Three out of 10 patients were diagnosed at an age >35 years. A high index of suspicion in clinically compatible situations is key.
Introduction
Wilson’s disease is a rare, autosomal-recessive, inherited disorder of hepatic copper metabolism with a worldwide prevalence of 1:30,000 [1–4]. It is caused by mutations of the ATP7B gene, leading to copper accumulation in hepatocytes and extrahepatic organs such as the brain and the cornea. The ATP7B gene encodes a copper-transporting P-type adenosine triphosphatase, which is expressed in hepatocytes and transports copper into the trans-Golgi network where it is either incorporated into apo-ceruloplasmin for the synthesis of functional ceruloplasmin or secreted into the bile. Under normal conditions, copper homeostasis is ensured by biliary excretion, since dietary consumption and absorption exceed metabolic needs. Loss of ATP7B function results in hepatic copper accumulation, leading to liver and/or neurological damage, with a fatal outcome possible if left untreated [1–4].
The clinical hallmark of Wilson’s disease is the combination of liver disease, neurologic symptoms and a Kayser-Fleischer corneal ring. However, the diagnosis of Wilson’s disease can be difficult, as clinical presentations vary widely and a Kayser-Fleischer ring is often absent, especially in the case of predominant liver disease [5]. Determination of intrahepatic copper content is considered the gold standard to reach a diagnosis of Wilson’s disease, but also has certain limitations. Molecular testing for ATP7B mutations plays a key role in the diagnosis of Wilson’s disease but is difficult due to the length of the gene and the large number of mutations identified so far. The most common mutation in Europe is H1069Q. Nevertheless, the ATP7B genotype does not allow predictions of the natural history and severity of Wilson’s disease [1, 2].
Histology is highly variable, ranging from mild to severe chronic hepatitis, from simple steatosis to prominent steatohepatitis features, or showing confluent necrosis in fulminant cases. Progressively, increasing stages of fibrosis as well as macronodular cirrhosis are observed. Glycogenated nuclei, portal inflammation and steatosis are almost always present, although sometimes very mild, and there is often marked variability in nuclear size. Copper deposition, as identified by special stains, does not rule out the diagnosis and is not always found [6, 7].
Medical therapy with chelators (D-penicillamine or trientine) or zinc prevents disease progression and the development of hepatic and/or neurological complications. Liver transplantation represents a curative option [8].
Here, we describe the clinical manifestations, results of diagnostic tests, management and outcomes of the adult patients with a new diagnosis of hepatic Wilson’s disease made in our centre between September 2004 and August 2016. We demonstrate that the clinical presentation of hepatic Wilson’s disease is highly variable. Thus, improved awareness is key to the early recognition of the disease and, therefore, its optimal management and prognosis.
Patients and methods
Study population and design
Using standardised medical records, we collected clinical, laboratory and histological data of all patients with a new diagnosis of Wilson’s disease made in the Division of Gastroenterology and Hepatology of the University Hospital Lausanne (Centre Hospitalier Universitaire Vaudois, CHUV) between September 2004 and August 2016. The CHUV is a tertiary referral centre, with approximately 10,000 gastroenterology and hepatology outpatient consultations per year, as well as a very active inpatient consultation service. We closely collaborate with the Geneva University Hospitals (Hôpitaux Universitaires de Genève) in a joint liver transplant program. This study was approved by the Institutional Ethics Committee of the University of Lausanne and informed consent was obtained from the participants.
Clinical and laboratory analyses
Diagnosis of Wilson’s disease was based on a combination of clinical symptoms and signs, laboratory tests, including genetic analyses, and histological evaluation. Physical examination included neurological and psychiatric evaluation. Moreover, all patients were examined for the presence of a Kayser-Fleischer ring by an expert ophthalmologist.
Standard clinical chemistry and haematological analyses were complemented by serologic and, if appropriate, molecular testing for hepatitis A, B, C and E; determination of autoantibodies related to autoimmune hepatitis; and measurement of serum ferritin and transferrin saturation as well as α1-antitrypsin. Abdominal ultrasound, including Doppler evaluation, was also performed systematically. Liver biopsies were performed either percutaneously or, where appropriate, via the transjugular route.
The Leipzig score [9] was used to ascertain Wilson’s disease diagnosis. It comprises the following parameters: Kayser-Fleischer ring (2 or 0 points for present or absent respectively), neurological symptoms (2, 1 or 0 points for severe, mild or absent respectively), serum ceruloplasmin (2, 1 or 0 points for <0.1 g/l, 0.1–0.2 g/l or normal [>0.2 g/l] respectively), Coombs-negative haemolytic anaemia (1 or 0 for present or absent respectively), liver copper content (2, 1 or −1 points for >250 µg/g, 50–250 µg/g or normal [<50 µg/g] respectively), urinary copper excretion (2, 1 or 0 points for >1.6 µmol/24 h, 0.8–1.6 µmol/24 h or normal [<0.8 µmol/24 h]), and ATP7B mutation analysis (4, 1 or 0 points for mutations detected on both chromosomes, a mutation detected on one chromosome or no mutations detected respectively). A score of four was considered diagnostic for Wilson’s disease.
The Dhawan score [10], based on serum bilirubin, international normalised ratio, aspartate aminotransferase (AST), albumin and white blood cell count, was used to determine the prognosis and probability of survival without liver transplantation in patients with acute liver failure. A score ≥11 predicts a high probability of death without liver transplantation.
Mutation analysis
ATP7B mutation H1069Q, which is the most common European mutation, was detected by PCR [11]. In the absence of a homozygote mutation, we analysed all ATP7B exons using amplification with published primers [12] by denaturing high performance liquid chromatography (WAVE MD System Model 4000; Transgenomics, Crewe, UK). All exons with possible mutations were Sanger sequenced using the ABI Prism 310 Genetic Analyzer (Perkin Elmer, Norwalk, CT) until 2012 and using the 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA) thereafter. For the patients in whom we did not identify any mutation, all exons were sequenced. Multiplex ligation-dependent probe amplification was performed with SALSA MLPA P098 for Wilson’s disease (MRC-Holland, Amsterdam, NL) for the detection of large deletions.
Histological evaluation
After 24 h of formalin fixation followed by paraffin embedding, biopsies were cut at 4 µm and routinely stained with haematoxylin-eosin, Masson's trichrome, Prussian blue, periodic acid Schiff-diastase and reticulin. The presence of inflammation, necrosis, fat, iron, architectural distortion and fibrosis was recorded. In addition, rhodanine and orcein stains were performed in order to identify copper granules and copper-binding protein respectively.
Results
Clinical features, laboratory results and the Leipzig score at the time of presentation are summarised in table 1. Ten adult patients were newly diagnosed with Wilson’s disease between September 2004 and August 2016. All presented with exclusive hepatic forms of the disease.
Table 1 Characteristics of patients with Wilson disease.
|
Pt
|
Sex
|
Age at P/D
|
Clinical presentation
|
Kayser-Fleischer ring
|
CP
(g/l)
|
S Cu
(μg/l)
|
24 h U Cu (μmol)
|
H Cu (μg/g)
|
APT7B mutation
|
Leipzig score
|
| 1 |
F |
31/37 |
Cirrhosis |
No |
0.30 |
393 |
2.5 |
284 |
R969Q/R969Q |
8 |
| 2 |
M |
22/22 |
Acute liver failure |
Yes |
0.13 |
546 |
60 |
789 |
H1069Q/n.i. |
9 |
| 3 |
F |
22/22 |
Acute liver failure |
No |
0.11 |
n.a. |
77.3 |
n.a. |
H1069Q/n.i. |
(5) |
| 4 |
M |
25/25 |
Elevated LFTs |
No |
0.03 |
14 |
2.2 |
402 |
M645R/D1306Y |
10 |
| 5 |
F |
30/30 |
Acute liver failure |
No |
0.18 |
433 |
62 |
1049 |
R616Q/A1140V |
10 |
| 6 |
M |
18/18 |
Elevated LFTs |
No |
0.17 |
95 |
22 |
757 |
L979Q/n.i. |
6 |
| 7 |
M |
36/56 |
Elevated LFTs |
No |
<0.03 |
n.a. |
0.3 |
426 |
T977M/A1074T |
8 |
| 8 |
F |
25/27 |
Elevated LFTs |
No |
0.11 |
163 |
1.9 |
911 |
H1069Q/n.i. |
6 |
| 9 |
F |
18/18 |
Acute liver failure |
No |
0.05 |
499 |
n.a. |
1677 |
n.i. |
(5) |
| 10 |
M |
37/44 |
Cirrhosis |
No |
0.17 |
337 |
2.8 |
852 |
n.i. |
5 |
Clinical presentation
Five patients were women and five men. Median age at diagnosis was 26 years (range 18–56 years). Notably three patients were older than 35 years at diagnosis. Clinical presentation was highly variable. Four patients presented with acute liver failure, four with persistently elevated liver function tests, and two with decompensated cirrhosis. Patient 8 is the sister of patient 3. None of the patients had neurological manifestations. Only one patient presented a Kayser-Fleischer corneal ring. Time from initial presentation to diagnosis varied between a few hours for patients presenting with typical laboratory features in a context of acute liver failure and 20 years for patient 7.
Laboratory parameters
Standard laboratory tests
Median AST at diagnosis was 122 U/l (range 32–161 U/l) and median ALT was 90 U/l (range 40–317 U/l). AST:ALT ratio was >1 in six patients. Median alkaline phosphatase (ALP) was 94 U/l (range 15–253 U/l). In the cases with acute liver failure, ALP was low in all patients and inferior to the normal range in two; transaminases were only mildly elevated, with an AST:ALT ratio >1 in all patients and a ratio >2 in two (table 2).
Table 2 Laboratory results for patients with acute liver failure.
|
Pt
|
AST
(U/l)
|
ALT (U/l)
|
AST:ALT Ratio
|
ALP (U/l)
|
Bilirubin (μmol/l)
|
ALP:total bilirubin ratio
|
INR
|
Hb (g/l)
|
Dhawan score
|
| 2 |
154 |
140 |
1.1 |
60 |
364 |
0.2 |
3.6 |
103 |
13 |
| 3 |
123 |
42 |
2.9 |
23 |
250 |
0.1 |
2.4 |
85 |
13 |
| 5 |
156 |
97 |
1.6 |
109 |
69 |
1.6 |
2.2 |
98 |
10 |
| 9 |
161 |
59 |
2.7 |
15 |
175 |
0.1 |
3.5 |
66 |
16 |
Copper metabolism
Median serum ceruloplasmin level at diagnosis was 0.13 g/l (range <0.03–0.30 g/l); only one patient had a normal level. Median 24-h urinary copper excretion was 2.8 μmol (range 0.3–77.3 µmol); only one patient had a urinary copper excretion <1.6 µmol/24 h. Median hepatic copper content was 789 μg/g (range, 284–1677 µg/g) and all patients had a concentration >250 μg/g. Median non-ceruloplasmin-bound serum copper was 365 μg/l (range 14–546 µg/l) and it was <150 μg/l in two patients.
Genetic testing
Molecular analysis of the ATP7B gene was performed in all patients. At least one mutation in the ATP7B gene was identified in eight patients. Allelic frequency of the common H1069Q mutation was 19%. In patient 9, a young lady presenting with the typical clinical, laboratory and histological features of Wilson’s disease-related acute liver failure, and in patient 10, a 44-year-old man presenting with decompensated cirrhosis, no ATP7B mutation could be identified.
Histology
The histological patterns observed in this series are summarised in table 3. Five patients showed chronic hepatitis as a predominant pattern, ranging from mild to moderate, or even severe in one patient (fig. 1A), associated with advanced fibrosis in one and with cirrhosis in three patients. Macrovesicular steatosis was prominent in two patients, including one showing features of steatohepatitis with mild fibrosis (fig. 1B). One patient had a pattern of multifocal necrosis on a background of fibrosis (fig. 1C). Copper granules and copper-binding proteins were observed in this latter case as well as in the three patients with cirrhosis. Glycogenated nuclei were present in all cases.
Table 3 Histological lesional patterns.
|
Pt
|
Clinical presentation
|
Histology
|
| 1 |
Cirrhosis |
Cirrhosis and moderate chronic hepatitis, copper + |
| 2 |
Acute liver failure |
Advanced fibrosis and severe chronic hepatitis |
| 3 |
Acute liver failure |
n.a. |
| 4 |
Elevated LFTs |
Steatosis |
| 5 |
Acute liver failure |
Cirrhosis and moderate chronic hepatitis, copper + |
| 6 |
Elevated LFTs |
Mild chronic hepatitis, no fibrosis |
| 7 |
Elevated LFTs |
n.a. |
| 8 |
Elevated LFTs |
Steatohepatitis |
| 9 |
Acute liver failure |
Multifocal necrosis, advanced fibrosis, copper + |
| 10 |
Cirrhosis |
Cirrhosis and moderate chronic hepatitis, copper + |
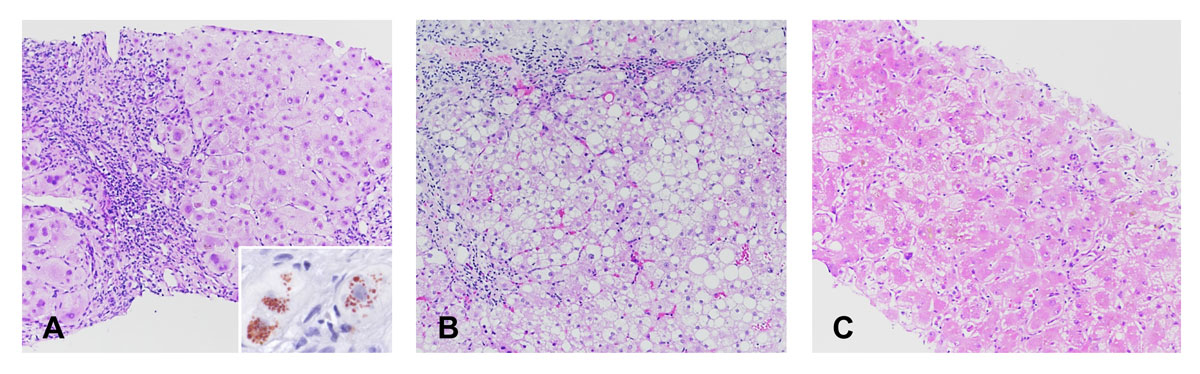
Fig. 1
Histological patterns observed in patients with Wilson’s disease. (A) Severe chronic hepatitis characterised by numerous inflammatory cells in the portal tracts, interface hepatitis and advanced fibrosis (patient 10, haematoxylin-eosin, × 10). Copper granules can be identified in the cytoplasm of periportal hepatocytes (insert, rhodanine stain, × 40). (B) Steatohepatitis with macrovesicular steatosis, ballooned hepatocytes and mild inflammation and fibrosis (patient 8, haematoxylin-eosin, × 10). (C) Multifocal necrosis (patient 9, hematoxylin-eosin, × 10).
Diagnosis
The Leipzig score was ≥5 in all patients, confirming the diagnosis of Wilson’s disease.
Management and outcome
The treatment and outcomes of the patients are summarised in table 4. All patients are alive. Of the four patients presenting with acute liver failure, three were transplanted with excellent outcomes. Patient 5 was treated with a copper chelating agent and recovered without liver transplantation, as predicted by a Dhawan score <11 (see table 2).
Table 4 Management and outcome of patients with Wilson’s disease.
|
Pt
|
Medical treatment
|
Liver transplantation
|
Outcome
|
| 1 |
D-penicillamine → trientine |
Yes |
Excellent |
| 2 |
D-penicillamine |
Yes |
Excellent |
| 3 |
none |
Yes |
Excellent |
| 4 |
Zinc acetate → D-penicillamine |
No |
Normal LFTs |
| 5 |
D-penicillamine → trientine |
No |
Excellent |
| 6 |
D-penicillamine |
No |
Normal LFTs |
| 7 |
Zinc acetate → D-penicillamine |
No |
Normal LFTs |
| 8 |
D-penicillamine |
No |
Normal LFTs |
| 9 |
none |
Yes |
Excellent |
| 10 |
D-penicillamine → trientine |
Yes |
Excellent |
Of the six patients presenting with chronic forms of the disease, four were treated with D-penicillamine and two with zinc as first-line treatment. D-penicillamine had to be replaced by trientine in three patients because of hematologic adverse effects (predominantly neutropenia). Chelation therapy did not allow for recovery of the two patients who presented initially with decompensated cirrhosis, both of whom were >35 years old at the time of diagnosis. Thus, patients 1 and 10 underwent liver transplantation. Patient 7, in whom diagnosis was made at age 56 in the context of persistently elevated liver function tests, stopped treatment and is currently stable.
Discussion
Ten adult patients with hepatic Wilson’s disease were diagnosed in the Division of Gastroenterology and Hepatology of the CHUV between September 2004 and August 2016. Our study underlines the challenge of establishing Wilson’s disease diagnosis, not only because it is a rare condition but also because the clinical, laboratory and histological manifestations are highly variable. As shown in our study, prompt recognition of the disease plays a key role in the management of patients, as hepatic and neurological damage can develop rapidly [1–4].
In our cohort, all patients had exclusive hepatic presentation. Median age was 26 years. The oldest patient, patient 7, was 56 years old at diagnosis. Patients with Wilson’s disease usually become symptomatic between the ages of 5 and 35 years, with earlier onset of symptoms for hepatic disease, and neurological and psychiatric manifestations appearing later [1, 2]. However, cases of later onset Wilson’s disease have been described [13, 14]. Given its highly variable presentation, Wilson’s disease should be considered in any patient presenting with unclear hepatic or neurologic disease, independently of age [14].
Four patients presented with acute liver failure and the other six with chronic forms (persistently elevated liver function tests or decompensated cirrhosis). In a retrospective study including 30 patients with Wilson’s disease, 22 presented with chronic forms and 8 with acute liver failure, with a mean age at diagnosis of 21 years [15]. In another study including 163 patients with WD, 137 were symptomatic at diagnosis, 26 were diagnosed by family screening and 8 presented with acute liver failure [16].
As stated above, the diagnosis of Wilson’s disease is not always straightforward. In our patients, Wilson’s disease was suspected in the presence of a clinical presentation and compatible laboratory tests, and confirmed by the results of intrahepatic copper determination and genetic analyses. Intrahepatic copper is considered the best biochemical test for Wilson’s disease [1, 2]. All our patients presented with an intrahepatic copper content >250 μg/g. Another instrument that was shown to provide good diagnostic accuracy in the diagnosis of Wilson’s disease is the Leipzig score [1]. It is based on clinical symptoms and signs, laboratory tests and mutation analysis. A score >4 is considered diagnostic for Wilson’s disease [17]. The Leipzig score was ≥5 in all our patients.
In the cases with acute liver failure due to Wilson’s disease, the decision to start chelation treatment or to evaluate patients for liver transplantation should not await the results of laboratory investigations documenting disturbed copper metabolism [18]. Indeed, Korman et al. demonstrated that an ALP:total bilirubin ratio <4 in patients with acute liver failure is, as an initial screening tool, highly sensitive and specific for Wilson’s disease (94% and 96%, respectively) [19]. Moreover, when combined with an AST:ALT ratio >2.2, sensitivity and specificity increase up to 100%. In our cohort, we observed that all patients with acute liver failure presented an ALP:total bilirubin ratio <4 and two of the four had an AST:ALT ratio >2.2.
Genetic analyses revealed at least one ATP7B mutation in eight patients. Allelic frequency of H1069Q, the most common mutation in Europe [20], was 19%. In a retrospective study of 117 patients with Wilson’s disease, the most frequent ATP7B mutation was H1069Q, with an allelic frequency of 54% [21]. However, there are more than 500 reported mutations, and most patients are compound heterozygotes. On this background, genetic analyses are sometimes inconclusive [22].
No mutation was identified in patients 9 and 10, as described in earlier studies [16]. One explanation could be that genes other than ATP7B have a role in copper metabolism, resulting in copper accumulation when mutated. Moreover, mutations in introns or regulatory elements may affect ATP7B gene expression [23]. More advances in this area are needed.
Most of our patients presented an excellent outcome. However, the two patients who presented with decompensated cirrhosis at the age of 37 and 44 years (patients 1 and 10 respectively) did not recover on chelation therapy, indicating that the disease was too advanced. Both were successfully transplanted. Wilson’s disease, if treated early, has a good prognosis, with survival rates similar to the general population [16, 22]. As first-line treatment, six patients were treated with D-penicillamine and two patients with zinc. Treatment was readjusted in five patients. Three of the six patients (50%) treated initially with D-penicillamine developed hematologic adverse effects and had to be switched to trientine. Adverse effects of D-penicillamine requiring a change in therapy have been reported to occur in approximately 30% of patients [24]. We did not register any significant adverse effect of trientine.
In conclusion, Wilson’s disease is a rare but potentially fatal disease that should always be included in the differential diagnosis of patients with acute or chronic liver disease. Our study underlines the variability in presentation and the challenge in establishing a diagnosis of Wilson’s disease in some patients. The Leipzig score was confirmed to be very useful for this purpose. Timely diagnosis is important to avert progression of liver disease and to stratify patients presenting with acute liver failure for liver transplantation.
References
1
European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012;56(3):671–85. doi:.https://doi.org/10.1016/j.jhep.2011.11.007
2
Roberts
EA
,
Schilsky
ML
; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–111. doi:.https://doi.org/10.1002/hep.22261
3
Gitlin
JD
. Wilson disease. Gastroenterology. 2003;125(6):1868–77. doi:.https://doi.org/10.1053/j.gastro.2003.05.010
4
Ala
A
,
Walker
AP
,
Ashkan
K
,
Dooley
JS
,
Schilsky
ML
. Wilson’s disease. Lancet. 2007;369(9559):397–408. doi:.https://doi.org/10.1016/S0140-6736(07)60196-2
5
Steindl
P
,
Ferenci
P
,
Dienes
HP
,
Grimm
G
,
Pabinger
I
,
Madl
C
, et al.
Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997;113(1):212–8. doi:.https://doi.org/10.1016/S0016-5085(97)70097-0
6
Stromeyer
FW
,
Kamal
GI
,
Gerber
MA
,
Mathew
T
. Ground-glass cells in hepatocellular carcinoma. Am J Clin Pathol. 1980;74(3):254–8. doi:.https://doi.org/10.1093/ajcp/74.3.254
7
Johncilla
M
,
Mitchell
KA
. Pathology of the liver in copper overload. Semin Liver Dis. 2011;31(3):239–44. doi:.https://doi.org/10.1055/s-0031-1286055
8
Schilsky
ML
. Wilson disease: Diagnosis, treatment, and follow-up. Clin Liver Dis. 2017;21(4):755–67. doi:.https://doi.org/10.1016/j.cld.2017.06.011
9
Ferenci
P
,
Caca
K
,
Loudianos
G
,
Mieli-Vergani
G
,
Tanner
S
,
Sternlieb
I
, et al.
Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139–42. doi:.https://doi.org/10.1034/j.1600-0676.2003.00824.x
10
Dhawan
A
,
Taylor
RM
,
Cheeseman
P
,
De Silva
P
,
Katsiyiannakis
L
,
Mieli-Vergani
G
. Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation. Liver Transpl. 2005;11(4):441–8. doi:.https://doi.org/10.1002/lt.20352
11
Maier-Dobersberger
T
,
Ferenci
P
,
Polli
C
,
Balać
P
,
Dienes
HP
,
Kaserer
K
, et al.
Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med. 1997;127(1):21–6. doi:.https://doi.org/10.7326/0003-4819-127-1-199707010-00004
12
Thomas
GR
,
Forbes
JR
,
Roberts
EA
,
Walshe
JM
,
Cox
DW
. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9(2):210–7. doi:. Erratum in: Nat Genet. 1995;9:451.https://doi.org/10.1038/ng0295-210
13
Ala
A
,
Borjigin
J
,
Rochwarger
A
,
Schilsky
M
. Wilson disease in septuagenarian siblings: Raising the bar for diagnosis. Hepatology. 2005;41(3):668–70. doi:.https://doi.org/10.1002/hep.20601
14
Ferenci
P
,
Członkowska
A
,
Merle
U
,
Ferenc
S
,
Gromadzka
G
,
Yurdaydin
C
, et al.
Late-onset Wilson’s disease. Gastroenterology. 2007;132(4):1294–8. doi:.https://doi.org/10.1053/j.gastro.2007.02.057
15
Gow
PJ
,
Smallwood
RA
,
Angus
PW
,
Smith
AL
,
Wall
AJ
,
Sewell
RB
. Diagnosis of Wilson’s disease: an experience over three decades. Gut. 2000;46(3):415–9. doi:.https://doi.org/10.1136/gut.46.3.415
16
Merle
U
,
Schaefer
M
,
Ferenci
P
,
Stremmel
W
. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56(1):115–20. doi:.https://doi.org/10.1136/gut.2005.087262
17
Ferenci
P
. Phenotype-genotype correlations in patients with Wilson’s disease. Ann N Y Acad Sci. 2014;1315(1):1–5. doi:.https://doi.org/10.1111/nyas.12340
18
O’Brien
A
,
Williams
R
. Rapid diagnosis of Wilson disease in acute liver failure: no more waiting for the ceruloplasmin level?
Hepatology. 2008;48(4):1030–2. doi:.https://doi.org/10.1002/hep.22587
19
Korman
JD
,
Volenberg
I
,
Balko
J
,
Webster
J
,
Schiodt
FV
,
Squires
RH, Jr
, et al.; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology. 2008;48(4):1167–74. doi:.https://doi.org/10.1002/hep.22446
20
Ferenci
P
. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006;120(2):151–9. doi:.https://doi.org/10.1007/s00439-006-0202-5
21
Bruha
R
,
Marecek
Z
,
Pospisilova
L
,
Nevsimalova
S
,
Vitek
L
,
Martasek
P
, et al.
Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int. 2011;31(1):83–91. doi:.https://doi.org/10.1111/j.1478-3231.2010.02354.x
22
Ferenci
P
. Wilson’s Disease. Clin Gastroenterol Hepatol. 2005;3(8):726–33. doi:.https://doi.org/10.1016/S1542-3565(05)00484-2
23
Gupta
A
,
Das
S
,
Ray
K
. A glimpse into the regulation of the Wilson disease protein, ATP7B, sheds light on the complexity of mammalian apical trafficking pathways. Metallomics. 2018;10(3):378–87. doi:.https://doi.org/10.1039/C7MT00314E
24
Medici
V
,
Trevisan
CP
,
D’Incà
R
,
Barollo
M
,
Zancan
L
,
Fagiuoli
S
, et al.
Diagnosis and management of Wilson’s disease: results of a single center experience. J Clin Gastroenterol. 2006;40(10):936–41. doi:.https://doi.org/10.1097/01.mcg.0000225670.91722.59