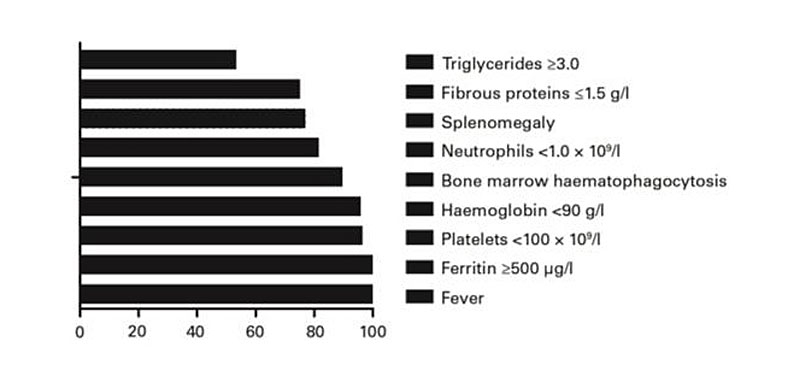
Figure 1 Relative frequency of the clinical and laboratory parameters characteristic of HLH patients.
DOI: https://doi.org/10.4414/smw.2018.14641
Haemophagocytic lymphohistiocytosis (HLH) is a serious life-threatening immune disorder, characterised by excessive activation of lymphocytes and macrophages leading to cytokine storm. The main clinical manifestations include fever, hepatosplenomegaly, pancytopenia, hyperglycaemia, hypofibrinogenaemia, neurological symptoms, and haemophagocytic phenomena in bone marrow, liver, spleen, lymphoid tissue [1–3]. In addition, examination of a bone marrow smear often fails to detect the phenomenon of phagocytosis: multiple bone marrow punctures may be required in order to detect phagocytosis [4, 5].
The main obstacle to the initiation of treatment of HLH is the inability to diagnose it early and the risk of misdiagnosis. In part because of the low incidence and the multiple changes during the course of the disease, diagnosis in the early stages can be difficult; thus, diagnosis requires a certain amount of time [6]. HLH in adults accounts for 40% of cases and, compared with childhood HLH, is more difficult to diagnose, because it is often secondary to cancer, autoimmune diseases and infections [7, 8].
Early diagnosis of secondary HLH is essential to avoid fast fatal outcomes. Both the internationally accepted diagnostic criteria, HLH-2004 defined by the Histiocyte Society [4], and the widely used specific treatment programme [9] for HLH derive from experience with paediatric patients with congenital HLH. Although the aetiology and clinical features of adult HLH are different from those of children [10], the current diagnostic criteria are still widely used in adult patients.
Secondary HLH lacks specificity. Outcomes in adult HLH are heterogeneous; mortality ranges from 20.4 to 88%, depending on the primary disease. The study with the lowest mortality rate assessed only 30-day mortality in order to limit the impact of underlying disease [11]. Most of the previous research data came from the departments of rheumatism or internal medicine [7, 12]. Secondary HLH has many different causes, and the proportion of HLH cases associated with malignant tumours is less than 50%. The highest mortality rate is in patients with underlying lymphoma, which is consistently an adverse prognostic factor [11, 13–18]. Mortality is generally lowest in autoimmune disease, followed by infection-associated and idiopathic HLH. Malignancy, particularly lymphoma, is a prominent adverse prognostic marker, correlating with poorer survival in multiple large studies [11, 13–17]. Thus, from the haematology perspective, secondary HLH is usually considered a rapidly fatal disease, for which active treatment may not be worthwhile. Recently, different diagnostic criteria for adult secondary HLH have been published [19, 20], the latest being HScore [21].
In this study, we report a case series of adult-acquired HLH in our Department of Haematology, the highest proportion of which were lymphoma-associated HLH. We compare use of the HScore diagnostic criteria with the older HLH-04 criteria to determine which are more effective. Clinical characteristics and prognostic factors were also analysed in order to identify potentially curable patient subgroups.
The First Affiliated Hospital of Zhejiang University Medical College Ethics Committee reviewed and approved this retrospective study and waived the request for written informed consent. The protocol was approved by the local ethics committee.
From January 2010 to December 2015, 174 of our central haematology patients were eventually diagnosed with adult secondary HLH (sHLH) according to the 2004 HLH Diagnostic Criteria for Tissue Cytology (HLH-04) diagnostic criteria. Reasons for referring patients included unexplained cytopenia, lymph node or splenic enlargement, fever of unexplained origin and suspected secondary HLH in patients already diagnosed with conditions potentially complicated by HLH.
In accordance with the HLH-04 [4], we extracted diagnostic and related clinical and laboratory data from medical records. For a diagnosis of HLH, the patient needs to meet at least five of the following eight criteria: (i) fever; (ii) splenomegaly; (iii) cytopenia involving at least two of the three lineages in the peripheral blood – haemoglobin <90 g/l, platelets <100×109/l, neutrophils <1.0×109/l; (iv) hypertriglyceridaemia (≥3.0 mM) and/or hypofibrinogenaemia (≤1.5 g/l); (v) haemophagocytosis in bone marrow, spleen, or lymph nodes; (vi) low or absent natural killer (NK) cell activity; (vii) hyperferritinaemia (≥500 µg/l); and (viii) increased soluble CD25 levels (≥2400 U/ml). Because the tests of soluble CD25 levels and NK cell activity were not readily available in our institution, the sHLH patients who fulfilled five or more of the six other criteria were enrolled. None of these patients underwent genetic testing. All patients who met the HLH-04 criteria were assessed with the HScore diagnostic criteria [19].
In order to analyse the factors that may affect the prognosis, the following variables were retrieved and considered: sex, age, underlying haematological disease, severity of anaemia (<9 g/dl), lymphocytopenia (<0.5×109/l), neutropenia (<1×109/l) and thrombocytopenia (<100×109/l), hypertriglyceridaemia (>265 mg/dl), urea nitrogen >20 mg/dl, hypofibrinogenaemia (<150 mg/dl), ferritin >10000 ng/ml, bilirubin >2mg/dl, lactate dehydrogenase (LDH) >2 × the upper limit of normal, hepatosplenomegaly, concurrent viral infection and response to treatment.
Responses to HLH treatment are defined as: reduction of blood cell recovery, no fever, recovery of hyperlipidaemia and hypofibrinogenaemia, splenomegaly returned to normal.
The following treatment regimens were used: glucocorticoid; glucocorticoid + ciclosporin; glucocorticoid + intravenous immunoglobulin; glucocorticoid + chemotherapy with etoposide, vindesine, cyclophosphamide and nordoxorubicin); glucocorticoid + intravenous immunoglobulin + chemotherapy; chemotherapy with etoposide, dexamethasone, vindesine, cyclophosphamide and nordoxorubicin). Antiviral drugs were included in the supportive therapy of patients with viral infection. Blood component transfusions, including erythrocyte suspension, platelets, fresh-frozen plasma and cryoprecipitate, were used.
All statistical analyses were performed using SPSS (Statistical Package for Social Sciences) software, version 17 (SPSS Inc., Chicago, IL, USA). Patients’ characteristics were summarised as standard descriptive statistics. The total lifetime (overall survival) was defined as the time from diagnosis to the last follow-up or death. The Kaplan-Meier method and log-rank test were used for survival analysis. The difference in survival was assessed with the log-rank test in univariate analysis. Univariate analysis of variables in the single factor analysis using Cox proportional hazards regression models for multivariate analysis. A p-value <0.05 was considered statistically significant for all analyses.
We included 174 adult patients with HLH in the analysis. There were 111 males and 63 females; the median age was 51 years (range 17–90). In 92/174 patients (52.9%) HLH was combined with potential blood system diseases (4 acute leukaemia, 1 thrombotic thrombocytopenic purpura, 3 Hodgkin’s lymphoma [HL], 17 B-cell non-Hodgkin’s lymphoma [NHL], 67 T cell NHL including 22 with NK/T-cell NHL); 6 (3.4%) patients had autoimmune diseases (2 haemolytic anaemia, 2 undifferentiated connective tissue disease, 1 systemic lupus erythematosus, 1 Still’s disease); in 76 patients (43.7%) the underlying disease could not be determined. Viral infections (Epstein-Barr virus [EBV] 44, 25.3%; cytomegalovirus [CMV] 11, 6.3%; hepatitis B virus [HBV] 9. 5.2%; human immunodeficiency virus [HIV] 3, 1.7%, diagnosed from the presence of circulating viral genome) or syphilis (1, 0.5%), were present in 57 patients, specifically in 7/17 B-cell NHL cases, in 1/3 HL and in 25/67 T-cell NHL cases, and in 24/76 patients without underlying diseases. Of these, fifteen patients had more than one viral infection.
The clinical data and laboratory data of 174 patients with HLH are shown in figure 1. Nearly all of our patient subjects had hyperferritinaemia, high LDH, fever and low albumin, with bone marrow haemophagocytosis in 89.1% of cases.
Figure 1 Relative frequency of the clinical and laboratory parameters characteristic of HLH patients.
According to the HLH-04 standard diagnostic criteria for HLH, after retrospective application of HScore, one patient showed non- conformity with the HScore. By the HScore, 4/174 patients had a >50% and 16/174 patients had a >90% probability of not having HLH. All 174 patients fulfilled more than five HLH-04 diagnostic criteria, but 16 of them had a low probability of HLH by the HScore (table 1).
Table 1 HLH diagnostic criteria fulfilled according to HLH-04 and to the Hscore.
| n | HLH-04 score | HScore | Probability of HLH according to HScore |
|---|---|---|---|
| 4 | ≥5 | 145 (133–154) | <50% |
| 12 | ≥5 | 189 (173–203) | 50–90% |
| 5 | ≥5 | 207 (204–213) | 90–95% |
| 41 | ≥5 | 229 (218–243) | 95–99% |
| 112 | ≥5 | 277 (245–337) | >99% |
Treatment of the 174 patients included intravenous immunoglobulin, corticosteroids, ciclosporin, the HLH-04 regimen [20], antiviral agents, and/or antilymphoma treatment, alone or in combination. The treatments given, the response of HLH and the blood disease to treatment, and the results are shown in table 2. Ninety-two patients with lymphoma received antitumour therapy.
Table 2 Treatment, responses of HLH and the underlying haematological disease to treatment, and HLH outcome.
| Treatment | Underlying disease | n | Resolution of HLH |
Survival
(mouths) |
|---|---|---|---|---|
| GC | 29 | 14 | ||
| T-cell NHL (except NK/T-cell NHL) | 9 | 1 | 2 (0.5–7) | |
| B-cell NHL | 1 | 0 | 1 | |
| Autoimmune disorder | 2 | 2 | 17.5 (7–28) | |
| No underlying disease was identified | 17 | 11 | 17.4 (0.5–52) | |
| GC + IVIG | 14 | 7 | ||
| T-cell NHL (except NK/T-cell NHL) | 2 | 0 | 0.7 (0.5–1) | |
| Autoimmune disorder | 2 | 2 | 37.5 (33–42) | |
| No underlying disease was identified | 8 | 5 | 21.9 (0.5–47) | |
| AML | 1 | 0 | 1 | |
| TTP | 1 | 0 | 0.5 | |
| GC + CsA | 1 | 0 | 9 | |
| GC + CHT | 67 | 22 | ||
| T-cell NHL (except NK/T-cell NHL) | 27 | 5 | 4.2 (0.5–35) | |
| B-cell NHL | 14 | 11 | 25 (0.5–47) | |
| NK/T-cell NHL | 16 | 4 | 2.6 (0.5–8) | |
| No underlying disease was identified | 8 | 2 | 6.6 (1–34) | |
| AML | 1 | 0 | 5 | |
| GC + IVIG + CHT | 11 | 5 | ||
| T-cell NHL (except NK/T-cell NHL) | 3 | 2 | 23.3 (1–35) | |
| B-cell NHL | 1 | 0 | 1 | |
| NK/T-cell NHL | 3 | 1 | 16.7 (2–46) | |
| Autoimmune disorder | 1 | 1 | 2 | |
| No underlying disease was identified | 3 | 1 | 19.8 (0.5–41) | |
| CHT | 10 | 1 | ||
| T-cell NHL (except NK/T-cell NHL) | 3 | 0 | 0.8 (0.5–1) | |
| B-cell NHL | 1 | 1 | 42 | |
| NK/T-cell NHL | 2 | 0 | 3 (2–4) | |
| HL | 2 | 0 | 3 (1–5) | |
| AML | 2 | 0 | 1.3 (0.5–2) | |
| IVIG | 6 | 1 | ||
| NK/T-cell NHL | 1 | 0 | 1 | |
| No underlying disease was identified | 5 | 1 | 3.3 (0.5–10) |
AML = acute myeloid leukaemia; TTP = thrombotic thrombocytopenic purpura; HL = Hodgkin’s lymphoma; B-cell NHL = B-cell non-Hodgkin’s lymphoma; T-cell NHL = T-cell non-Hodgkin’s lymphoma; NK/T-cell NHL = T/NK-cell non-Hodgkin’s lymphoma; GC = glucocorticoid; IVIG = intravenous immunoglobulin; CsA = ciclosporin; CHT = chemotherapy.
Overall, 50 patients showed a clinical resolution of HLH. HLH secondary to connective tissue disease has the best prognosis; HLH secondary to lymphoma has the worst prognosis, especially with NK/T-cell lymphoma. The best treatment for NK/T-cell lymphoma was a glucocorticoid + intravenous immunoglobulin + chemotherapy.
Seventy-two of the 174 patients (41.4%) are alive. Five-year overall survival and HLH relapse-free survival were 41.8%, and 4-year HLH-free survival was 30.1%.
Multivariate analysis showed that age ≥60 years, male sex, lymphocytopenia, hypofibrinogenaemia, bilirubin >2mg/dl and blood urea nitrogen >20 mg/dl were poor prognostic factors. Patients with B-cell NHL had a better survival.
Through multivariate analysis, it was confirmed that lymphopenia and fibrinogenaemia are predictors of death; the presence of any viral infection did not have a statistically significant effect on prognosis (table 3). Compared with B-cell NHL, NK/T- cell NHL was confirmed to be associated with a worse prognosis (p = 0.036).
Table 3 Univariate and multivariate analysis of HLH survival.
| Variable | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% Cl | p-value | HR | 95%Cl | p-value | |
| Age ≥60 years | 1.708 | 1.153–2.530 | 0.008 | 1.446 | 0.940–2.224 | 0.093 |
| Male sex | 1.771 | 1.140–2.752 | 0.011 | 1.568 | 0.989–2.487 | 0.056 |
| Autoimmune disease | 0.208 | 0.029–1.490 | 0.118 | – | – | – |
| B-cell lymphoma | 0.383 | 0.156–0.941 | 0.036 | 0.539 | 0.228–1.274 | 0.159 |
| T-cell lymphoma (except NK/T-cell NHL) | 2.038 | 1.352–3.073 | 0.001 | 1.584 | 1.032–2.432 | 0.036 |
| NK/T-cell NHL | 1.432 | 0.850–2.414 | 0.177 | – | – | – |
| Fever | 1.724 | 0.425–6.991 | 0.446 | – | – | – |
| Hepatomegaly | 1.304 | 0.792–2.148 | 0.296 | – | – | – |
| Splenomegaly | 0.896 | 0.571–1.407 | 0.634 | – | – | – |
| Hb <9 g/dl | 1.229 | 0.687–2.200 | 0.487 | – | – | – |
| Plt <100×109/ | 4.656 | 0.649–33.396 | 0.126 | – | – | – |
| Neutrophils <1×109/l | 1.176 | 0.792–1.744 | 0.422 | – | – | – |
| Lymphocytes <0.5×109/l | 1.936 | 1.238–3.027 | 0.004 | 1.691 | 1.062–2.692 | 0.027 |
| Triglicerides >265 mg/dl | 1.227 | 0.832–1.810 | 0.301 | – | – | – |
| Fibrinogen <150 mg/dl | 1.979 | 1.174–3.334 | 0.010 | 1.925 | 1.111–3.336 | 0.020 |
| Ferritin >10,000 mg/ml | 1.398 | 0.944–2.069 | 0.094 | – | – | – |
| LDH 2 × ULRR | 1.316 | 0.535–3.237 | 0.549 | – | – | – |
| Bilirubin >2 mg/dl | 1.721 | 1.120–2.663 | 0.015 | 1.090 | 0.680–1.749 | 0.719 |
| Urea nitrogen >20 mg/dl | 1.865 | 1.234–2.820 | 0.003 | 1.235 | 0.787–1.940 | 0.359 |
| Viral infection | 1.220 | 0.815–1.828 | 0.334 | – | – | – |
CI = confidence interval; Hb = haemoglobin; HR = hazard ratio; LDH = lactate dehydrogenase; NK/T-cell NHL = natural killer T-cell non-Hodgkin lymphoma; Plt = platelets; ULRR = upper limit of the reference range
The 3-year survival rate of B-cell NHL and T-cell NHL patients was significantly better than that of NK/T-cell NHL patients (p = 0.0056) (fig. 2).
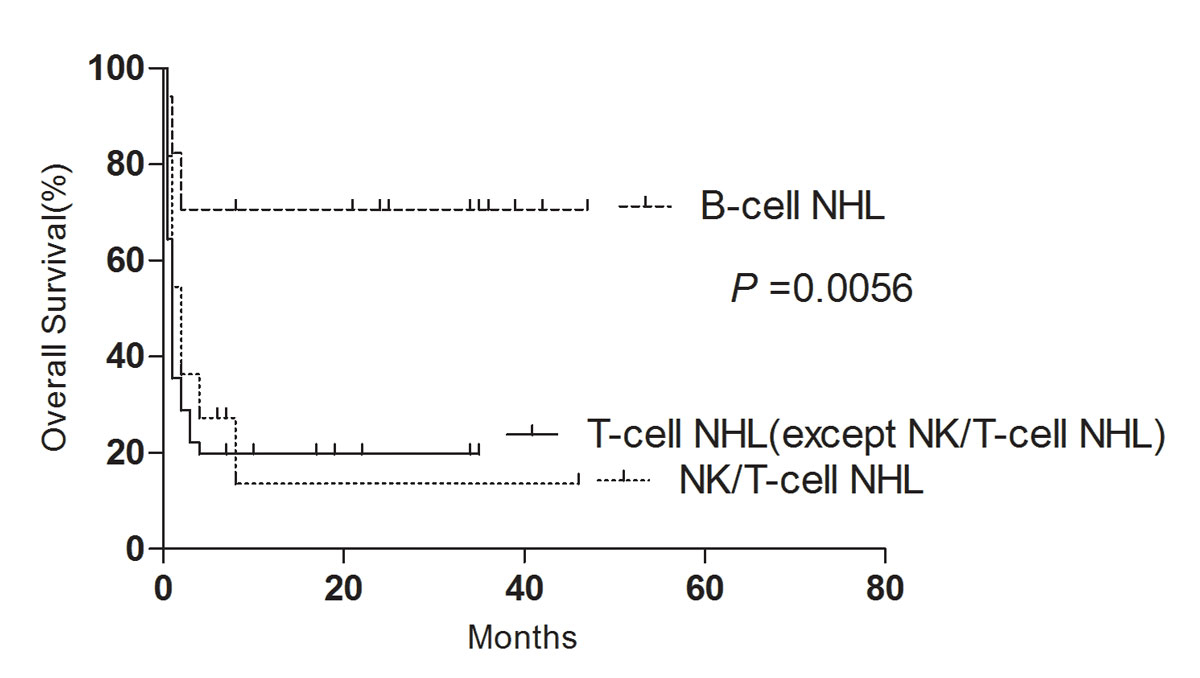
Figure 2 Impact of adverse pathology on overall survival.
In this single-centre study in adult patients with secondary HLH, we found that the clinical manifestations, laboratory indicators and underlying diseases are closely related to prognosis, and that the prognosis for some subgroups of secondary adult HLH is good.
The patient's initial diagnosis was based on the HLH-04 standard. The more recent HScore has been designed specifically to determine the probability of secondary adult HLH, and in this system the primary indicator is the more commonly used clinical presentation rather than laboratory indicators. These indicators are can be obtained more easily and earlier. Patients diagnosed according to the HLH-04 standard were analysed by retrospective application of the HScore. By the HScore, 4/174 patients had a >50% and 16/174 >90% probability of not having HLH. The patients fulfilled five HLH-04 diagnostic criteria, but some with a low probability of HLH with the HScore. All patients met the diagnostic criteria of the HLH-04, but some did not meet the HScore diagnostic criteria.
Of the various forms of secondary adult HLH among our haematology clinic patients, tumour-related HLH was the most common, accounting for about 52.9%. HLH may be associated with adult malignant lymphoma [22]. Adult HLH is associated with a variety of diseases. Our study was conducted in the haematological ward and may have underestimated other causes of HLH, such as connective tissue disease and infectious diseases. In other recent series of unselected patients with HLH, the proportion associated with malignancy ranged from just 23 to 52% [7, 12, 15, 21]. Takahashi et al. [18] reported that HLH associated with lymphomas had a significantly higher mortality than HLH caused by infections and autoimmune diseases (overall survival 8 vs 83%). Patients with T-cell lymphoma generally had worse outcomes than those with B cell lymphoma [17, 23, 24]. The lowest survival rate was in NK/T-cell cell lymphoma-associated HLH [12]. A recent study from the Mayo Clinic on 62 adult patients with HLH found that malignancy was a predictor of poor survival on multivariate analysis (overall survival 1.4 vs 22.8 months) [12]. Gavand et al. [25] reported that elevated LDH, aspartate transaminase, C-reactive protein, ferritin and procalcitonin were associated with poor prognosis. Ruscittia et al. [26] observed that the increased number of CD68/H-ferritin positive cells significantly correlated with the mortality rate. We found that malignancy, lymphocytopenia and hypofibrinogenaemia were associated with poor prognosis on multivariate analysis. In our study, tumour-associated HLH had a worse prognosis than non-tumour-associated HLH. Most of the cases were associated with NK/T-cell NHL.
In our study, of the 87 cases of lymphoma-associated HLH, 67 were related to T-cell NHL (22 to NK/T-cell NHL), 17 to B-cell NHL and 3 to Hodgkin’s lymphoma. The results are closely related to the success of lymphoma chemotherapy and immunotherapy, which is greater in B-cell than in T-cell NHL. The HLH recurrence rate was very low. Thirteen patients had recurrence of lymphoma combined with HLH recurrence, suggesting that cure of underlying disease is usually associated with lasting remission of the HLH.
Many patients, particularly in Asia, have been treated with doxorubicin-based therapy such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), often in the context of aggressive lymphoma / lymphoma-associated haemophagocytic syndrome (LAHS). In a retrospective study examining treatment outcomes with CHOP in 17 adult patients with HLH secondary to lymphoma, EBV or unknown cause, overall response rate was 58.8% (5/7 with lymphoma, 3/5 with EBV, 2/5 with idiopathic HLH), with 2-year overall survival of 43.9%, which suggests possible benefit of CHOP in adult HLH of different aetiologies [27]. Therefore, we suggest that lymphoma-associated HLH should be rapidly treated with specific anti-lymphoma therapy, rather than therapy for HLH itself. The combination of standard CHOP regimens [27] or other regimens [28] has been effective in the treatment of lymphoma-associated HLH independently of its aetiology.
HLH related to viral infection has been reported to have a poor prognosis by Wang et al. [29]. Viral infection can stimulate lymphocyte activation and cytokine storms are recognised [29]. Riviere et al. [15] reported that haematological malignancies associated with HLH have a worse prognosis than viral infection-related HLH (on the basis of survival at 1 month). Our patients with viral infections did not have a worse prognosis.
Our study has limitations, primarily its retrospective design and differences in treatment between patients, although the patients were treated in the same medical institution throughout the study period and the same diagnostic criteria were used. However, HLH is a very rare disease, and prospective studies are impractical.
In conclusion, our study emphasises that in clinical work HLH-04 is still valuable for the diagnosis of secondary adult HLH. In addition, studies have in the haematology department, lymphoma is still the most common cause of secondary HLH, and has a worse prognosis than HLH associated with other diseases. However, although the prognosis of HLH associated with T-cell NHL is poor, the prognosis of B-cell NHL-associated HLH prognosis is similar to that of other causes. For lymphoma-associated HLH, anti-lymphoma chemotherapy should be rapidly started, because the prognosis of these patients is closely related to lymphoma response.
The authors state that they have no interests which might be perceived as posing a conflict or bias.
1 Brisse E , Wouters CH , Matthys P . Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 2015;26(3):263–80. doi:.https://doi.org/10.1016/j.cytogfr.2014.10.001
2 George MR . Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69–86. doi:.https://doi.org/10.2147/JBM.S46255
3 Shabbir M , Lucas J , Lazarchick J , Shirai K . Secondary hemophagocytic syndrome in adults: a case series of 18 patients in a single institution and a review of literature. Hematol Oncol. 2011;29(2):100–6. doi:.https://doi.org/10.1002/hon.960
4 Henter JI , Horne A , Aricó M , Egeler RM , Filipovich AH , Imashuku S , et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. doi:.https://doi.org/10.1002/pbc.21039
5 Gupta A , Tyrrell P , Valani R , Benseler S , Weitzman S , Abdelhaleem M . The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(3):402–4. doi:.https://doi.org/10.1002/pbc.21564
6 Jordan MB , Allen CE , Weitzman S , Filipovich AH , McClain KL . How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–52. doi:.https://doi.org/10.1182/blood-2011-03-278127
7 Ramos-Casals M , Brito-Zerón P , López-Guillermo A , Khamashta MA , Bosch X . Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–16. doi:.https://doi.org/10.1016/S0140-6736(13)61048-X
8 Freeman HR , Ramanan AV . Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96(7):688–93. doi:.https://doi.org/10.1136/adc.2009.176610
9 Trottestam H , Horne A , Aricò M , Egeler RM , Filipovich AH , Gadner H , et al.; Histiocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–84. doi:.https://doi.org/10.1182/blood-2011-06-356261
10 Janka GE . Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63(1):233–46. doi:.https://doi.org/10.1146/annurev-med-041610-134208
11 Arca M , Fardet L , Galicier L , Rivière S , Marzac C , Aumont C , et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015;168(1):63–8. doi:.https://doi.org/10.1111/bjh.13102
12 Ishii E , Ohga S , Imashuku S , Yasukawa M , Tsuda H , Miura I , et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58–65. doi:.https://doi.org/10.1532/IJH97.07012
13 Parikh SA , Kapoor P , Letendre L , Kumar S , Wolanskyj AP . Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89(4):484–92. doi:.https://doi.org/10.1016/j.mayocp.2013.12.012
14 Rivière S , Galicier L , Coppo P , Marzac C , Aumont C , Lambotte O , et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127(11):1118–25. doi:.https://doi.org/10.1016/j.amjmed.2014.04.034
15 Otrock ZK , Eby CS . Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90(3):220–4. doi:.https://doi.org/10.1002/ajh.23911
16 Li J , Wang Q , Zheng W , Ma J , Zhang W , Wang W , et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93(2):100–5. doi:.https://doi.org/10.1097/MD.0000000000000022
17 Li F , Li P , Zhang R , Yang G , Ji D , Huang X , et al. Identification of clinical features of lymphoma-associated hemophagocytic syndrome (LAHS): an analysis of 69 patients with hemophagocytic syndrome from a single-center in central region of China. Med Oncol. 2014;31(4):902. doi:.https://doi.org/10.1007/s12032-014-0902-y
18 Takahashi N , Chubachi A , Kume M , Hatano Y , Komatsuda A , Kawabata Y , et al. A clinical analysis of 52 adult patients with hemophagocytic syndrome: the prognostic significance of the underlying diseases. Int J Hematol. 2001;74(2):209–13. doi:.https://doi.org/10.1007/BF02982007
19 Fardet L , Galicier L , Lambotte O , Marzac C , Aumont C , Chahwan D , et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–20. doi:.https://doi.org/10.1002/art.38690
20 Ravelli A , Minoia F , Davì S , Horne A , Bovis F , Pistorio A , et al.; Paediatric Rheumatology International Trials Organisation; Childhood Arthritis and Rheumatology Research Alliance; Pediatric Rheumatology Collaborative Study Group; Histiocyte Society. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016;75(3):481–9. doi:.https://doi.org/10.1136/annrheumdis-2015-208982
21 Schram AM , Comstock P , Campo M , Gorovets D , Mullally A , Bodio K , et al. Haemophagocytic lymphohistiocytosis in adults: a multicentre case series over 7 years. Br J Haematol. 2016;172(3):412–9. doi:.https://doi.org/10.1111/bjh.13837
22 Emmenegger U , Schaer DJ , Larroche C , Neftel KA . Haemophagocytic syndromes in adults: current concepts and challenges ahead. Swiss Med Wkly. 2005;135(21-22):299–314.
23 Yu JT , Wang CY , Yang Y , Wang RC , Chang KH , Hwang WL , et al. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol. 2013;92(11):1529–36. doi:.https://doi.org/10.1007/s00277-013-1784-3
24 Han AR , Lee HR , Park BB , Hwang IG , Park S , Lee SC , et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86(7):493–8. doi:.https://doi.org/10.1007/s00277-007-0278-6
25 Gavand PE , Serio C , Larroche N , Costedoat-Chalumeau C , Lavigne L , Mouthon M , et al. Association syndrome lymphohystiocytaire (HLH) et lupus systémique: étude multicentrique française de 103 épisodes chez 89 patients adultes. Rev Med Interne. 2016;37:A122–3. doi:.https://doi.org/10.1016/j.revmed.2016.10.106
26 Ruscitti P , Cipriani P , Di Benedetto P , Ciccia F , Liakouli V , Carubbi F , et al. Increased level of H-ferritin and its imbalance with L-ferritin, in bone marrow and liver of patients with adult onset Still’s disease, developing macrophage activation syndrome, correlate with the severity of the disease. Autoimmun Rev. 2015;14(5):429–37. doi:.https://doi.org/10.1016/j.autrev.2015.01.004
27 Shin HJ , Chung JS , Lee JJ , Sohn SK , Choi YJ , Kim YK , et al. Treatment outcomes with CHOP chemotherapy in adult patients with hemophagocytic lymphohistiocytosis. J Korean Med Sci. 2008;23(3):439–44. doi:.https://doi.org/10.3346/jkms.2008.23.3.439
28 Ravelli A , Grom AA , Behrens EM , Cron RQ . Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–98. doi:.https://doi.org/10.1038/gene.2012.3
29 Wang Y , Huang W , Wei N , Zeng X , Zhang J , Wang J , et al. [Clinical study of DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis]. Zhonghua Xue Ye Xue Za Zhi. 2014;35(10):901–4. Article in Chinese.
Qiaolei Zhang and Li Li Contributed equally in this manuscript.
The authors state that they have no interests which might be perceived as posing a conflict or bias.