
Figure 1 Study recruitment.
DOI: https://doi.org/10.4414/smw.2018.14682
Endogenous hyperinsulinaemic hypoglycaemia (EHH) is defined as inappropriate endogenous insulin secretion leading to hypoglycaemia and associated symptoms. Biochemically, EHH is characterised by low glucose concentrations in the presence of inappropriately increased C-peptide and insulin levels. Importantly, the administration of exogenous insulin secretagogues has to be excluded (negative sulfonylurea screening). Causes of EHH in adults include insulinoma – sporadic benign or in association with multiple endocrine neoplasia type 1 (MEN-1) – and adult nesidioblastosis. Insulinomas are well-differentiated, generally benign, neuroendocrine tumours derived from the pancreatic beta cells, whereas nesidioblastosis is characterised by beta-cell hypertrophy and hyperplasia, with enlarged and hyperchromatic nuclei and a lobulated islet pattern [1]. The incidence of insulinoma is estimated to be four cases per one million person-years [2], but there are no formal epidemiological studies analysing the incidence of nesidioblastosis. This pathology is considerably rarer [1, 3] outside the setting of gastric bypass.
In order to proceed to biochemical confirmation of EHH by means of a standardised fasting test, Whipple’s triad has to be fulfilled, namely, symptoms consistent with hypoglycaemia, documented low plasma glucose levels and improvement of symptoms after ingestion of carbohydrates [4]. However, hypoglycaemic symptoms vary considerably. This is reflected by a wide range of reported intervals between the first hypoglycaemic symptoms and final diagnosis. Previous studies documented a time interval from initial symptoms until diagnosis ranging from 20 days to 180 months [5, 6]. There is some evidence that neuroglycopenic symptoms in particular may be of clinical diagnostic value [5–7]; however, in the literature, sympathicoadrenal symptoms are also identified as important indicators of EHH [6]. An additional controversial issue is the biochemical cut-offs that confirm, at the end of the fasting test, the diagnosis of EHH [8]. In particular, a glucose level <2.5 mmol/l has been suggested to be a reliable cut-off to end the fasting test. However, Wiesli et al. [7] showed that terminating the fasting test at a glucose level <2.5 mmol/l does not allow confirmation or exclusion of an insulinoma. In contrast, neuroglycopenic symptoms are the prerequisite criterion to end the fasting test before 72 hours.
In summary, the current literature suggests that clinical symptoms, which lead to the investigation and documentation of EHH, are essentially neurological in nature and/or of sympathicoadrenal origin. However, they vary considerably and are ill defined, which may lead to a wide range of disease durations until the diagnosis is formally established. We, therefore, aimed at retrospectively analyse the main clinical symptoms, the time interval between first symptoms and final diagnosis, and the characteristics of the biochemical investigation (duration of fasting test; glucose, C-peptide and insulin levels at the end of the fasting test) in patients with EHH. This analysis was in the framework of two prospective studies investigating a new molecular imaging technique using glucagon-like peptide-1 (GLP-1) receptor imaging.
All endocrinologists with patients with EHH, who had referred patients from European endocrine centres for prospective studies investigating GLP-1 receptor imaging to the University Hospital of Basel between 2008 and 2011 [9] and 2014 and 2017 [10], were asked to report the main symptoms and duration of symptoms that led to the investigation of EHH. In addition, they were to document details of the biochemical assessment of EHH (duration of fasting test; plasma glucose, insulin and C-peptide levels at the end of the fasting test). These data were prospectively collected and retrospectively analysed. Inclusion criteria for both prospective studies [9, 10] were positive Whipple’s triad and biochemically proven EHH (criteria for end of fasting test: glucose level <2.5 mmol/l and neuroglycopenic symptoms) (fig. 1). Exclusion criteria included concomitant medications that influence glucose levels and evidence for a malignant insulinoma on conventional imaging. The local ethics committee approved both studies and participants gave written consent ([9] NCT 00937079, [10] NCT02127541). Following the GLP-1-receptor imaging study, the patients were treated locally and the clinical (absence of hypoglycaemic episodes) and histological outcomes were documented [9, 10]. A total of 84 patients were included in these two prospective studies [9, 10]. For the current analysis, 30 patients were excluded, 8 because of malignant insulinoma and 22 because of insufficient or unreliable documentation of main clinical symptoms. The remaining 54 patients were included in the analysis (15 patients from the first and 39 patients from the second study).
Figure 1 Study recruitment.
The symptoms of all included patients were analysed and categorised into three classes. The classification was based on the Textbook of Clinical Neurology (3rd edition) chapter 38 [11] and slightly adapted according to the reported symptoms. The first category included neurological symptoms and was subdivided in moderately impaired consciousness (confusion, dizziness, somnolence and delirium) visual, speech and sensorimotor impairment, severely impaired consciousness (loss of consciousness and apathy), attention deficit, seizures and personality changes. The second category contained sympathicoadrenal symptoms, namely sweating, tremor, palpitations, hunger, shivering and pallor, and the third category included unspecific other symptoms, such nonspecific asthenia, weight gain, gastrointestinal symptoms and headaches.
This was an observational study. Therefore, mainly descriptive statistics were applied. Data are presented as mean ± standard deviation or median, where applicable. Absolute numbers and percentages are given. The duration of symptoms was compared between patients with and without sympathicoadrenal symptoms using unpaired t-tests.
Fifty-four patients referred from secondary European endocrine centres and endocrinologists, with full documentation of EHH and its main symptoms, from Switzerland, UK, Germany, France, Romania, Spain and Belgium were included in the analysis. The clinical characteristics are summarised in table 1.
Table 1 Clinical characteristics of the patients with endogenous hyperinsulinaemic hypoglycaemia (EHH).
| Number of patients, number (%) | 54 (100%) |
| Age in years, median (range) | 54 (22–84) |
| Gender | |
| Female, number (%) | 40 (74%) |
| Male, number (%) | 14 (26%) |
| Pathological findings in histopathological report | |
| Single benign insulinoma, number (%) | 37 (68.5%) |
| Multiple insulinomas, number (%) | 3 (5.6%) |
| Nesidioblastosis, number (%) | 5 (9.3%) |
| Histological report not available*, number (%) | 9 (16.7%) |
| Confirmed MEN-1, number (%) | 3 (5.6%) |
MEN-1 = multiple endocrine neoplasia * No surgery due to lack of unambiguous localisation (n = 3), no histopathological lesion was found in the surgical specimen despite biochemically proven EHH (n = 2), or patients refused surgery despite clear localisation (n = 4)
The main symptoms are documented in table 2. Briefly, 50 (92.6%) of 54 patients presented with nonspecific neurological symptoms and more than half of these patients suffered from two or more neurological symptoms (table 2).
Table 2 Overview of main symptoms in 54 patients with confirmed endogenous hyperinsulinaemic hypoglycaemia.
| Symptoms | Number | % |
|---|---|---|
| Overall neurological symptoms | 50 | 92.6 |
| Moderately impaired consciousness | 25 | 46.3 |
| Visual, speech and sensorimotor function impairment | 24 | 44.4 |
| Severely impaired consciousness | 20 | 37 |
| Disturbance of attention | 17 | 31.5 |
| Seizures | 9 | 16.7 |
| Personality changes | 7 | 13 |
| Overall sympathicoadrenal symptoms | 33 | 61.1 |
| Sweating | 27 | 50 |
| Tremor | 11 | 20.4 |
| Palpitations | 7 | 13 |
| Hunger | 5 | 9.3 |
| Shivering | 2 | 3.7 |
| Various symptoms | 36 | 66.7 |
| Asthenia | 30 | 61.1 |
| Weight gain | 21 | 38.9 |
| Gastrointestinal symptoms | 7 | 13 |
| Headaches | 4 | 7.4 |
Main sympathicoadrenal symptoms are also summarised in table 2. They were documented in 33 (61.1%) of all patients. Twenty-nine patients (54%) presented with two or more sympathicoadrenal symptoms.
Nonspecific other symptoms occurred in 36 (66.7%) of 54 patients and are summarised in table 2 as well.
Importantly, 43 (79.6%) patients presented with symptoms of at least two categories (table 3).
Table 3 Combination of symptoms at the time of diagnosis in 54 patients with endogenous hyperinsulinaemic hypoglycaemia.
| Number | % | |
|---|---|---|
| Symptom complex of at least two different categories | 43 | 79.6 |
| Only neurological symptoms | 8 | 14.8 |
| Only sympathicoadrenal symptoms | 2 | 3.7 |
| Only various nonspecific symptoms | 1 | 1.9 |
The duration of the interval between onset of symptoms and diagnosis of EHH, was reliably documented in 35 patients (figs 2A and 2B ), and showed a median of 12 months (interquartile range [IQR] 6–36; range 0–120). More than 50% were diagnosed within the first year after onset of symptoms and in two patients (5.7%) the time interval was more than 5 years. (fig. 2). We analysed the time to diagnosis in patients with and without sympathicoadrenal symptoms. Due to the small sample size of patients with the interval from disease onset to diagnosis reliably documented, this analysis did not result in significantly different interval between first symptoms and final diagnosis (p = 0.9).
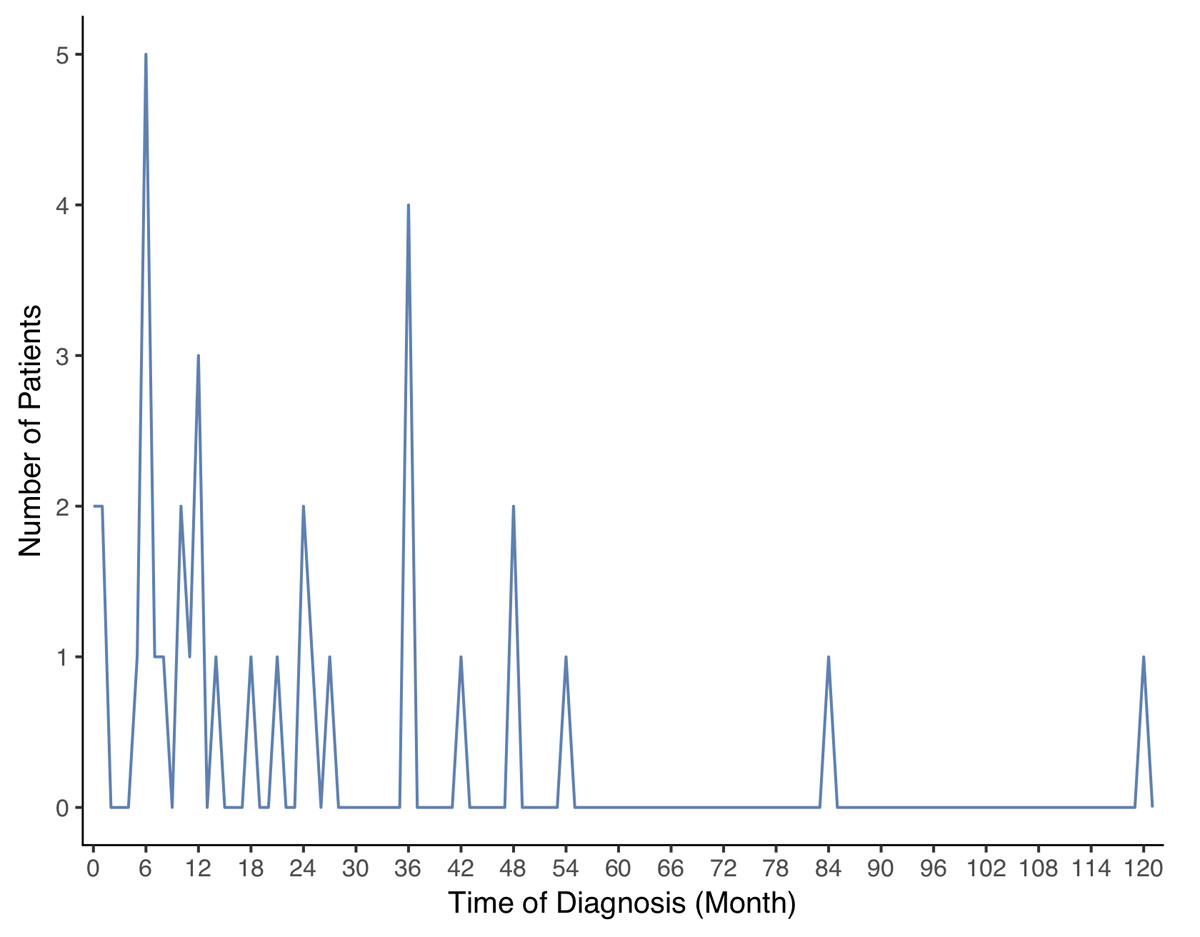
Figure 2A Interval between onset of symptoms and diagnosis: distribution of the time to diagnosis. On the y-axis the number of patients which had been diagnosed at a given time (x-axis).

Figure 2B Interval between onset of symptoms and diagnosis: cumulative share of the patients diagnosed with EHH in the course of time.
Detailed data on the fasting test was available in 49 patients (90.7%). In the other five (9.3%) patients, blood samples were drawn during a symptomatic episode in the emergency room. Fasting tests lasted 23.7 ± 18 hours, mean ± standard deviation SD, (range 2–72 hours). In over 70% of the tests, there were pathological findings within the first 24 hours. After 48 hours, the test was stopped with a diagnostic result in 97% of the patients (figs 3A and 3B ). At the end of the fasting test, plasma glucose, insulin and C-peptide concentrations were 2.0 ± 0.4 mmol/l, 22.5 ± 42.2 mU/l (156.3 ± 293.1 pmol/l) and 985.1 ± 587.7 pmol/l, respectively. Interestingly, although recommended by current guidelines [12], proinsulin and beta-hydroxybutyrate concentrations were measured in only 8 and 12 patients, respectively, at the end of the fasting test. Similarly, the glucagon test was rarely performed.
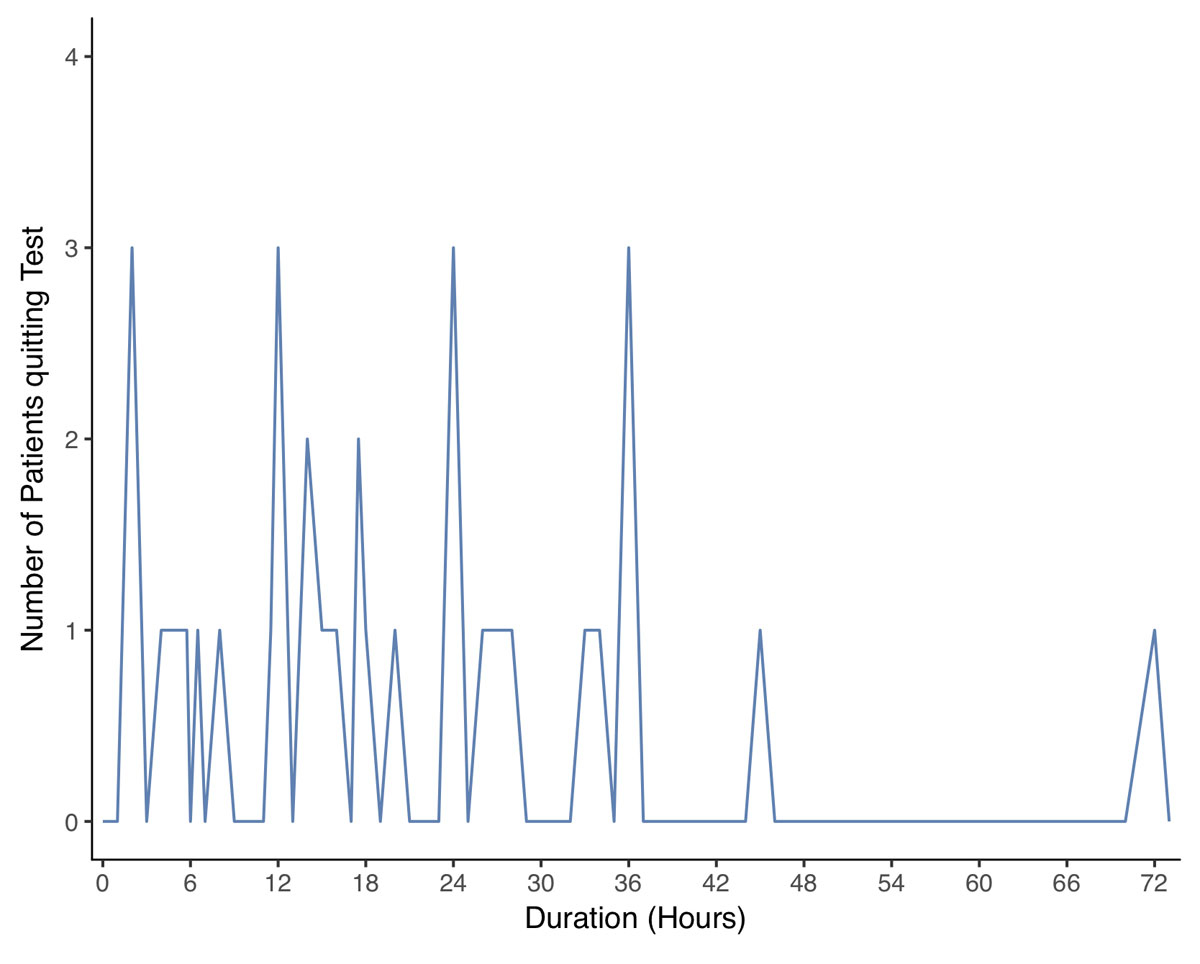
Figure 3A 72-hour fasting test: distribution of the time until the end of fasting. On the y-Axis the number of patients which had been diagnosed at a given time (x-axis).
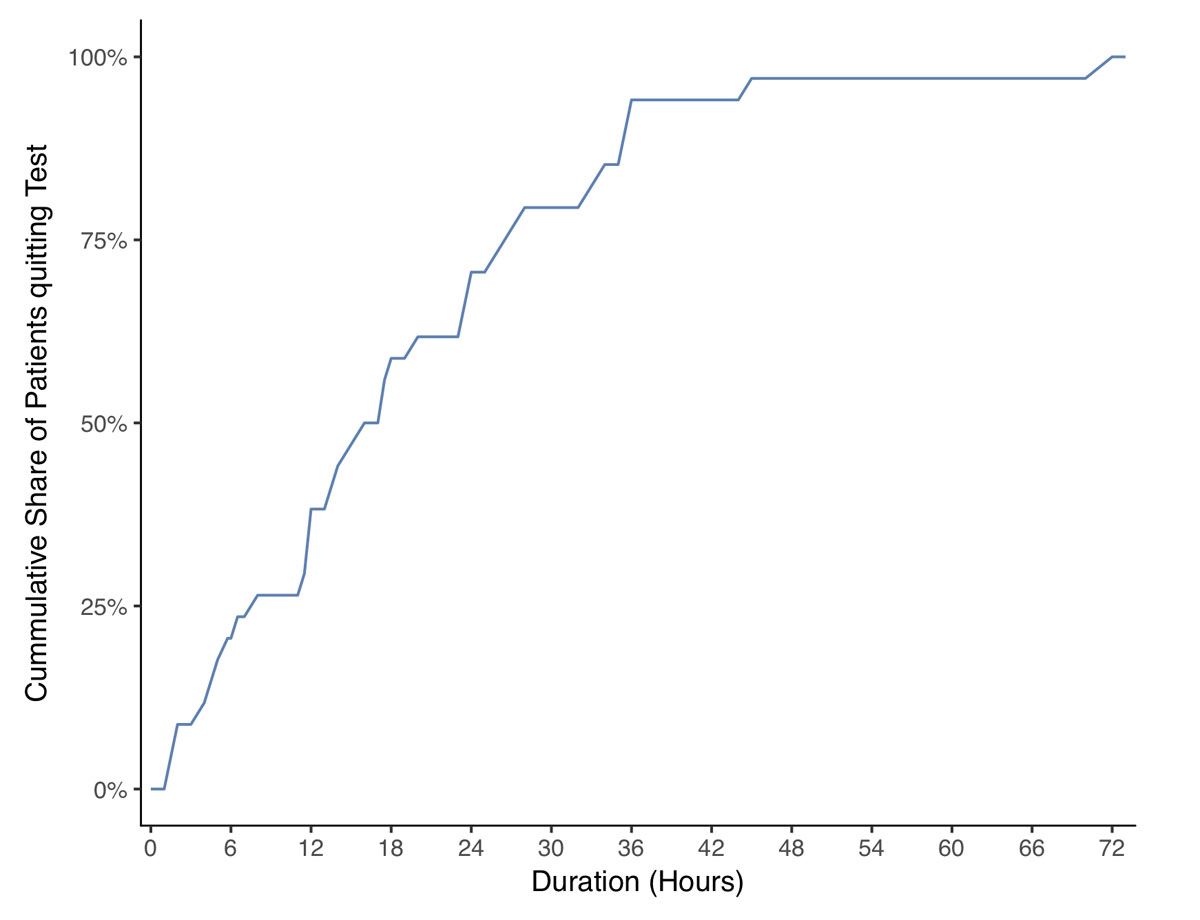
Figure 3B 72-hour fasting test: cumulative share of the patients diagnosed with during the fasting test.
The major findings of this study can be summarised as follows:
The demographic findings in the current study (mean age, female preponderance) are consistent with the literature [2, 5, 6, 13]. In addition, an underlying genetic context – MEN-1 – was diagnosed in 5.6% of the patients in the current study, in keeping with the literature [2, 6]. Based on these observations, the current patient cohort can be considered as representative for EHH.
Over 90% of all patients presented with neurological symptoms, which suggests that EHH is a differential diagnosis in a wide variety of neurological disorders. Importantly, over 40% of the patients presented with severe neurological symptoms (severely impaired consciousness and seizures), either in combination or alone. Although the detailed analysis of the main neurological symptoms of EHH is new, there are data in the literature indicating that symptoms in EHH are frequently neurological in nature [5, 6, 13], consistent with our findings.
Sympathicoadrenal symptoms were reported in >60% of the patients, the most prevalent symptom of this category being sweating. The comprehensive analysis of symptoms in this category in patients with EHH is novel. However, previous data revealed a higher prevalence of sympathicoadrenal symptoms [6]. This is an intriguing finding and the underlying explanation remains to be established. A possible reason may be a more prevalent hypoglycaemia unawareness [14] in our cohort. However, in this case we would expect a longer duration from initial symptoms until diagnosis than reported in the literature. The latter cannot be confirmed by our data.
Nonspecific other symptoms were recorded in about two-thirds of the patients, of which nonspecific asthenia was the most frequently mentioned (61.1%), followed by weight gain (38.9%). Previous reports included only weight gain as a nonspecific finding in EHH. The prevalence of weight gain in the literature corroborate our findings [6].
A remarkable finding in our survey was a rather long interval between disease onset and the time of diagnosis, with a median duration of 12 months (range 0–120), in keeping with previous reports [5, 6]. This is possibly because the clinical symptoms in EHH are nonspecific.
All the patients fulfilled Whipple's triad. In five patients, EHH was documented during a symptomatic episode in the emergency room. The remaining patients underwent a fasting test, as described before [15]. In more than 70% of the patients, the fasting test was diagnostic within the first 24 hours. There was only one patient, who was asymptomatic after 48 hours, but after 72 hours in this case also clear biochemical evidence for EHH was found. A previous report published by Dizon et al. [6] suggests that biochemical documentation of EHH after 24 hours of fasting is found in 84% of cases, slightly higher than in the current study. However, the levels of glucose, insulin and C-peptide at the end of the fasting test are consistent with previous findings [15]. Interestingly, the biochemical work-up differed considerably across the different endocrine centres and endocrinologists. Only measurements of plasma glucose, insulin and C-peptide concentrations and sulfonylurea screening were performed everywhere, whereas proinsulin and beta-hydroxybutyrate levels – as recommended by current guidelines [12] – were inconsistently assessed. Similarly, the glucagon test at the end of the fasting test was rarely performed, indicating that the work-up of EHH does not rely on current guidelines and rather depends on local experience and availability of laboratory measurements.
A strength of the current findings is the quite large cohort, considering the low incidence of EHH. In addition, the patients were prospectively recruited in the context of two prospective imaging studies with previously defined inclusion criteria [9, 10]. However, there are several limitations: (1) the main clinical symptoms were retrospectively analysed and susceptible to bias like all retrospective studies; (2) the fasting tests were performed in the respective referral centres including laboratory analysis and have, therefore, to be interpreted with caution; (3) nonspecific symptoms such as weakness and weight gain are much more frequent in the general population without EHH and related to a broad spectrum of differential diagnosis. The only difference is that these unspecific symptoms occurred in combination with the Whipple’s triad and the biochemical documentation of an EHH. A positive fasting test, however, is a very sensitive and specific tool for detecting EHH and for its differential diagnosis (most frequently insulinoma) [16].
In conclusion, these data indicate that a wide variety of primarily neurological symptoms (“neurological chameleon”) followed by sympathicoadrenal symptoms are characteristic for EHH. Therefore, general practitioners and neurologists should consider EHH as a differential diagnosis in many neurological disorders. In view of the recent improvements of the available systems for continuous glucose monitoring in the context of anti-diabetic therapy, these tools should be generously used to confirm significant hypoglycaemic episodes when EHH is suspected [17, 18].
We thank all the patients who participated in the trial, the referring physicians and the local investigators who contributed to the trial.
The study was supported by the Swiss National Science Foundation (grant number 320030-152938) and the Desirée and Niels Yde’s Foundation (grant number 389-12) which had no role in study design, data collection, analysis, interpretation, or writing of the report.
The authors confirm no conflict of interest with regard to the presented data.
1 Anlauf M , Wieben D , Perren A , Sipos B , Komminoth P , Raffel A , et al. Persistent hyperinsulinemic hypoglycemia in 15 adults with diffuse nesidioblastosis: diagnostic criteria, incidence, and characterization of beta-cell changes. Am J Surg Pathol. 2005;29(4):524–33. doi:.https://doi.org/10.1097/01.pas.0000151617.14598.ae
2 Service FJ , McMahon MM , O’Brien PC , Ballard DJ . Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66(7):711–9. doi:.https://doi.org/10.1016/S0025-6196(12)62083-7
3 Harrison TS , Fajans SS , Floyd JC, Jr , Thompson NW , Rasbach DA , Santen RJ , et al. Prevalence of diffuse pancreatic beta islet cell disease with hyperinsulinism: problems in recognition and management. World J Surg. 1984;8(4):583–7. doi:.https://doi.org/10.1007/BF01654942
4 Whipple AO . The surgical therapy of hyperinsulinism. J Int Chir. 1938;3:237–76.
5 Service FJ , Dale AJ , Elveback LR , Jiang NS . Insulinoma: clinical and diagnostic features of 60 consecutive cases. Mayo Clin Proc. 1976;51(7):417–29.
6 Dizon AM , Kowalyk S , Hoogwerf BJ . Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med. 1999;106(3):307–10. doi:.https://doi.org/10.1016/S0002-9343(99)00021-2
7 Wiesli P , Brändle M , Schwegler B , Lehmann R , Spinas GA , Schmid C . A plasma glucose concentration below 2.5 mmol L-1 is not an appropriate criterion to end the 72-h fast. J Intern Med. 2002;252(6):504–9. doi:.https://doi.org/10.1046/j.1365-2796.2002.01060.x
8 De León DD , Stanley CA . Determination of insulin for the diagnosis of hyperinsulinemic hypoglycemia. Best Pract Res Clin Endocrinol Metab. 2013;27(6):763–9. doi:.https://doi.org/10.1016/j.beem.2013.06.005
9 Christ E , Wild D , Ederer S , Béhé M , Nicolas G , Caplin ME , et al. Glucagon-like peptide-1 receptor imaging for the localisation of insulinomas: a prospective multicentre imaging study. Lancet Diabetes Endocrinol. 2013;1(2):115–22. doi:.https://doi.org/10.1016/S2213-8587(13)70049-4
10 Antwi K , Fani M , Heye T , Nicolas G , Rottenburger C , Kaul F , et al. Comparison of glucagon-like peptide-1 receptor (GLP-1R) PET/CT, SPECT/CT and 3T MRI for the localisation of occult insulinomas: evaluation of diagnostic accuracy in a prospective crossover imaging study. Eur J Nucl Med Mol Imaging. 2018. doi:.https://doi.org/10.1007/s00259-018-4101-5
11Ferrante MA. Endogenous Metabolic Disorders. In: Goetz CG, editor. Textbook of Clinical Neurology (3rd edition). Chicago: Saunders; 2007. pp. 827–864.
12 Cryer PE , Axelrod L , Grossman AB , Heller SR , Montori VM , Seaquist ER , et al.; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94(3):709–28. doi:.https://doi.org/10.1210/jc.2008-1410
13 Stefanini P , Carboni M , Patrassi N , Basoli A . Beta-islet cell tumors of the pancreas: results of a study on 1,067 cases. Surgery. 1974;75(4):597–609.
14 Mitrakou A , Fanelli C , Veneman T , Perriello G , Calderone S , Platanisiotis D , et al. Reversibility of unawareness of hypoglycemia in patients with insulinomas. N Engl J Med. 1993;329(12):834–9. doi:.https://doi.org/10.1056/NEJM199309163291203
15 Service FJ . Hypoglycemic disorders. N Engl J Med. 1995;332(17):1144–52. doi:.https://doi.org/10.1056/NEJM199504273321707
16 Service FJ , Natt N . The prolonged fast. J Clin Endocrinol Metab. 2000;85(11):3973–4. doi:.https://doi.org/10.1210/jcem.85.11.6934
17 Munir A , Choudhary P , Harrison B , Heller S , Newell-Price J . Continuous glucose monitoring in patients with insulinoma. Clin Endocrinol (Oxf). 2008;68(6):912–8. doi:.https://doi.org/10.1111/j.1365-2265.2007.03161.x
18 Kawasaki A , Suzuki K , Miyamoto M , Miyamoto T , Yanagi K , Shimizu M , et al. Disruptive nocturnal behavior due to insulinoma revealed by continuous glucose monitoring. Eur J Neurol. 2014;21(5):e46–7. doi:.https://doi.org/10.1111/ene.12388
The study was supported by the Swiss National Science Foundation (grant number 320030-152938) and the Desirée and Niels Yde’s Foundation (grant number 389-12) which had no role in study design, data collection, analysis, interpretation, or writing of the report.
The authors confirm no conflict of interest with regard to the presented data.