Synovial sarcoma: when epigenetic changes dictate tumour development
DOI: https://doi.org/10.4414/smw.2018.14667
Nicolo
Riggi, Luisa
Cironi, Ivan
Stamenkovic
Experimental Pathology Service, Institute of Pathology, Centre Hospitalier Universitaire Vaudois, University of Lausanne, Switzerland
Summary
Synovial sarcoma is a highly aggressive soft tissue malignancy that often affects adolescents and young adults. It is associated with a unique chromosomal translocation that results in the formation and expression of the fusion gene SS18-SSX, which underlies its pathogenesis. Although SS18-SSX provides a potentially unique therapeutic target, all attempts to neutralise it have been unsuccessful thus far. When complete surgical removal of the tumour fails, therapy is limited to largely ineffective cytotoxic drug regimens. Nevertheless, recent discoveries about the mechanisms of SS18-SSX protein function have provided insight into potential alternative therapeutic strategies. SS18-SSX displays oncogenic activity through protein-protein interactions and participation in chromatin remodelling complexes. This review summarises our current understanding of the function of SS18-SSX and the mechanisms by which it alters the epigenetic landscape of permissive cells to induce transformation and the subsequent development of synovial sarcoma.
Introduction
Synovial sarcoma accounts for 10 to 20% of all soft tissue sarcomas in the adolescent and young adult population and is the most common soft tissue sarcoma after rhabdomyosarcoma [1]. Its salient biological feature is the nonrandom chromosomal translocation t(X:18; p11:q11), which generates fusion between the nearly entire coding sequence of the SS18 gene and a portion of the coding sequence of an SSX gene (SSX1 and SSX2 being the two most commonly implicated). Similarly to numerous paediatric malignancies, synovial sarcoma is genetically quiescent and typically displays few genetic mutations other than the chromosomal translocation. Transformation of primary cells and subsequent tumour development are therefore driven by SS18-SSX, which functions as a regulator of gene expression despite lacking DNA-binding motifs. SS18-SSX interacts with numerous proteins, several of which play a central role in the regulation of the cellular epigenetic status and cumulating evidence suggests that most of the oncogenic properties of SS18-SSX stem from its ability to orchestrate epigenetic changes in the affected cells.
Incidence and clinical features
Although it occurs most frequently in young individuals, with a median age of 35 years, synovial sarcoma afflicts patients from 5 to more than 80 years of age [2]. A recent study on synovial sarcoma incidence and survival in the USA covering three decades (from 1983 to 2012) revealed that the incidence per 1,000,000 increased from 0.9 to 1.5 in the total population within this time span and peaked within the 15 to 29 age range, reaching 2.2 per 1,000,000 in the third decade of the study [3]. The same study found that 5-year survival rates of synovial sarcoma did not improve over the three decades and in fact decreased from 69.4% between 1983 and 1992 to 60.1% between 2003 and 2012 [3]. Although these observations may at least in part reflect an increase in diagnostic accuracy, they nevertheless underscore the need to elucidate the pathogenesis of synovial sarcoma and develop effective, mechanism-based therapeutic strategies. There are no data as to the precise incidence of synovial sarcoma in Switzerland, but based on the number of new cases each year in university hospitals, the incidence can be estimated to be comparable to that in the USA.
Synovial sarcoma was first reported in 1865 [1] and described as a tumour proximal to a joint with a histological resemblance to developing synovium, leading to its label. Later immunohistochemical and ultrastructural analyses refuted the notion that the tumour bears synovial features [4], but the label has remained despite the widely held view that it is a misnomer. Consistent with this notion are reports that synovial sarcoma can arise in tissues unrelated to joints including the lung [5], digestive tract [6], bone marrow [7], kidney [8] and heart [9].
About 75% of synovial sarcomas arise in the extremities, predominantly in the lower limbs [10–12]. Clinical signs are non-specific, the tumour most commonly appearing as a slowly growing, palpable, tender mass, whose radiological features resemble an oval, soft tissue opacity associated with calcifications in about 25% of cases [13]. On magnetic resonance imaging (MRI), synovial sarcoma displays an image that has been described as the triple signal pattern, which reflects a combination of calcifications, cystic changes due to necrosis and haemorrhage, and air fluid levels [12, 13].
Synovial sarcoma falls into the category of high-grade tumours based on its propensity for local invasion and high metastatic proclivity [10, 14]. Roughly 50% of adult patients have metastatic disease at diagnosis, the lung being the most common metastatic site [10, 15, 16]. In contrast, only 5 to 11% of paediatric synovial sarcomas are associated with metastasis at diagnosis [10, 16]. Metastatic tumours in both adults and children have complex chromosomal aberrations. Current therapy is the same for both local and metastatic synovial sarcoma and consists of multiple cycles of adjuvant or neoadjuvant chemotherapy and wide surgical resection, as well as removal of metastatic tumours if present as a single or a few well-defined and accessible lesions. Surgical excision may be followed by adjuvant or neo-adjuvant radiation therapy [17]. Sensitivity to cytotoxic drugs is relatively modest and, despite seemingly complete surgical removal of local disease, early and late recurrences are common [10]. Late local recurrences and pulmonary metastases more than 5 years after initial diagnosis are more typical of synovial sarcoma than other sarcomas [15] and the current 10-year disease-free survival following excision of local disease is about 50% [10].
Histopathology
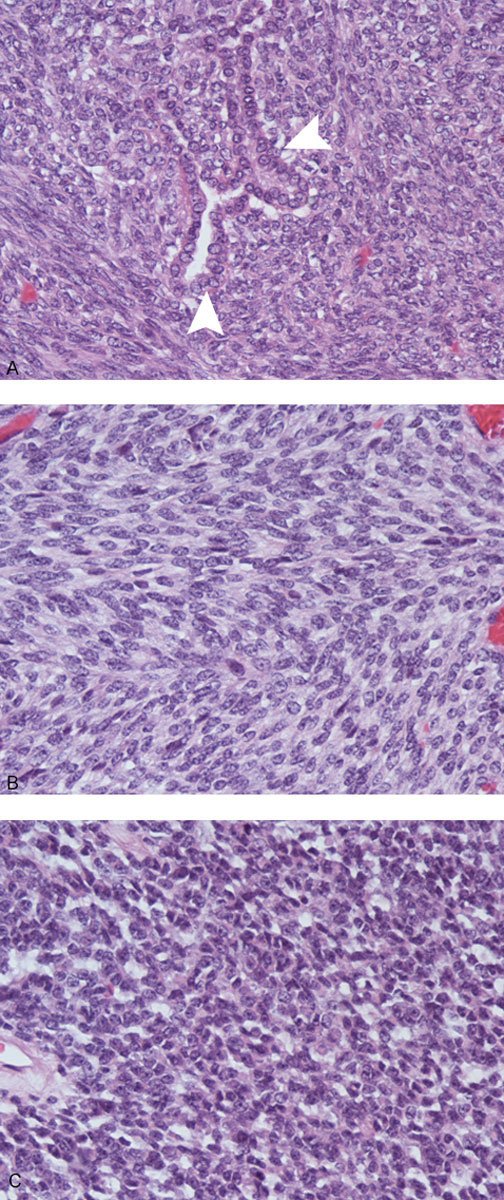
Synovial sarcoma has the unusual property among soft tissue tumours of displaying epithelial differentiation. There are two major synovial sarcoma subtypes: the monophasic subtype, which comprises about 75% of tumours, is composed of spindle cells without any detectable epithelial features. The biphasic subtype, which comprises the remaining 25% of tumours and is composed of spindle cells as well as cells with epithelial features, which often form glandular structures (fig. 1). The monophasic subtype can display poor differentiation and resemble small round blue cell tumours, which include Ewing sarcoma, alveolar rhabdomyosarcoma and lymphoma [18]. Most tumours express CD99 and endothelial growth factor receptor (EGFR) [19, 20] as well as platelet-derived growth factor receptor-alpha (PDGFRα) [21], whereas c-Met expression is associated with particularly aggressive SS phenotypes [22, 23]. However, there are currently no specific immunohistochemical synovial sarcoma markers and the only diagnostic molecular feature is the chromosomal translocation t(X:18).
The t(X:18) translocation – the diagnostic feature and pathogenic hallmark of synovial sarcoma
The balanced chromosomal translocation t(X:18; p11:q11), which is observed in virtually all cases, is unique to synovial sarcoma and is not associated with any other human tumour. The translocation generates an in-frame fusion of the SS18 gene (formerly referred to as SYT) to SSX1 or SSX2 (or SSX4 in a small fraction of cases [24, 25]), in which sequences encoding all but the 8 carboxy terminal (C-terminal) amino acids of SS18 become fused to sequences encoding the C-terminal 78 amino acids of the SSX partner. At least two observations support the notion that SS18-SSX is the key genetic driver in synovial sarcoma: first, it reflects the only cytogenetic abnormality in as many as a third of cases [26, 27]; second, in conditional mouse models, SS18-SSX alone can induce tumours with histological features and a gene expression profile reminiscent of those of human synovial sarcoma [28]. It has been suggested that synovial sarcoma cell viability decreases upon SS18-SSX depletion [29], but there is substantial variability among available synovial sarcoma cell lines, some of which remain viable and continue to proliferate following SS18-SSX removal.
Cell of origin and models of synovial sarcoma
As is the case with most sarcomas, the cell of origin of synovial sarcoma has not been easy to identify. A transgenic mouse model of synovial sarcoma was generated by expression of human SS18-SSX in an early myoblast population bearing the Myf5 marker, which was unexpected as neither human nor mouse synovial sarcomas express muscle differentiation markers [28]. Tumours arose with 100% penetrance and displayed a phenotype reminiscent of monophasic synovial sarcoma. The path toward developing the transgenic model suggested a narrow window of permissiveness for SS18-SSX-mediated transformation, as SS18-SSX expression in later stage Myf6+ myoblasts caused myopathy but no tumours, whereas conditional expression in earlier myoblast populations (Pax3+ or Pax7+) was embryonic lethal [30]. Embryonic lethality was also observed upon SS18-SSX expression in bone, endothelial and neural precursor populations as well as in early ectoderm [30]. Somewhat surprisingly, tamoxifen-induced conditional expression of SS18-SSX resulted in the development of more indolent tumours in atypical anatomical locations, including facial and paraspinal sites, suggesting that cells other than Myf5+ myoblasts may be permissive for SS18-SSX-mediated transformation. Consistent with this notion, a mouse embryonic cell line, C3H10T1/2, which displays mesenchymal stem cell (MSC) features, was observed not only to be permissive for SS18-SSX expression but also to acquire a gene expression profile with significant resemblance to that of primary human synovial sarcoma [31]. The argument that a MSC subpopulation may harbour the cell of origin of synovial sarcoma has been strengthened by the observation that silencing of SS18-SSX in two synovial sarcoma cell lines caused them to transit from growth in suspension, as spheres, to growth as adherent monolayers, adopt a spindle cell phenotype, express mesenchymal markers and display a broad differentiation potential to osteocytes, chondrocytes and adipocytes [32].
SS18-SSX can be expressed in many primary human cells without inducing senescence or apoptosis and primary human MSCs display gene expression profiles in response to SS18-SSX expression that overlap with the synovial sarcoma transcriptome [33]. However, the response was divergent among different batches of MSCs, some of which upregulated numerous genes associated with synovial sarcoma, whereas others displayed only minimal transcriptome changes despite allowing SS18-SSX protein expression. These observations support the notion that SS18-SSX may have a narrow window of opportunity to exert its oncogenic functions in primary cells, which is determined by host cell properties, most likely including their differentiation state. MSCs are defined by the expression of a set of common cell surface markers, the lack of lineage-specific markers, and the ability to differentiate along a variety of mesenchymal lineages under appropriate culture conditions [34]. However, these cells are heterogeneous such that any bulk MSC population may harbour subpopulations at discrete stages of differentiation, whose permissiveness for defined oncogenic events may differ markedly. It is therefore likely that permissiveness for oncogenic fusion protein expression, be it SS18-SSX or a fusion protein that underlies the pathogenesis of other sarcomas [35, 36], is the property of only some of these putative subpopulations. Currently, the lack of selective markers for discrete stages of MSC differentiation is a limitation in MSC biology in general and in determining the origin of a variety of sarcomas in particular. Furthermore, bone marrow and peripheral tissue MSCs display the same markers but differ in their expression of a variety of mediators [37] and neural crest derived MSCs display different features still. It will be highly instructive to determine the composition of MSC populations from different anatomical sites and how they respond to diverse sarcoma-specific oncogenic events.
Similar to other solid tumours, synovial sarcomas display intratumour heterogeneity. Although the observed heterogeneity appears to arise according to the cancer stem cell (CSC) model, synovial sarcoma CSCs remain to be characterised. Primary synovial sarcoma cells grow in suspension as spheres in serum-depleted medium and low attachment plates. The spheres are enriched in cells expressing stem cells markers Oct3/4, Nanog and Sox2 and some can initiate tumour growth in serial transplantation assays [32], which is consistent with enrichment in CSCs. However, with few exceptions [38], these cells do not express markers that are commonly associated with, although not specific for, CSCs. Although expression of the chemokine receptor CXCR4 has recently been reported to be associated with tumour-initiating properties in synovial sarcoma cells [39], the observation was made in cell lines, which may deviate phenotypically from primary cells endowed with self-renewal and tumour-initiating properties. The putative cell hierarchy in synovial sarcoma will therefore have to be addressed using alternative approaches in primary tumour cells, such as reporter systems based on the expression of stem cell-associated transcription factors and repression of differentiation-inducing factors, including key microRNAs (miRNAs).
SS18 and SSX proteins
Although a fusion protein typically displays unique features, including oncogenic properties, which its wt component proteins may lack, the physiological properties of each of its constituent proteins should at least partially explain its functions. The SS18-SSX fusion protein is a case in point.
SS18 and its implication in the BAF complex
SS18 encodes a 387 amino acid protein that is ubiquitously expressed in normal tissues from embryogenesis [40] to adulthood [24] and whose deletion causes early embryonic lethality. Although its physiological function remains to be fully elucidated, SS18 is implicated in chromatin modifications and in the regulation of gene expression. The wild type SS18 protein can be subdivided into three functional regions: an N-terminal 54 amino acid SYT N-terminal homology domain (SNH domain); a domain containing the nuclear localisation sequence; and the carboxy terminal (C-terminal) transcriptional activation region known as the QPGY domain (fig. 2). SS18 is observed in the cytoplasm but also forms unique nuclear bodies that appear as distinct nuclear speckles [41]. SS18 does not contain a DNA-binding domain and appears to regulate transcription by associating with proteins that form part of the SWI/SNF (SWItch/sucrose non-fermenting) or BAF (Brg/Brm-associated factor) chromatin remodelling complex, including BRG1 (Brahma-related gene 1 or SMARCA4) and BRM (human Brahma, or SMARCA2) [42–44].
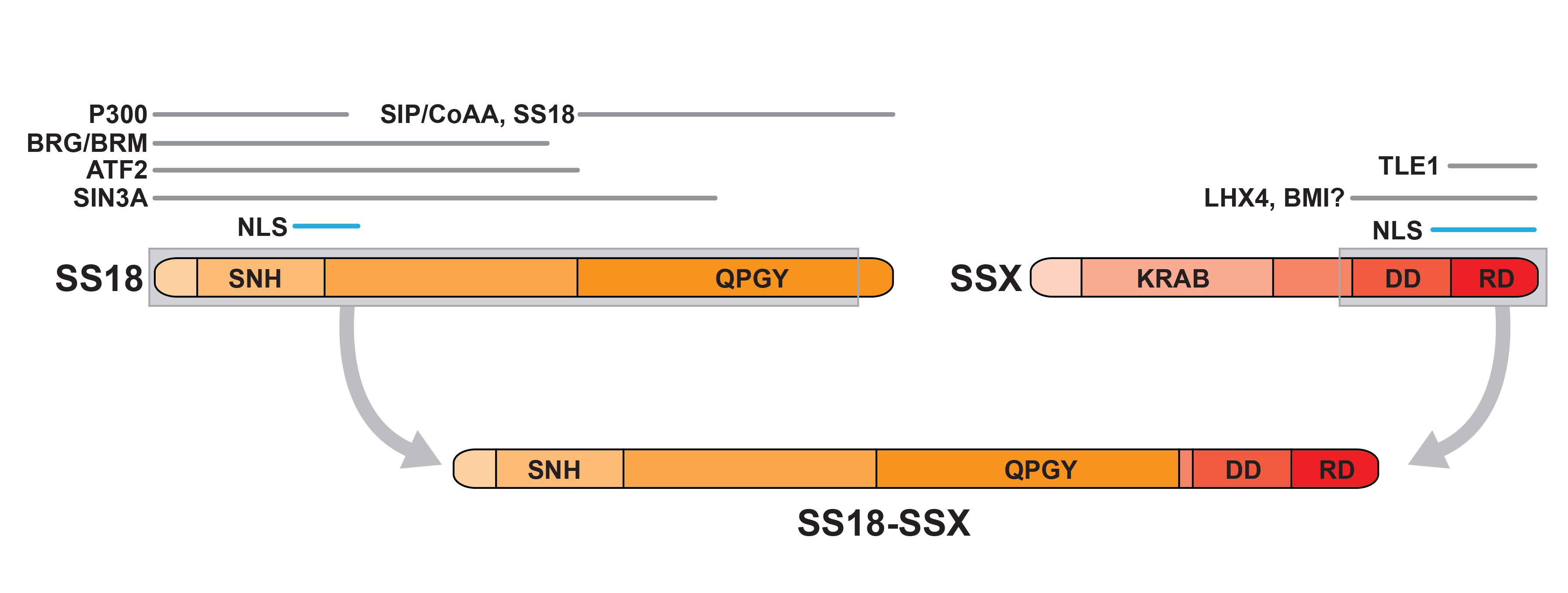
Figure 2
The SS18-SSX fusion protein. Illustration of the domain structure of SS18 and SSX and the regions within each that support interaction with the indicated proteins. The grey encasing indicates the segments of each partner that form part of the fusion protein (below). The indicated fusion partner domains are: SNH, SS18 N-terminal homology; QPGY, glutamine-proline-glycine-tyrosine-rich domain; KRAB, Kruppel-associated box domain; DD, divergent domain; RD, repression domain; NLS, nuclear localisation signal.
Chromatin is the macromolecular complex of DNA and histone proteins in which the heritable material of eukaryotic cells is stored. Its basic functional unit is the nucleosome, which contains 147 bases of DNA wrapped around a single octamer of histone proteins [45, 46]. Whereas the stability of the nucleosomal structure safeguards the genome, temporal and spatial DNA accessibility requires dynamic changes to the nucleosome architecture. Such changes are orchestrated by a variety of chromatin remodelling complexes, which modify histones to induce nucleosomal alterations [45]. Chromatin remodelling complexes are composed of multiple protein subunits at the core of which is an ATPase, which hydrolyses ATP to alter nucleosomal structures. The additional subunits fulfill specialised roles required for the function of the complex. The remodelling activity regulates binding of transcription factors to functional DNA elements, including promoters and enhancers. Chromatin relaxation, induced by activating histone modifications often referred to as “marks”, augments DNA accessibility to transcription factors, facilitating gene expression. Conversely, chromatin compaction triggered by repressive histone marks, renders DNA inaccessible to transcription factors and results in gene silencing [45]. BAF complexes mobilise nucleosomes on genomic DNA [47] to create nucleosome-depleted regions at core promoters distal regulatory elements and render DNA accessible to transcription factors. BAF activity drives changes in chromatin states across the genome and is associated with the activation of a broad range of transcription programmes, including stemness maintenance and differentiation [48, 49]. The mammalian BAF chromatin remodelling complex comprises 15 protein subunits and interacts with a range of nuclear proteins that help target the complex to specific regulatory elements where they can further recruit factors that regulate gene expression. SS18 has been found to be an integral component of the BAF complex [44] and to interact with several of its subunits [50].
SS18 binds to hBRM and BRG1, two mutually exclusive catalytic ATPase subunits of BAF [51] at a conserved region known as the SNF11-binding domain [43, 52] and associates with hSNF5/BAF47 [53]. SS18 interacts with mSin3A (mammalian switch-independent 3A) [54], a core component of the histone deacetylase (HDAC) complex [55], which is associated with BAF [56] and affects chromatin through co-repressors and methyl-CpG-binding proteins resulting in repression of gene transcription [57]. A region within the N-terminal 250 amino acids of SS18 recognises the histone acetyltransferase p300, whose activity relaxes chromatin, thereby acting as a transcriptional coactivator [58, 59]. One effect of the SS18/p300 complex is to activate β1 integrin and promote adhesion of cells that have undergone cell cycle arrest by contact inhibition [60]. All of these interactions are supported by the SNH domain and/or the nuclear localisation signal-containing domain (fig. 2).
The QPGY domain of SS18 contains a 100 amino acid sequence composed of degenerate repeats of glutamine, proline, glycine and tyrosine residues that mediates multimerisation of the SS18 protein [52]. It interacts with SS18-interacting protein / co-activator activator protein (SIP/CoAA) [61], which associates with p300 and several hormone response elements [62].
SSX proteins and gene repression
Nine members compose the SSX gene family and encode 188 amino acid proteins, with the exception of SSX8, which encodes a smaller, 142 amino acid protein [63]. Roughly 40% of each SSX protein is composed of charged amino acids and each SSX protein contains an acidic carboxy terminal tail with consensus motifs for posttranslational modifications, including tyrosine phosphorylation and N-linked glycosylation [64]. SSX contains two principal functional domains: KRAB (Krüppel-associated box) and SSXRD (SSX repression domain) [65]. A third domain of unknown function, located immediately upstream of SSXRD, displays the highest degree of divergence among SSX family members and is referred to as the SSX divergent domain (SSXDD) [66] (fig. 2). Unlike the broad distribution of SS18, SSX in normal adult tissues is confined to the testis and the thyroid, albeit at very low levels in the latter [63, 64]. In contrast, SSX is expressed in divergent tumour types, including breast carcinoma, osteosarcoma, melanoma and multiple myeloma [67–71], where its functional role remains to be elucidated.
The SSX proteins are potent transcriptional repressors. Repression is mediated both by the KRAB domain and by the SSX repressor domain (SSXRD), which is highly conserved among SSX1-5 proteins and is responsible for SSX nuclear localisation. In the nucleus, SSX co-localises with several members of the polycomb group proteins, including human polycomb group protein 2 (HPC2), B cell-specific moloney murine leukaemia virus insertion site 1 protein (BMI1) and ring finger proteins 1 and 2 (RING1 and RING2) [71]. PcG proteins form two multiprotein complexes, polycomb repressive complex 1 and 2 (PRC1 and PRC2), which respectively direct histone HA2 ubiquitination (H2AK119ub1) and trimethylation of histone H3 at lysine 27 (H3K27me3), leading to chromatin compaction that renders regulatory regions inaccessible to RNA polymerase II [72–74]. It appears that intact SSX is required for the recruitment of BMI1, as its amino-terminal domain is necessary for BMI1 interaction [75].
Known interactors of SSX proteins include RAB3IP, a Ras-like GTPase, which participates in vesicular transport and synovial sarcoma X-breakpoint 2 interacting protein (SSX2IP), which is implicated in cell cycle regulation [76]. Both proteins recognise the KRAB domain of SSX but the RAB3IP interaction is restricted to SSX2, whereas SSX2IP binds SSX2 and SSX3, suggesting selectivity among SSX family members for functional partners [76]. The carboxy termini of SSX1, SSX2 and SSX4 interact with LIM homeobox protein 4 (LHX4) which displays transcription factor features [65] and whose expression appears to be deregulated in acute and chronic leukaemias, possibly promoting leukaemia cell survival [77].
The SS18-SSX fusion protein
The SS18-SSX fusion protein is unique to synovial sarcoma and is both its signature feature and the driver of its pathogenesis. The fusion protein retains all of the SS18 protein with the exception of carboxy-terminal eight amino acids, which is fused to the highly polar carboxy-terminal SSX region. Whereas the SSXDD and SSXRD domains are preserved in the fusion protein, the KRAB DNA-binding domain is lost along with its binding partners, allowing novel protein-protein interactions. Thus, the fusion protein interacts with the epithelial-to-mesenchymal transition-inducing transcription factors Snail and Slug, resulting in partial alleviation of E-cadherin repression [78]. Retention of the carboxy-termini of SSX1, SSX2 and SSX4 in the fusion proteins ensures their interaction in vivo with proteins recognised by their wt counterparts [77].
Chromatin remodelling associated with SS18-SSX
Similar to wt SS18, SS18-SSX co-precipitates and co-sediments with hBRM and BRG1 of the BAF chromatin remodelling complex and HDAC [31, 43, 79]. SS18-SSX has been suggested to replace wt SS18 in the BAF complex and alter its composition by ejecting the tumour suppressor hSNF5/BAF47 [53] (Figure 3). Interestingly, wt SS18 can in turn replace the SS18-SSX fusion protein, suggesting that the two proteins may compete for incorporation into BAF complexes. SS18-SSX also retains some of the PcG binding properties of SSX and localises at discrete nuclear foci within BMI-labelled polycomb bodies [80]. The SS18-SSX fusion protein therefore employs two distinct domains to associate with chromatin remodelling complexes that display opposing functions.
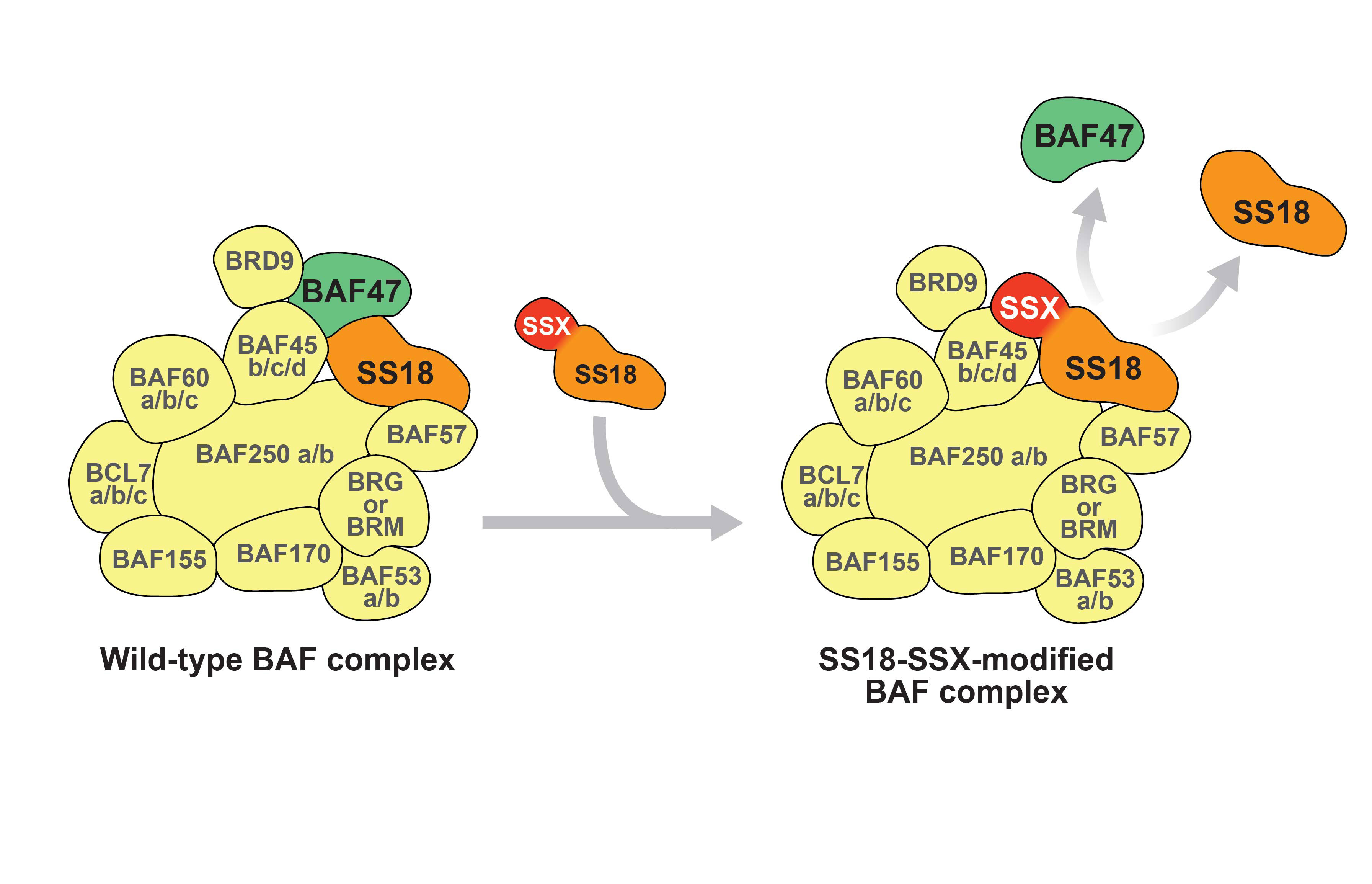
Figure 3
Relationship between SS18, SS18-SSX and the BAF complex. SS18 is an integral part of the wt BAF complex (left). The SS18-SSX fusion protein displaces wt SS18 and BAF47 within the complex (right).
In normal cells, the PRC2 methyltransferase enhancer of zeste homologue 2 (EZH2) induces trimethylation of histone H3, leading to chromatin compaction [72, 74]. Chromatin immunoprecipitation sequencing (ChIP-seq) results from tagged SS18-SSX expressed in C2C12 mouse myoblasts uncovered a relationship between SS18-SSX binding and repressive trimethylated histone H3K27 nucleosome marks [81]. SS18-SSX2 was reported to occupy H3K27me3 labelled regions within 70% of positively regulated and 40% of negatively regulated genes in these cells. The association of SS18-SSX2 with H3K27me3 marks suggests that the fusion protein does not randomly target to regions of open chromatin but rather occupies a subset of PcG loci [81]. It has also been suggested that H3K27me3 motifs represent one of the dominant epigenetic markers associated with SS18-SSX binding and gene repression [82] and that elevated EZH2 expression is observed in poorly differentiated synovial sarcomas where it correlates clinically with a worse outcome [83].
However, the role of PRC-dependent marks in synovial sarcoma pathogenesis remains to be determined in view of the notion that the BAF complex antagonises its PcG counterparts. Several mechanisms have been proposed to underlie the functional opposition of BAF toward PcG. BAF directly recruits RNA PolII, thereby opposing PcG complexes that interact with RNA PolII complexes to promote transcriptional elongation [84, 85]. Loss of hSNF5/BAF47 is reported to cause augmented expression of EZH2 [85], suggesting that the antagonistic action of BAF toward PcG requires that hSNF5/BAF47 counteract EZH2 activity by downregulating its expression. More recent observations suggest that BAF can rapidly evict PRCs in the absence of RNA PolII occupancy, transcription and cell replication [86] and propose that BAF complexes fulfill instructive functions by preparing PcG-repressed loci for accessibility to transcription factors (fig. 4). Thus, the SS18-SSX-modified BAF complex is reported to be recruited to the inactive SOX2 locus where it reverses PcG-mediated repression by removing Polycomb resulting in the activation of the transcription factor SOX2, which is highly expressed in synovial sarcoma and contributes to the proliferation of synovial sarcoma cells [32, 53]. The modified BAF complex activity may therefore promote oncogenesis by a combination of imbalanced PRC activity and aberrant epigenetic activation of PcG targets, which may lead to oncogene activation, tumour suppressor gene repression and induction of stem cell-associated programmes [85].
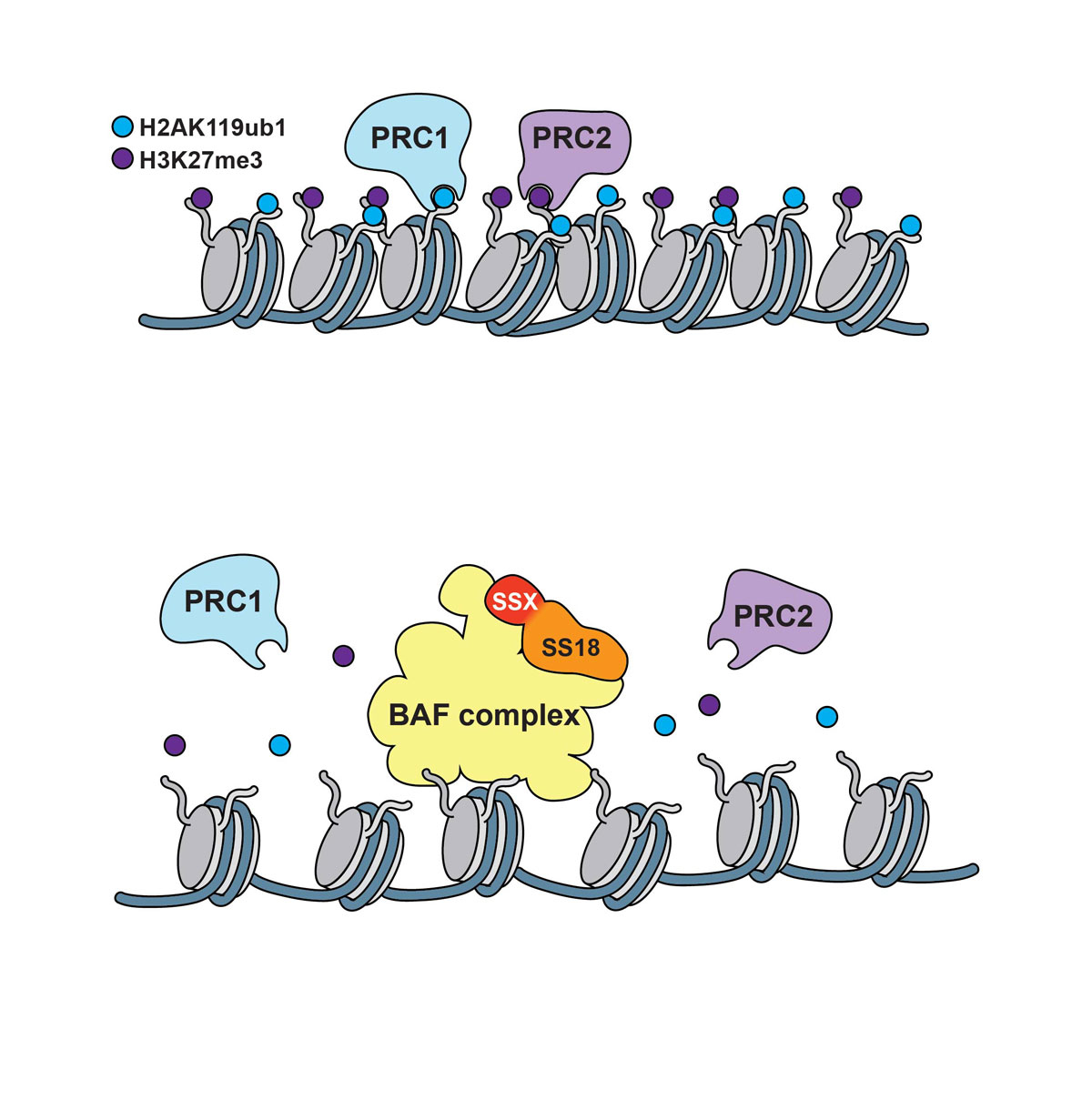
Figure 4 Polycomb repressor complexes induce chromatin compaction (upper panel); the BAF/SS18-SSX complex evicts PRC1/PRC2 causing displacement or decay of their marks and inducing chromatin relaxation (lower panel).
Signalling pathways and transcription factors in synovial sarcoma pathogenesis
At least two possible non-mutually exclusive mechanisms may underlie SS18-SSX/BAF-mediated induction of SOX2 and other target genes. One may be recruitment of the complex to DNA by a pioneer transcription factor that activates regulatory regions, including the creation of de novo enhancers; the other may be relatively nonspecific attachment of the complex to DNA, resulting in broad chromatin relaxation and accessibility of regulatory regions to a wide range of transcription factors devoid of pioneer function [86]. Transcription factors that may recruit SS18-SSX to DNA promoter or enhancer regions have largely remained elusive. However, recent work has identified two distinct sets of transcription factors that may fulfill the function of recruiting SS18-SSX to promoter and enhancer regions of candidate target genes. Thus, SS18-SSX has been proposed to serve as a scaffold that bridges activating transcription factor 2 (ATF2) and transducing-like enhancer of split 1 (TLE1). ATF2 binds to the SNH domain of SS18 whereas TLE1 is recruited to SSXRD [87]. SS18-SSX is suggested to be recruited to target promoters of ATF2, where the repressor activity of TLE1 causes ATF2 target gene silencing [87].
Potential recruitment of SS18-SSX to DNA may also be effected by transcription factors of the Wnt pathway, which is suggested to be implicated in synovial sarcoma pathogenesis [88–90]. The Wnt family of secreted proteins fulfills key evolutionarily conserved functions in normal development and adult tissue maintenance [91–93] and its deregulation by alteration of expression or mutation of its key components including β-catenin, Adenomateous Polyposis Coli (APC) and AXIN, is associated with development and progression of diverse cancer types [93–96]. Expression of SS18/SSX in HEK293 cells has been suggested to activate Wnt- β-catenin signalling. Studies on the MYF5-CRE SS18/SSX2 transgenic model of synovial sarcoma found that SS18-SSX2 aberrantly activates Wnt/β-catenin signaling and that genetic deletion of β-catenin blocks tumour formation [97]. They also suggested that SS18-SSX causes nuclear β-catenin accumulation, possibly by inducing autocrine signalling through its aberrant transcriptional effects. In contrast, introduction of SS18-SSX into NIH3T3 cells induced Wnt ligand-independent accumulation of β-catenin in the nucleus [88], suggesting an alternative mechanism of SS18-SSX-dependent deregulation of Wnt. Despite elevated expression of several Wnt target genes, particularly AXIN2, increased nuclear localisation of β-catenin is seen in only 30 to 60% of synovial sarcomas [98–101] and genetic screens [100, 102] revealed low incidence of β-catenin, APC, AXIN1 and AXIN2 mutations [103]. The molecular mechanisms whereby SS18-SSX may alter Wnt signalling and target gene expression thus remain to be fully elucidated.
Recent work has shown that SS18-SSX may interact with and even hijack the Wnt pathway in C3H10T1/2 cells [31]. One of the most highly upregulated genes in synovial sarcoma is AXIN2, which is also one of the most context-independent targets of Wnt. Expression of SS18-SSX in C3H10T1/2 cells induced robust AXIN2 expression consistent with activation of the Wnt pathway. Surprisingly, whereas SS18-SSX was found to interact with Tcf4/Lef, the core Wnt transcription factors, it did not recruit β-catenin to the complex but rather appeared to exclude and replace β-catenin. SS18-SSX also recruited HDAC and TLE to the complex, potentially inactivating both [31]. Tcf4/Lef may constitute important transcription factors that help recruit SS18-SSX to relevant promoters and possibly enhancers. Alternatively, SS18-SSX in association with BAF may help open chromatin and allow recruitment of Tcf/Lef to promoter and enhancers that in its absence may be inaccessible. In either case, however, SS18-SSX interacts with the transcription factors, possibly regulating their activity in the absence of β-catenin.
Epithelial differentiation in synovial sarcoma
An intriguing observation in synovial sarcoma is that the tumour phenotype (monophasic versus biphasic histology) correlates with the underlying gene fusion type [104]. Thus, almost all biphasic synovial sarcomas have been found to express the SS18-SSX1 fusion [104, 105]. Biphasic histology occurs in 30 to 40% of tumours with SS18-SSX1 and only about 3% of synovial sarcomas bearing SS18-SSX2 [2].
Functional differences between SS18-SSX1 and SS18-SSX2 are thought to account for the distinct differentiation profiles among tumours bearing these fusions. E-cadherin expression is observed in a subset of synovial sarcomas and can even be heterogeneous in the same tumour, as can cytokeratin expression. SS18-SSX1 and SS18-SSX2 can interfere selectively with Snail and Slug, respectively, and release repression of E-cadherin expression [78]. SS18-SSX1 interacts with Snail, which is a stronger repressor of E-cadherin than Slug, and dissociates Snail from the E-cadherin promoter, resulting in stronger de-repression of E-cadherin transcription [78]. This process also implicates hyperacetylation of histones H3 and H4 induced by SS18-SSX, dissociating Snail form the E-cadherin promoter.
A potentially interesting model has been proposed according to which the apparent mesenchymal to epithelial transition (MET) in synovial sarcoma may be better viewed as an EMT based on the notion that all synovial sarcoma progenitor cells with the chromosomal translocation t(X;18)(p11.2;q11.2) are theoretically capable of some epithelial differentiation. However, the majority of the cells lose such capability as a result of a variety of signals [106, 107], including extracellular matrix (ECM) remodelling [108].
Future perspectives
Elucidation of synovial sarcoma pathogenesis is confronted by several major obstacles. First, the cell of origin remains unknown. Although it is widely believed that synovial sarcoma arises in mesenchymal stem cells, MSCs are heterogeneous and discrete degrees of differentiation may suffice to render them permissive or prohibitive for SS18-SSX expression and function. Expression of SS18-SSX in human MSCs derived from different healthy donors had markedly different effects in terms of target gene expression despite cell purification and maintenance in culture by identical methods [33]. A mouse MSC, C3H10T1/2, has shown to be permissive for SS18-SSX expression and to upregulate numerous SS18-SSX target genes, particularly Wnt pathway genes, recapitulating, at least in part, the synovial sarcoma gene signature [31]. However, primary human MSCs have yet to show a comparable level of response to SS18-SSX expression and the epithelial differentiation observed in about a third of synovial sarcomas raises the possibility that the cell of origin may display greater plasticity than most MSCs as currently defined. One hope is that single cell RNA sequencing of MSCs from different anatomical sites and age groups may uncover hitherto unsuspected markers for different subsets and facilitate division of bulk MSCs into subpopulations that can be tested separately for their permissiveness and response to SS18-SSX.
The second major obstacle is the lack of SS18-SSX-specific antibodies that can be used to assess SS18-SSX complexes and epigenetic changes by ChIP-seq. Currently, all available antibodies recognise both wt SS18 and the fusion protein. Although tagged SS18-SSX has been used to conduct ChIP-seq experiments, the approach has two major limitations. First, it relies on SS18-SSX overexpression, which may fail to recapitulate naturally occurring fusion protein expression levels. Second, the overexpression is typically conducted in heterologous cells whose response to SS18-SSX may only partially mimic that of synovial sarcoma cells of origin. One solution would be to genomically tag endogenous SS18-SSX in primary synovial sarcoma cells and to subsequently conduct ChIP-seq studies. However, even such an approach may provide only a snapshot of a process that has evolved over time and that may not provide an explanation as to how the image obtained came to be. In other words, the mechanism that govern the antagonism between the SS18-SSX-modified BAF complex and PcG, which is thought to be at the heart of the epigenetic modifications that characterise synovial sarcoma and underlie its pathogenesis, may not be resolved. The ideal approach would be to conditionally induce the translocation in the synovial sarcoma cell of origin so as to be able to follow the early events that most likely play a crucial role in transformation and establishment of synovial sarcoma features.
The mechanisms that ensure SS18-SSX-associated chromatin remodelling complex recruitment to DNA require further investigation. ATF2 and Tcf4/Lef provide conceptually attractive transcription factors to recruit SS18-SSX to relevant regulatory regions but if they indeed do so, it is likely that other transcription factors, some of which display pioneer functions, participate in SS18-SSX recruitment to promoter and enhancer regions. It will then be essential to determine the repertoire of transcription factors that display such capability. Conversely, it is possible that SS18-SSX is not recruited by transcription factors but rather by other mechanisms that may include a combination of chromatin marks. In association with chromatin remodelling complexes, SS18-SSX may then merely open chromatin in a manner that allows diverse transcription factors to activate target genes whose concerted action transforms target cells.
Another intriguing issue is the relative contribution of HDAC and TLE1. Both display repressor functions and our own results suggest that both are inactivated by SS18-SSX. The relative contribution of BAF, HDAC, TLE, PcG and other putative regulators of gene expression needs to be better understood to obtain clear insight into how SS18-SSX transforms target cells along with clues toward designing effective mechanism-based therapeutic options.
References
1
Herzog
CE
. Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol. 2005;27(4):215–8. doi:.https://doi.org/10.1097/01.mph.0000161762.53175.e4
2
Ladanyi
M
,
Antonescu
CR
,
Leung
DH
,
Woodruff
JM
,
Kawai
A
,
Healey
JH
, et al.
Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62(1):135–40.
3
Wang
S
,
Song
R
,
Sun
T
,
Hou
B
,
Hong
G
,
Mallampati
S
, et al.
Survival changes in Patients with Synovial Sarcoma, 1983-2012. J Cancer. 2017;8(10):1759–68. doi:.https://doi.org/10.7150/jca.17349
4
Smith
ME
,
Fisher
C
,
Wilkinson
LS
,
Edwards
JC
. Synovial sarcoma lack synovial differentiation. Histopathology. 1995;26(3):279–81. doi:.https://doi.org/10.1111/j.1365-2559.1995.tb01444.x
5
Falkenstern-Ge
RF
,
Kimmich
M
,
Grabner
A
,
Horn
H
,
Friedel
G
,
Ott
G
, et al.
Primary pulmonary synovial sarcoma: a rare primary pulmonary tumor. Lung. 2014;192(1):211–4. doi:.https://doi.org/10.1007/s00408-013-9521-1
6
Billings
SD
,
Meisner
LF
,
Cummings
OW
,
Tejada
E
. Synovial sarcoma of the upper digestive tract: a report of two cases with demonstration of the X;18 translocation by fluorescence in situ hybridization. Mod Pathol. 2000;13(1):68–76. doi:.https://doi.org/10.1038/modpathol.3880011
7
Hiraga
H
,
Nojima
T
,
Isu
K
,
Yamashiro
K
,
Yamawaki
S
,
Nagashima
K
. Histological and molecular evidence of synovial sarcoma of bone. A case report. J Bone Joint Surg Am. 1999;81(4):558–63. doi:.https://doi.org/10.2106/00004623-199904000-00014
8
Schoolmeester
JK
,
Cheville
JC
,
Folpe
AL
. Synovial sarcoma of the kidney: a clinicopathologic, immunohistochemical, and molecular genetic study of 16 cases. Am J Surg Pathol. 2014;38(1):60–5. doi:.https://doi.org/10.1097/PAS.0b013e31829b2d0d
9
Wang
JG
,
Li
NN
. Primary cardiac synovial sarcoma. Ann Thorac Surg. 2013;95(6):2202–9. doi:.https://doi.org/10.1016/j.athoracsur.2013.01.030
10
Sultan
I
,
Rodriguez-Galindo
C
,
Saab
R
,
Yasir
S
,
Casanova
M
,
Ferrari
A
. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009;115(15):3537–47. doi:.https://doi.org/10.1002/cncr.24424
11
Spillane
AJ
,
A’Hern
R
,
Judson
IR
,
Fisher
C
,
Thomas
JM
. Synovial sarcoma: a clinicopathologic, staging, and prognostic assessment. J Clin Oncol. 2000;18(22):3794–803. doi:.https://doi.org/10.1200/JCO.2000.18.22.3794
12
Siegel
HJ
,
Sessions
W
,
Casillas
MA, Jr
,
Said-Al-Naief
N
,
Lander
PH
,
Lopez-Ben
R
. Synovial sarcoma: clinicopathologic features, treatment, and prognosis. Orthopedics. 2007;30(12):1020–5, quiz 1026–7.
13
Jaganathan
S
,
Goyal
A
,
Gadodia
A
,
Rastogi
S
,
Mittal
R
,
Gamanagatti
S
. Spectrum of synovial pathologies: a pictorial assay. Curr Probl Diagn Radiol. 2012;41(1):30–42. doi:.https://doi.org/10.1067/j.cpradiol.2011.07.002
14
Ray
A
,
Huh
WW
. Current state-of-the-art systemic therapy for pediatric soft tissue sarcomas. Curr Oncol Rep. 2012;14(4):311–9. doi:.https://doi.org/10.1007/s11912-012-0243-y
15
Bakri
A
,
Shinagare
AB
,
Krajewski
KM
,
Howard
SA
,
Jagannathan
JP
,
Hornick
JL
, et al.
Synovial sarcoma: imaging features of common and uncommon primary sites, metastatic patterns, and treatment response. AJR Am J Roentgenol. 2012;199(2):W208–15. doi:.https://doi.org/10.2214/AJR.11.8039
16
Ferrari
A
,
De Salvo
GL
,
Oberlin
O
,
Casanova
M
,
De Paoli
A
,
Rey
A
, et al.
Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer. 2012;48(9):1370–5. doi:.https://doi.org/10.1016/j.ejca.2012.01.013
17
Dantonello
TM
,
Int-Veen
C
,
Harms
D
,
Leuschner
I
,
Schmidt
BF
,
Herbst
M
, et al.
Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol. 2009;27(9):1446–55. doi:.https://doi.org/10.1200/JCO.2007.15.0466
18
Riggi
N
,
Cironi
L
,
Suvà
ML
,
Stamenkovic
I
. Sarcomas: genetics, signalling, and cellular origins. Part 1: The fellowship of TET. J Pathol. 2007;213(1):4–20. doi:.https://doi.org/10.1002/path.2209
19
Folpe
AL
,
Schmidt
RA
,
Chapman
D
,
Gown
AM
. Poorly differentiated synovial sarcoma: immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol. 1998;22(6):673–82. doi:.https://doi.org/10.1097/00000478-199806000-00004
20
Sato
O
,
Wada
T
,
Kawai
A
,
Yamaguchi
U
,
Makimoto
A
,
Kokai
Y
, et al.
Expression of epidermal growth factor receptor, ERBB2 and KIT in adult soft tissue sarcomas: a clinicopathologic study of 281 cases. Cancer. 2005;103(9):1881–90. doi:.https://doi.org/10.1002/cncr.20986
21
Fleuren
EDG
,
Vlenterie
M
,
van der Graaf
WTA
,
Hillebrandt-Roeffen
MHS
,
Blackburn
J
,
Ma
X
, et al.
Phosphoproteomic Profiling Reveals ALK and MET as Novel Actionable Targets across Synovial Sarcoma Subtypes. Cancer Res. 2017;77(16):4279–92. doi:.https://doi.org/10.1158/0008-5472.CAN-16-2550
22
Yamada
S
,
Imura
Y
,
Nakai
T
,
Nakai
S
,
Yasuda
N
,
Kaneko
K
, et al.
Therapeutic potential of TAS-115 via c-MET and PDGFRα signal inhibition for synovial sarcoma. BMC Cancer. 2017;17(1):334. doi:.https://doi.org/10.1186/s12885-017-3324-3
23
Imura
Y
,
Nakai
T
,
Yamada
S
,
Outani
H
,
Takenaka
S
,
Hamada
K
, et al.
Functional and therapeutic relevance of hepatocyte growth factor/c-MET signaling in synovial sarcoma. Cancer Sci. 2016;107(12):1867–76. doi:.https://doi.org/10.1111/cas.13092
24
Clark
J
,
Rocques
PJ
,
Crew
AJ
,
Gill
S
,
Shipley
J
,
Chan
AM
, et al.
Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7(4):502–8. doi:.https://doi.org/10.1038/ng0894-502
25
Skytting
B
,
Nilsson
G
,
Brodin
B
,
Xie
Y
,
Lundeberg
J
,
Uhlén
M
, et al.
A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst. 1999;91(11):974–5. doi:.https://doi.org/10.1093/jnci/91.11.974
26
Panagopoulos
I
,
Mertens
F
,
Isaksson
M
,
Limon
J
,
Gustafson
P
,
Skytting
B
, et al.
Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer. 2001;31(4):362–72. doi:.https://doi.org/10.1002/gcc.1155
27
Joseph
CG
,
Hwang
H
,
Jiao
Y
,
Wood
LD
,
Kinde
I
,
Wu
J
, et al.
Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosomes Cancer. 2014;53(1):15–24. doi:.https://doi.org/10.1002/gcc.22114
28
Haldar
M
,
Hancock
JD
,
Coffin
CM
,
Lessnick
SL
,
Capecchi
MR
. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell. 2007;11(4):375–88. doi:.https://doi.org/10.1016/j.ccr.2007.01.016
29
Carmody Soni
EE
,
Schlottman
S
,
Erkizan
HV
,
Uren
A
,
Toretsky
JA
. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin Orthop Relat Res. 2014;472(3):874–82. doi:.https://doi.org/10.1007/s11999-013-3065-9
30
Haldar
M
,
Hedberg
ML
,
Hockin
MF
,
Capecchi
MR
. A CreER-based random induction strategy for modeling translocation-associated sarcomas in mice. Cancer Res. 2009;69(8):3657–64. doi:.https://doi.org/10.1158/0008-5472.CAN-08-4127
31
Cironi
L
,
Petricevic
T
,
Fernandes Vieira
V
,
Provero
P
,
Fusco
C
,
Cornaz
S
, et al.
The fusion protein SS18-SSX1 employs core Wnt pathway transcription factors to induce a partial Wnt signature in synovial sarcoma. Sci Rep. 2016;6(1):22113. doi:.https://doi.org/10.1038/srep22113
32
Naka
N
,
Takenaka
S
,
Araki
N
,
Miwa
T
,
Hashimoto
N
,
Yoshioka
K
, et al.
Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28(7):1119–31.
33
Cironi
L
,
Provero
P
,
Riggi
N
,
Janiszewska
M
,
Suva
D
,
Suva
ML
, et al.
Epigenetic features of human mesenchymal stem cells determine their permissiveness for induction of relevant transcriptional changes by SYT-SSX1. PLoS One. 2009;4(11):e7904. doi:.https://doi.org/10.1371/journal.pone.0007904
34
Dominici
M
,
Le Blanc
K
,
Mueller
I
,
Slaper-Cortenbach
I
,
Marini
F
,
Krause
D
, et al.
Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7. doi:.https://doi.org/10.1080/14653240600855905
35
Riggi
N
,
Cironi
L
,
Provero
P
,
Suvà
ML
,
Kaloulis
K
,
Garcia-Echeverria
C
, et al.
Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005;65(24):11459–68. doi:.https://doi.org/10.1158/0008-5472.CAN-05-1696
36
Riggi
N
,
Cironi
L
,
Provero
P
,
Suvà
ML
,
Stehle
JC
,
Baumer
K
, et al.
Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res. 2006;66(14):7016–23. doi:.https://doi.org/10.1158/0008-5472.CAN-05-3979
37
Galland
S
,
Vuille
J
,
Martin
P
,
Letovanec
I
,
Caignard
A
,
Fregni
G
, et al.
Tumor-Derived Mesenchymal Stem Cells Use Distinct Mechanisms to Block the Activity of Natural Killer Cell Subsets. Cell Reports. 2017;20(12):2891–905. doi:.https://doi.org/10.1016/j.celrep.2017.08.089
38
Liu
A
,
Feng
B
,
Gu
W
,
Cheng
X
,
Tong
T
,
Zhang
H
, et al.
The CD133+ subpopulation of the SW982 human synovial sarcoma cell line exhibits cancer stem-like characteristics. Int J Oncol. 2013;42(4):1399–407. doi:.https://doi.org/10.3892/ijo.2013.1826
39
Kimura
T
,
Wang
L
,
Tabu
K
,
Tsuda
M
,
Tanino
M
,
Maekawa
A
, et al.
Identification and analysis of CXCR4-positive synovial sarcoma-initiating cells. Oncogene. 2016;35(30):3932–43. doi:.https://doi.org/10.1038/onc.2015.461
40
de Bruijn
DR
,
Baats
E
,
Zechner
U
,
de Leeuw
B
,
Balemans
M
,
Olde Weghuis
D
, et al.
Isolation and characterization of the mouse homolog of SYT, a gene implicated in the development of human synovial sarcomas. Oncogene. 1996;13(3):643–8.
41
Brett
D
,
Whitehouse
S
,
Antonson
P
,
Shipley
J
,
Cooper
C
,
Goodwin
G
. The SYT protein involved in the t(X;18) synovial sarcoma translocation is a transcriptional activator localised in nuclear bodies. Hum Mol Genet. 1997;6(9):1559–64. doi:.https://doi.org/10.1093/hmg/6.9.1559
42
Middeljans
E
,
Wan
X
,
Jansen
PW
,
Sharma
V
,
Stunnenberg
HG
,
Logie
C
. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS One. 2012;7(3):e33834. doi:.https://doi.org/10.1371/journal.pone.0033834
43
Thaete
C
,
Brett
D
,
Monaghan
P
,
Whitehouse
S
,
Rennie
G
,
Rayner
E
, et al.
Functional domains of the SYT and SYT-SSX synovial sarcoma translocation proteins and co-localization with the SNF protein BRM in the nucleus. Hum Mol Genet. 1999;8(4):585–91. doi:.https://doi.org/10.1093/hmg/8.4.585
44
Kato
H
,
Tjernberg
A
,
Zhang
W
,
Krutchinsky
AN
,
An
W
,
Takeuchi
T
, et al.
SYT associates with human SNF/SWI complexes and the C-terminal region of its fusion partner SSX1 targets histones. J Biol Chem. 2002;277(7):5498–505. doi:.https://doi.org/10.1074/jbc.M108702200
45
Suvà
ML
,
Riggi
N
,
Bernstein
BE
. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–70. doi:.https://doi.org/10.1126/science.1230184
46
Dawson
MA
,
Kouzarides
T
. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi:.https://doi.org/10.1016/j.cell.2012.06.013
47
Mueller-Planitz
F
,
Klinker
H
,
Becker
PB
. Nucleosome sliding mechanisms: new twists in a looped history. Nat Struct Mol Biol. 2013;20(9):1026–32. doi:.https://doi.org/10.1038/nsmb.2648
48
St. Pierre
R
,
Kadoch
C
. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. 2017;42:56–67. doi:.https://doi.org/10.1016/j.gde.2017.02.004
49
Masliah-Planchon
J
,
Bièche
I
,
Guinebretière
JM
,
Bourdeaut
F
,
Delattre
O
. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10(1):145–71. doi:.https://doi.org/10.1146/annurev-pathol-012414-040445
50
Zöllner
SK
,
Rössig
C
,
Toretsky
JA
. Synovial sarcoma is a gateway to the role of chromatin remodeling in cancer. Cancer Metastasis Rev. 2015;34(3):417–28. doi:.https://doi.org/10.1007/s10555-015-9575-z
51
Wang
W
,
Xue
Y
,
Zhou
S
,
Kuo
A
,
Cairns
BR
,
Crabtree
GR
. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996;10(17):2117–30. doi:.https://doi.org/10.1101/gad.10.17.2117
52
Perani
M
,
Ingram
CJ
,
Cooper
CS
,
Garrett
MD
,
Goodwin
GH
. Conserved SNH domain of the proto-oncoprotein SYT interacts with components of the human chromatin remodelling complexes, while the QPGY repeat domain forms homo-oligomers. Oncogene. 2003;22(50):8156–67. doi:.https://doi.org/10.1038/sj.onc.1207031
53
Kadoch
C
,
Crabtree
GR
. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153(1):71–85. doi:.https://doi.org/10.1016/j.cell.2013.02.036
54
Ito
T
,
Ouchida
M
,
Ito
S
,
Jitsumori
Y
,
Morimoto
Y
,
Ozaki
T
, et al.
SYT, a partner of SYT-SSX oncoprotein in synovial sarcomas, interacts with mSin3A, a component of histone deacetylase complex. Lab Invest. 2004;84(11):1484–90. doi:.https://doi.org/10.1038/labinvest.3700174
55
Dufau
ML
,
Liao
M
,
Zhang
Y
. Participation of signaling pathways in the derepression of luteinizing hormone receptor transcription. Mol Cell Endocrinol. 2010;314(2):221–7. doi:.https://doi.org/10.1016/j.mce.2009.05.005
56
Silverstein
RA
,
Ekwall
K
. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2005;47(1):1–17. doi:.https://doi.org/10.1007/s00294-004-0541-5
57
Nan
X
,
Ng
HH
,
Johnson
CA
,
Laherty
CD
,
Turner
BM
,
Eisenman
RN
, et al.
Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–9. doi:.https://doi.org/10.1038/30764
58
Bannister
AJ
,
Kouzarides
T
. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384(6610):641–3. doi:.https://doi.org/10.1038/384641a0
59
Ogryzko
VV
,
Schiltz
RL
,
Russanova
V
,
Howard
BH
,
Nakatani
Y
. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–9. doi:.https://doi.org/10.1016/S0092-8674(00)82001-2
60
Eid
JE
,
Kung
AL
,
Scully
R
,
Livingston
DM
. p300 interacts with the nuclear proto-oncoprotein SYT as part of the active control of cell adhesion. Cell. 2000;102(6):839–48. doi:.https://doi.org/10.1016/S0092-8674(00)00072-6
61
Perani
M
,
Antonson
P
,
Hamoudi
R
,
Ingram
CJ
,
Cooper
CS
,
Garrett
MD
, et al.
The proto-oncoprotein SYT interacts with SYT-interacting protein/co-activator activator (SIP/CoAA), a human nuclear receptor co-activator with similarity to EWS and TLS/FUS family of proteins. J Biol Chem. 2005;280(52):42863–76. doi:.https://doi.org/10.1074/jbc.M502963200
62
Iwasaki
T
,
Chin
WW
,
Ko
L
. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM). J Biol Chem. 2001;276(36):33375–83. doi:.https://doi.org/10.1074/jbc.M101517200
63
Güre
AO
,
Wei
IJ
,
Old
LJ
,
Chen
YT
. The SSX gene family: characterization of 9 complete genes. Int J Cancer. 2002;101(5):448–53. doi:.https://doi.org/10.1002/ijc.10634
64
Gure
AO
,
Türeci
O
,
Sahin
U
,
Tsang
S
,
Scanlan
MJ
,
Jäger
E
, et al.
SSX: a multigene family with several members transcribed in normal testis and human cancer. Int J Cancer. 1997;72(6):965–71. doi:.https://doi.org/10.1002/(SICI)1097-0215(19970917)72:6<965::AID-IJC8>3.0.CO;2-N
65
de Bruijn
DR
,
van Dijk
AH
,
Willemse
MP
,
van Kessel
AG
. The C terminus of the synovial sarcoma-associated SSX proteins interacts with the LIM homeobox protein LHX4. Oncogene. 2008;27(5):653–62. doi:.https://doi.org/10.1038/sj.onc.1210688
66
dos Santos
NR
,
Torensma
R
,
de Vries
TJ
,
Schreurs
MW
,
de Bruijn
DR
,
Kater-Baats
E
, et al.
Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines. Cancer Res. 2000;60(6):1654–62.
67
Mischo
A
,
Kubuschok
B
,
Ertan
K
,
Preuss
KD
,
Romeike
B
,
Regitz
E
, et al.
Prospective study on the expression of cancer testis genes and antibody responses in 100 consecutive patients with primary breast cancer. Int J Cancer. 2006;118(3):696–703. doi:.https://doi.org/10.1002/ijc.21352
68
Naka
N
,
Araki
N
,
Nakanishi
H
,
Itoh
K
,
Mano
M
,
Ishiguro
S
, et al.
Expression of SSX genes in human osteosarcomas. Int J Cancer. 2002;98(4):640–2. doi:.https://doi.org/10.1002/ijc.10277
69
dos Santos
NR
,
de Bruijn
DR
,
van Kessel
AG
. Molecular mechanisms underlying human synovial sarcoma development. Genes Chromosomes Cancer. 2001;30(1):1–14. doi:.https://doi.org/10.1002/1098-2264(2000)9999:9999<::AID-GCC1056>3.0.CO;2-G
70
Taylor
BJ
,
Reiman
T
,
Pittman
JA
,
Keats
JJ
,
de Bruijn
DR
,
Mant
MJ
, et al.
SSX cancer testis antigens are expressed in most multiple myeloma patients: co-expression of SSX1, 2, 4, and 5 correlates with adverse prognosis and high frequencies of SSX-positive PCs. J Immunother. 2005;28(6):564–75. doi:.https://doi.org/10.1097/01.cji.0000175685.36239.e5
71
dos Santos
NR
,
de Bruijn
DR
,
Kater-Baats
E
,
Otte
AP
,
van Kessel
AG
. Delineation of the protein domains responsible for SYT, SSX, and SYT-SSX nuclear localization. Exp Cell Res. 2000;256(1):192–202. doi:.https://doi.org/10.1006/excr.2000.4813
72
Francis
NJ
,
Kingston
RE
,
Woodcock
CL
. Chromatin compaction by a polycomb group protein complex. Science. 2004;306(5701):1574–7. doi:.https://doi.org/10.1126/science.1100576
73
Sauvageau
M
,
Sauvageau
G
. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7(3):299–313. doi:.https://doi.org/10.1016/j.stem.2010.08.002
74
Margueron
R
,
Reinberg
D
. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9. doi:.https://doi.org/10.1038/nature09784
75
Wang
J
,
Wang
H
,
Hou
W
,
Liu
H
,
Zou
Y
,
Zhang
H
, et al.
Subnuclear distribution of SSX regulates its function. Mol Cell Biochem. 2013;381(1-2):17–29. doi:.https://doi.org/10.1007/s11010-013-1684-9
76
de Bruijn
DR
,
dos Santos
NR
,
Kater-Baats
E
,
Thijssen
J
,
van den Berk
L
,
Stap
J
, et al.
The cancer-related protein SSX2 interacts with the human homologue of a Ras-like GTPase interactor, RAB3IP, and a novel nuclear protein, SSX2IP. Genes Chromosomes Cancer. 2002;34(3):285–98. doi:.https://doi.org/10.1002/gcc.10073
77
Kawamata
N
,
Sakajiri
S
,
Sugimoto
KJ
,
Isobe
Y
,
Kobayashi
H
,
Oshimi
K
. A novel chromosomal translocation t(1;14)(q25;q32) in pre-B acute lymphoblastic leukemia involves the LIM homeodomain protein gene, Lhx4. Oncogene. 2002;21(32):4983–91. doi:.https://doi.org/10.1038/sj.onc.1205628
78
Saito
T
,
Nagai
M
,
Ladanyi
M
. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: a potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res. 2006;66(14):6919–27. doi:.https://doi.org/10.1158/0008-5472.CAN-05-3697
79
Nagai
M
,
Tanaka
S
,
Tsuda
M
,
Endo
S
,
Kato
H
,
Sonobe
H
, et al.
Analysis of transforming activity of human synovial sarcoma-associated chimeric protein SYT-SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha. Proc Natl Acad Sci USA. 2001;98(7):3843–8. doi:.https://doi.org/10.1073/pnas.061036798
80
Soulez
M
,
Saurin
AJ
,
Freemont
PS
,
Knight
JC
. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene. 1999;18(17):2739–46. doi:.https://doi.org/10.1038/sj.onc.1202613
81
Garcia
CB
,
Shaffer
CM
,
Eid
JE
. Genome-wide recruitment to Polycomb-modified chromatin and activity regulation of the synovial sarcoma oncogene SYT-SSX2. BMC Genomics. 2012;13(1):189. doi:.https://doi.org/10.1186/1471-2164-13-189
82
Lubieniecka
JM
,
de Bruijn
DR
,
Su
L
,
van Dijk
AH
,
Subramanian
S
,
van de Rijn
M
, et al.
Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res. 2008;68(11):4303–10. doi:.https://doi.org/10.1158/0008-5472.CAN-08-0092
83
Changchien
YC
,
Tátrai
P
,
Papp
G
,
Sápi
J
,
Fónyad
L
,
Szendrői
M
, et al.
Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J Transl Med. 2012;10(1):216. doi:.https://doi.org/10.1186/1479-5876-10-216
84
Mousavi
K
,
Zare
H
,
Wang
AH
,
Sartorelli
V
. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol Cell. 2012;45(2):255–62. doi:.https://doi.org/10.1016/j.molcel.2011.11.019
85
Wilson
BG
,
Wang
X
,
Shen
X
,
McKenna
ES
,
Lemieux
ME
,
Cho
YJ
, et al.
Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–28. doi:.https://doi.org/10.1016/j.ccr.2010.09.006
86
Kadoch
C
,
Williams
RT
,
Calarco
JP
,
Miller
EL
,
Weber
CM
,
Braun
SM
, et al.
Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2017;49(2):213–22. doi:.https://doi.org/10.1038/ng.3734
87
Su
L
,
Sampaio
AV
,
Jones
KB
,
Pacheco
M
,
Goytain
A
,
Lin
S
, et al.
Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell. 2012;21(3):333–47. doi:.https://doi.org/10.1016/j.ccr.2012.01.010
88
Pretto
D
,
Barco
R
,
Rivera
J
,
Neel
N
,
Gustavson
MD
,
Eid
JE
. The synovial sarcoma translocation protein SYT-SSX2 recruits beta-catenin to the nucleus and associates with it in an active complex. Oncogene. 2006;25(26):3661–9. doi:.https://doi.org/10.1038/sj.onc.1209413
89
Fukukawa
C
,
Nagayama
S
,
Tsunoda
T
,
Toguchida
J
,
Nakamura
Y
,
Katagiri
T
. Activation of the non-canonical Dvl-Rac1-JNK pathway by Frizzled homologue 10 in human synovial sarcoma. Oncogene. 2009;28(8):1110–20. doi:.https://doi.org/10.1038/onc.2008.467
90
Trautmann
M
,
Sievers
E
,
Aretz
S
,
Kindler
D
,
Michels
S
,
Friedrichs
N
, et al.
SS18-SSX fusion protein-induced Wnt/β-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene. 2014;33(42):5006–16. doi:.https://doi.org/10.1038/onc.2013.443
91
Cadigan
KM
,
Peifer
M
. Wnt signaling from development to disease: insights from model systems. Cold Spring Harb Perspect Biol. 2009;1(2):a002881. doi:.https://doi.org/10.1101/cshperspect.a002881
92
MacDonald
BT
,
Tamai
K
,
He
X
. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:.https://doi.org/10.1016/j.devcel.2009.06.016
93
Clevers
H
,
Nusse
R
. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–205. doi:.https://doi.org/10.1016/j.cell.2012.05.012
94
Logan
CY
,
Nusse
R
. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20(1):781–810. doi:.https://doi.org/10.1146/annurev.cellbio.20.010403.113126
95
Polakis
P
. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–51.
96
Reya
T
,
Clevers
H
. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–50. doi:.https://doi.org/10.1038/nature03319
97
Barham
W
,
Frump
AL
,
Sherrill
TP
,
Garcia
CB
,
Saito-Diaz
K
,
VanSaun
MN
, et al.
Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013;3(11):1286–301. doi:.https://doi.org/10.1158/2159-8290.CD-13-0138
98
Saito
T
,
Oda
Y
,
Sakamoto
A
,
Tamiya
S
,
Kinukawa
N
,
Hayashi
K
, et al.
Prognostic value of the preserved expression of the E-cadherin and catenin families of adhesion molecules and of beta-catenin mutations in synovial sarcoma. J Pathol. 2000;192(3):342–50. doi:.https://doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH705>3.0.CO;2-R
99
Ng
TL
,
Gown
AM
,
Barry
TS
,
Cheang
MC
,
Chan
AK
,
Turbin
DA
, et al.
Nuclear beta-catenin in mesenchymal tumors. Mod Pathol. 2005;18(1):68–74. doi:.https://doi.org/10.1038/modpathol.3800272
100
Saito
T
,
Oda
Y
,
Sakamoto
A
,
Kawaguchi
K
,
Tanaka
K
,
Matsuda
S
, et al.
APC mutations in synovial sarcoma. J Pathol. 2002;196(4):445–9. doi:.https://doi.org/10.1002/path.1066
101
Horvai
AE
,
Kramer
MJ
,
O’Donnell
R
. Beta-catenin nuclear expression correlates with cyclin D1 expression in primary and metastatic synovial sarcoma: a tissue microarray study. Arch Pathol Lab Med. 2006;130(6):792–8.
102
Saito
T
,
Oda
Y
,
Yamamoto
H
,
Kawaguchi
K
,
Tanaka
K
,
Matsuda
S
, et al.
Nuclear beta-catenin correlates with cyclin D1 expression in spindle and pleomorphic sarcomas but not in synovial sarcoma. Hum Pathol. 2006;37(6):689–97. doi:.https://doi.org/10.1016/j.humpath.2006.01.017
103
Sato
H
,
Hasegawa
T
,
Kanai
Y
,
Tsutsumi
Y
,
Osamura
Y
,
Abe
Y
, et al.
Expression of cadherins and their undercoat proteins (alpha-, beta-, and gamma-catenins and p120) and accumulation of beta-catenin with no gene mutations in synovial sarcoma. Virchows Arch. 2001;438(1):23–30. doi:.https://doi.org/10.1007/s004280000318
104
Antonescu
CR
,
Kawai
A
,
Leung
DH
,
Lonardo
F
,
Woodruff
JM
,
Healey
JH
, et al.
Strong association of SYT-SSX fusion type and morphologic epithelial differentiation in synovial sarcoma. Diagn Mol Pathol. 2000;9(1):1–8. doi:.https://doi.org/10.1097/00019606-200003000-00001
105
Kawai
A
,
Woodruff
J
,
Healey
JH
,
Brennan
MF
,
Antonescu
CR
,
Ladanyi
M
. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338(3):153–60. doi:.https://doi.org/10.1056/NEJM199801153380303
106
Saito
T
,
Oda
Y
,
Kawaguchi
K
,
Sugimachi
K
,
Yamamoto
H
,
Tateishi
N
, et al.
E-cadherin mutation and Snail overexpression as alternative mechanisms of E-cadherin inactivation in synovial sarcoma. Oncogene. 2004;23(53):8629–38. doi:.https://doi.org/10.1038/sj.onc.1207960
107
Saito
T
,
Oda
Y
,
Sugimachi
K
,
Kawaguchi
K
,
Tamiya
S
,
Tanaka
K
, et al.
E-cadherin gene mutations frequently occur in synovial sarcoma as a determinant of histological features. Am J Pathol. 2001;159(6):2117–24. doi:.https://doi.org/10.1016/S0002-9440(10)63063-5
108
Saito
T
. The SYT-SSX fusion protein and histological epithelial differentiation in synovial sarcoma: relationship with extracellular matrix remodeling. Int J Clin Exp Pathol. 2013;6(11):2272–9.