Genetics and epigenetics of inflammatory bowel disease
DOI: https://doi.org/10.4414/smw.2018.14671
Marcin
Wawrzyniak, Michael
Scharl
Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Switzerland
Summary
The relevance of genetic and epigenetic alterations in the pathogenesis of inflammatory bowel disease (IBD) is still poorly understood. So far, 240 risk gene loci have been associated with IBD. They are mainly involved in regulating innate and adaptive immunity, as well as maintaining intestinal epithelial barrier function. However, the functional consequences of the identified genetic polymorphisms for IBD pathogenesis in vivo are often unknown. Even less is known about the role for epigenetic modifications in IBD pathogenesis. Though a number of epigenetic events seem to be causatively involved IBD pathogenesis, our knowledge about the functional relevance of those epigenetic modifications is scanty. This opens up a broad research field that generates novel insights into the pathophysiology of intestinal and chronic inflammatory disease. Patterns of DNA methylation and histone modifications might serve not only as biomarkers of disease activity or disease course, but also as new targets in therapeutic interventions in IBD patients.
Introduction
Crohn’s disease and ulcerative colitis are the main subtypes of inflammatory bowel disease (IBD). From a clinical perspective, they represent a chronic intestinal inflammation that often begins in young adulthood and is frequently relapsing. Crohn’s disease represents a discontinuous, transmural inflammation that can occur anywhere in the gastrointestinal tract, whereas ulcerative colitis is a continuous inflammation of the mucosal layer of the colon that always starts in the rectum. In addition to the gastrointestinal tract inflammation, so-called extraintestinal symptoms are common, affecting the joints, eyes, skin and liver [1]. In Crohn’s disease, fistulas and stenosis are a severe clinical problem that often require surgery [2, 3]. Since IBD is a life-long burden in many patients, it obviously impacts the quality of life of the affected patients and has severe socioeconomic consequences [1]. Importantly, the incidence of IBD is rising in Switzerland and also world-wide [4, 5].
IBD develops as a result of a combination of complex interactions between the individual’s genetic background, alterations in the composition of the intestinal microbiota on a qualitative as well as quantitative level, a dysregulated innate and adaptive immune system and environmental factors, such as diet, drugs and smoking [6]. To date, 240 susceptibility loci have been identified by genome-wide association studies (GWAS); however, only a minor part of disease risk and heritability can be explained by genetic factors alone [7–9]. It was proposed more than a decade ago that epigenetic regulation of gene expression might play a role in the development and regulation of IBD [10]. Today, several publications suggest that epigenetic mechanisms might help us to classify and diagnose patients, improve our understanding of IBD and, more importantly, provide new treatment opportunities.
Genetic factors contributing to IBD pathogenesis
Genetic factors have been widely considered as important risk factors for the onset of inflammatory bowel disease. GWAS performed in the last few years have been extremely successful in identifying genes that contribute to IBD susceptibility. NOD2 (located within the IBD1 locus) was the first gene to be associated with Crohn’s disease [11, 12]. Since then, several additional genes implicated in IBD have been identified. The strongest genetic effects were IL23R in IBD (odds ratio [OR] 2.01), NOD2 in Crohn’s disease (OR 3.01) and HLA in ulcerative colitis (OR 1.44). Most gene loci showed the same direction of effect in Crohn’s disease and ulcerative colitis, but there were some exceptions. For example, NOD2 and PTPN22 exhibited a significant protective effect in ulcerative colitis, but were risk factors for Crohn’s disease. Nevertheless, GWAS have helped us gain a better understanding of the genetic basis and their contribution with the pathogenesis of IBD. It is important to mention that many IBD risk loci are shared with other autoimmune or chronic inflammatory diseases, such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, type 1 diabetes, vitiligo or psoriasis. Further, there are IBD risk loci that are associated with both IBD subforms and some that are only associated with either Crohn’s disease or ulcerative colitis. This is particularly true for genes associated with epithelial barrier function (associated with ulcerative colitis) and genes involved in cellular innate immunity (Crohn’s disease) [8, 13–15].
For example, some genetic variations have been linked to particular pathways and certain disease phenotypes. Variations in genes associated with autophagy, phagocytosis or paneth cell failure, such as NOD2, IRGM, ATG16L1 or NCF4/NCF2 have been associated with segmental, early-onset disease or structuring disease [16]. Mutations in genes of the adaptive immune system, such as within the interleukin (IL)-10 / IL-10 receptor signalling pathway have been associated with a severe form of very early onset IBD [17]. A further challenge nowadays is to bring increasing knowledge about the role of intestinal microbiota in IBD pathogenesis together with the observed genetic alterations in IBD patients on a functional level.
The odds of individual single nucleotide polymorphisms (SNPs) on the risk of developing IBD have been characterised in some detail in recent years, largely with odds ratios only slightly over 1, indicating a rather mild clinical effect. However, there is still very little knowledge on the numbers and potential interplay of these mutations, and ultimately their impact on course of disease in patients with IBD. Although there is robust knowledge on the prevalence of individual SNPs in patients with Crohn’s disease and ulcerative colitis (or IBD overall) as compared with healthy subjects, we know less about the quantity and composition of SNPs in patients with IBD, including the frequency of patients carrying more than one SNP risk-allele. However, understanding of the functional and clinical consequences of the associated alleles is still an ongoing process. Some association signals correspond to nonsynonymous coding variations, but the majority of signals do not. They involve noncoding genetic variations mainly related to changes in gene expression. Moreover, it has been shown that many effects seem to be highly cell-type specific. The integration of genetic, transcriptomic and epigenetic studies should lead to more insight into IBD pathogenesis and new future treatment options [18].
A recent study by Cleynen et al. revealed that the genetic risk score representing all known risk alleles for IBD showed a strong association with the disease subphenotypes defined in the Montreal classification system for IBD. Said classification distinguishes IBD into three subphenotypes: ulcerative colitis, colonic Crohn’s disease and ileal Crohn’s disease. Furthermore, they found that disease location is an intrinsic aspect of a patient’s disease, in part genetically determined, and the major driver for changes in disease behaviour over time [19].
Interestingly, in twin pair studies only a 40 to 50% concordance in the onset of IBD was detected [20, 21]. On the one hand, this clearly underlines the importance of genetic susceptibility in the disease development. However, on the other hand, this also represents a clear limitation in the concept of a genetic cause of IBD and clearly points to the (additional) involvement of other factors, such as the intestinal microbiota and environmental factors, in IBD pathogenesis. This latter aspect is also supported by the observation that similar genetic risk factors or risk gene profiles for Crohn’s disease, ulcerative colitis and other chronic inflammatory or autoimmune diseases in one individual result in the development of Crohn’s disease or ulcerative colitis and in the other individual may result in the development of another disease. Further, the fact that the risk increase, namely the odds ratio, which is associated with a large number of these genetic variations, is only about 1.2, which means that having such a variant means the chance of developing this disease is only 20% higher than for anybody who is not carrying this particular risk variant. One the one hand, this demonstrates that many people who are carrying those risk genes are healthy, or at least not affected by such a disease. On the other hand, it means also that genetic testing is not likely to be useful for diagnosis of IBD, even though there is an increasing market for this approach.
Epigenetics in inflammatory bowel disease
Principles of epigenetic mechanisms
The term “epigenetics” was introduced in 1942 by Waddington to explain how a phenotype might be produced by interaction between genes and their environment [22]. The modern definition refers to heritable alternations of gene expression events that are caused independently of genetic information carried by the primary DNA sequence [23]. The main epigenetic mechanisms controlling gene expression include DNA methylation, histone modification that modulates chromatin structure, micro RNA (miRNA) interference that regulates posttranscriptional steps, and positioning of nucleosomes [24]. By controlling patterns of gene expression, epigenetic mechanisms are involved in correct cell development, differentiation, function and homeostasis. In addition, all mentioned mechanisms are influenced by exposure to environmental factors, persists through mitosis and meiosis, and, more importantly, can be reversed [25, 26]. It was proposed more than a decade ago that epigenetic regulation of gene expression might play a role in development and regulation of IBD [10]. Today, several publications suggest that epigenetic mechanisms might help us to classify and diagnose patients, improve our understanding of IBD and, more importantly, provide new treatment opportunities.
In the process of DNA methylation a methyl group is covalently added to 5′ carbon of cytosines that are part of cytosine-guanine dinucleotides (CpG) [27]. Full methylation occurs when cytosine residues on both DNA strands are methylated [28]. Representing less than 1% of all dinucleotides, CpG dinucleotides are rather rare in the genome, but are often concentrated in particular regions of the genome called “CpG islands”. CpG islands are typically, but not exclusively, associated with gene promoters or first exons of approximately two thirds of all genes [29]. CpG islands are mainly protected from methylation and remain unmethylated, whereas in other regions of the genome, CpGs are hypermethylated [30]. DNA methylation is catalysed by so-called DNA methyltransferases (DNMTs). Based on protein sequence homology, the DNMT protein family consisted initially of five members: DNMT1, DNMT2, DNMT3A, DNMT3B, DNMT3L [31]. After it was recognised that DNMT2 methylates RNA and DNMT3L lacks 5-cytosine-methyl-transferase activity, DNTM’s were subdivided into those maintaining DNA methylation patterns (DNMT1) and de novo methylating DNMTs (DNMT3A, DNMT3B) [32]. In general, hypermethylation of CpG islands in regulatory genetic elements such as promoters is transcriptionally repressive and leads to gene silencing [33]. Recently, the so-called ten-eleven translocation (TET) family of enzymes have been identified and proven to oxidise 5-hydroxymethylcytosine, which is a crucial step in the demethylation of previously methylated DNA regions [34].
An additional level of epigenetic regulation of gene expression is achieved with the process of posttranslational histone modifications. In eukaryotic cells, genomic DNA is wrapped around eight core histone proteins (two molecules of each H2A, H2B, H3B and H4 histone) to form a nucleosome, a basic subunit of chromatin. DNA compaction in chromatin is one of the most important mechanisms regulating gene expression. Chromatin that is loosely packed with lightly attached DNA, which favours active transcription of genes, is called euchromatin. In contrast, highly compacted heterochromatin is transcriptionally silent owing to limited accessibility by transcription factors. Posttranslational histone modifications occur mainly in histone tails and include acetylation, methylation, ubiquitination, phosphorylation, sumoylation and citrulination [35].
Histone acetylation, the addition of acetyl groups to lysine residues of histone is catalysed by histone acetyltransferases (HATs). Removal of acetyl groups is performed by histone deacetylases (HDACs). In general, chromatin opening during histone acetylation is associated with transcriptional gene activity, whereas increased activity of HDACs and histone deacetylation causes hypoacetylation, chromatin compacting and gene silencing (fig. 1) [36].
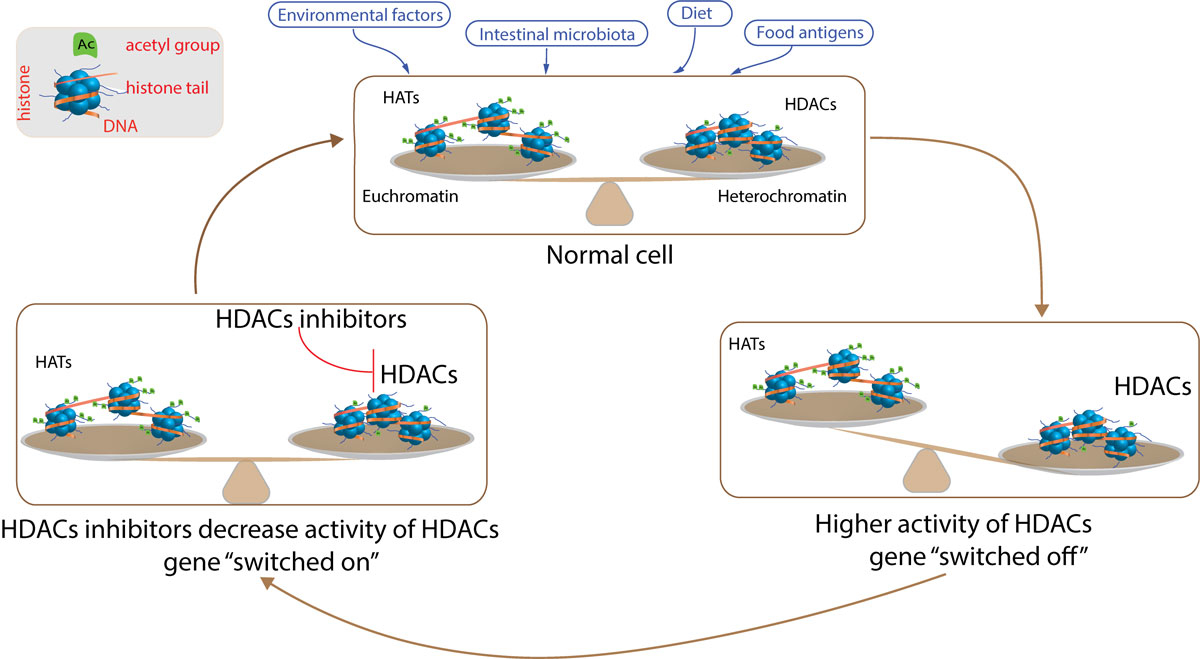
Figure 1
Epigenetic regulation of gene expression – histone acetylation. Cells in the gastrointestinal track are exposed to several environmental factors, dietary antigens, intestinal microbiota and microbiota derived-metabolites that may modulate epigenetic machinery. In the steady state, there is a balance between acetylation and deacetylation. Increased activity of histone deacetylases (HDACs) leads to higher histone deacetylation, which causes formation of heterochromatin and silences gene expression. On the other hand, treatment with HDAC inhibitors decreases HDAC activity and restores balance between acetylation and deacetylation in the cell, allowing genes to be transcribed. HAT = histone acetyltransferase
In contrast to methylation of cytosine residues in DNA, which leads to transcriptional repression, histone methylation by histone methyltransferases (HMTs) can be associated with either repression or active transcription [37]. The outcome depends on the position of targeted amino acid, the type of residues involved (arginine vs lysine) and the degree of methylation [30]. For example, methylation of lysine (K) in position 4, 36 and 79 of H3 is found in open chromatin whereas closed heterochromatin is enriched in methylation of H3K9 and H3K27.
In summary, it is becoming more and more clear that the epigenetic mechanisms that regulate gene expression are not separate but rather connected in such a way that DNA methylation may influence histone modification and vice versa [38].
Epigenetic modifications in IBD
In the first study trying to understand epigenetic mechanisms in IBD, Gloria et al. showed that, as compared with healthy controls, rectal mucosa from ulcerative colitis patients was characterised by global hypomethylation. A similar DNA global hypomethylation profile was observed in patients with active, inflamed ulcerative colitis as compared with patients with inactive ulcerative colitis [39]. In IBD, it has been demonstrated that local inflammation increases colonic epithelial cell turnover and accelerates DNA methylation changes [40]. Increased DNA methylation may result in genetic instability that leads to cancer development. As candidate genes referred to carcinogenesis are known, epigenetic changes could be easily investigated. That is the reason why the first DNA methylation studies were mainly focused on IBD related cancer development.
Genes that were already associated with carcinogenesis, namely CDKN2a/p16INK4A
, CDKN2a/p14ARF
, CDH1, MLH1, HPP1 and MYOD1 have been found differentially methylated in colonic mucosa from ulcerative colitis patients with dysplasia and/or carcinoma as compared with quiescent mucosa from the same patients [41–45]. Interestingly, as a consequence of hypermethylation lower levels of CDH1, encoding cell adhesion molecule E-cadherin, and MLH1 were observed in immunohistochemical stains of gut tissue [46].
Later, the research focus was directed to a potential role of epigenetic regulation events in IBD pathogenesis itself. GWAS analysis revealed an association between IBD and polymorphisms in a gene encoding an enzyme responsible for establishment of DNA methylation – DNMT3a [47]. Moreover, as compared with uninflamed paired samples, in inflamed mucosa from ulcerative colitis patients, higher levels of expression of DNMT1 and DNMT3b were reported [48]. Increased interest in the role of DNA methylation in the pathogenesis of IBD was followed with the development of advanced platform-based DNA methylation array technologies, which shifted interest from single, candidate gene approaches to broad, general methylation analysis.
Epigenome-wide methylation association studies (EWAS) were initially performed using peripheral blood. Nimmo et al. reported a methylation profile that is characteristic for Crohn’s disease, with 50 significantly altered methylation sites in Crohn’s disease patients compared with controls [49]. Differences in methylation were observed in genes important for immune responses, such as IL21R, S100A13, FASLG, MAPK13, RIPK3 or PRF1. Use of peripheral blood as a material to investigate IBD-related methylation changes was questioned by a study where peripheral blood mononuclear cells (PBMCs) from monozygotic twins and IBD patients did not showed differentially methylated genes with exception of hypermethylation of a locus of TEPP gene (which has no clear relevance in IBD pathogenesis) [50].
Obviously more relevant for IBD research are tissue-specific variations in DNA methylation. In an EWAS study using whole tissue intestinal biopsy specimens from monozygotic twins, Hasler et al. were able to identify 61 differently methylated loci, including several loci responsible for regulation of immune responses. Interestingly, differently methylated loci were later validated in other cohorts and showed differentially expressed transcripts (CFI, FLNA, HKDC1, IGHG1, MT1H, PTN, SLC7A7, SPINK4, THY1, TK1) [51]. A nice example of a gene whose promotor region is hypermethylated and whose corresponding transcript is downregulated in the rectal mucosa of ulcerative colitis patients without differences in methylation profile in circulating leucocytes, is BRINP3 [52]. As BRINP3 was never identified in GWAS studies, it also serves as perfect example that epigenetic studies can identify new genes relevant for IBD pathogenesis.
In a very interesting study, Cooke et al. compared DNA methylation profiles in isolated intestinal epithelial cells from inflamed and uninflamed rectal biopsies from ulcerative colitis and Crohn’s disease patients [53]. Differentially methylated genes identified during this study had already been reported in other EWAS [49] and as Crohn’s disease-associated (TAP1, IL8RP, PKLR, PTFR) or ulcerative colitis-associated (ICAM3, CDH1, CARD9, IL8RB, IL8RA) susceptibility genes in GWAS as well [54].
The comparison between different studies investigating the role of epigenetic mechanisms in IBD is difficult, as the main problem is reproducibility and lack of consistency regarding the type of tissue analysed (PBMCs, epithelial cells, biopsies), controls (healthy controls, unaffected tissue from the same patient) and heterogeneity of the analysed population. As the methylome signature is specific for a given cell type, changes in cell proportions in tissues due to inflammation might mimic true epigenetic changes and lead to a false understanding of the whole process. Despite the use of statistical algorithms for estimating cell proportions in tissues, methylation profiles should ideally be studied in sorted cell populations to allow proper conclusions about real epigenetic changes.
Methylation and acetylation events of histones have been studied in IBD to a lesser extent. In dextran sulphate sodium (DSS) and 2,4-trinitrobenzene sulfonic acid (TNBS) induced experimental rat models of colitis, histone acetylation was observed in colonic tissues [55]. In this study, an increase in histone 4 acetylation on lysine (K8 and K12) was reported in inflamed mucosa as compared with uninflamed mucosa. Identical pattern of acetylation were confirmed in biopsies from patients with Crohn’s disease [55]. However, most of our understanding of histone modifications and their influence on IBD pathogenesis come from the use of HDAC inhibitors and might be therefore somehow artificial.
Administration of HDAC inhibitors in DSS and TNBS-induced experimental colitis reduces disease severity and expression of pro-inflammatory cytokines [56]. Additionally, inhibition of HDAC9 prevents colitis in mice as a result of increased development and suppressive T regulatory cell (Treg) function [57]. HATs and HDACs do not act exclusively on histones, but can modulate acetylation of non-histone proteins including p53, STAT3 (signal transducer and activator of transcription-3) or NFκB (nuclear factor kappa B) [58].
Interestingly, short-chain fatty acids (SCFAs), bacterial metabolites that are formed as result of anaerobic fermentation of dietary fibre, possess HDAC inhibitory activity [59, 60]. Many bacteria from the Firmicutes and Bacteroides genera secret SCFAs (acetate, propionate, butyrate) at high concentration [61] and reduced numbers of bacteria that produce SCFAs have been reported in patients with IBD [62]. Conversely, application of Roseburia, a bacterium that is able to produce butyrate, showed positive effects on ulcerative colitis treatment [63, 64]. Possible mechanisms of action involve generation of Tregs from naïve CD4+ T cells. In an experimental setup, butyrate led to increased histone H3 acetylation within Foxp3 loci, the key transcription factor required for Treg cell differentiation [59, 60]. In addition, butyrate might modulate the function of intestinal macrophages [65]. Lipopolysaccharide-induced secretion of proinflammatory mediators such as IL-12 and IL-6, but not of tumour necrosis factor-alpha or monocyte chemoattractant protein-1 (MCP-1), was downregulated after treatment of macrophages with butyrate. As intestinal macrophages are the most abundant cells in lamina propria, bacterial-derived butyrate induces macrophages hyporesponsiveness and maintains tolerance.
The potential role of gut microbiota in the development of IBD, as well as of dysbiosis in gut microbiota composition in IBD patients, have been reported [66]. However, there is more and more evidence suggesting that, via epigenetic regulation of gene expression, commensal microbiota may play a beneficial role in IBD treatment.
Conclusion
Current knowledge about genetic and epigenetic involvement in IBD pathogenesis is still poor. Since the identification of the first IBD risk gene, NOD2 in 2001, GWAS have unravelled 240 risk gene loci involved in IBD pathogenesis, and many of them are involved in regulating innate and adaptive immune responses or intestinal barrier function. This strongly emphasises that, in addition to genetic alterations, the intestinal microbiota and environmental factors also play a critical role in IBD pathogenesis. However, there is still only a little knowledge about the direct consequences of the identified SNPs for IBD pathogenesis and human physiology overall, since the functional consequences of those genetic variations in vivo are often still unknown.
Even less information is available about the role for epigenetic modifications in IBD patients and their impact on IBD pathogenesis. EWAS and other approaches detected a number of epigenetic events that might be causatively involved in the onset of IBD, but our knowledge about the functional relevance of those epigenetic modifications is still scarce. This however opens up a broad research field that might help to obtain crucial novel insights into the pathophysiology of intestinal and chronic inflammatory disease. Specific patterns of DNA methylation and histone modifications might serve not only as biomarkers for disease activity or disease course, but also as new targets for therapeutic interventions in IBD patients. So it will be important to further unravel and elucidate the exact functional consequences of genetic and epigenetic alterations in IBD pathogenesis to pave the road for the development of novel therapeutic strategies.
Search strategy and selection criteria
References for this review were identified through searches of PubMed with the search terms genetics, epigenetics, IBD susceptibility genes, IBD risk genes as well as IBD and methylation from 1938 until July, 2018. Articles were also identified through searches of the authors’ own files. Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this review.
Author contributions
All authors participated sufficiently, intellectually or practically, in the work to take public responsibility for the content of the article, including the conception, design, data interpretation and writing of the manuscript. The final version of the manuscript was approved by all authors.
References
1
Cosnes
J
,
Gower-Rousseau
C
,
Seksik
P
,
Cortot
A
. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785–94.e4. doi:.https://doi.org/10.1053/j.gastro.2011.01.055
2
Siegmund
B
,
Feakins
RM
,
Barmias
G
,
Ludvig
JC
,
Teixeira
FV
,
Rogler
G
, et al.
Results of the Fifth Scientific Workshop of the ECCO (II): Pathophysiology of Perianal Fistulizing Disease. J Crohn’s Colitis. 2016;10(4):377–86. doi:.https://doi.org/10.1093/ecco-jcc/jjv228
3
Latella
G
,
Rogler
G
,
Bamias
G
,
Breynaert
C
,
Florholmen
J
,
Pellino
G
, et al.
Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohn’s Colitis. 2014;8(10):1147–65. doi:.https://doi.org/10.1016/j.crohns.2014.03.008
4
Braegger
CP
,
Ballabeni
P
,
Rogler
D
,
Vavricka
SR
,
Friedt
M
,
Pittet
V
; Swiss IBD Cohort Study Group. Epidemiology of inflammatory bowel disease: Is there a shift towards onset at a younger age?
J Pediatr Gastroenterol Nutr. 2011;53(2):141–4. doi:.https://doi.org/10.1097/MPG.0b013e318218be35
5
Ray
K
. IBD: The changing epidemiology of IBD. Nat Rev Gastroenterol Hepatol. 2017;14(12):690. doi:.https://doi.org/10.1038/nrgastro.2017.159
6
de Souza
HS
,
Fiocchi
C
. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. doi:.https://doi.org/10.1038/nrgastro.2015.186
7
Brant
SR
. Promises, delivery, and challenges of inflammatory bowel disease risk gene discovery. Clin Gastroenterol Hepatol. 2013;11(1):22–6. Published online November 03, 2012. doi:.https://doi.org/10.1016/j.cgh.2012.11.001
8
Jostins
L
,
Ripke
S
,
Weersma
RK
,
Duerr
RH
,
McGovern
DP
,
Hui
KY
, et al.; International IBD Genetics Consortium (IIBDGC). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24. doi:.https://doi.org/10.1038/nature11582
9
de Lange
KM
,
Moutsianas
L
,
Lee
JC
,
Lamb
CA
,
Luo
Y
,
Kennedy
NA
, et al.
Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–61. doi:.https://doi.org/10.1038/ng.3760
10
Petronis
A
,
Petroniene
R
. Epigenetics of inflammatory bowel disease. Gut. 2000;47(2):302–6. doi:.https://doi.org/10.1136/gut.47.2.302
11
Hugot
JP
,
Chamaillard
M
,
Zouali
H
,
Lesage
S
,
Cézard
JP
,
Belaiche
J
, et al.
Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi:.https://doi.org/10.1038/35079107
12
Ogura
Y
,
Bonen
DK
,
Inohara
N
,
Nicolae
DL
,
Chen
FF
,
Ramos
R
, et al.
A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–6. doi:.https://doi.org/10.1038/35079114
13
Khor
B
,
Gardet
A
,
Xavier
RJ
. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–17. doi:.https://doi.org/10.1038/nature10209
14
Lees
CW
,
Barrett
JC
,
Parkes
M
,
Satsangi
J
. New IBD genetics: common pathways with other diseases. Gut. 2011;60(12):1739–53. doi:.https://doi.org/10.1136/gut.2009.199679
15
Xavier
RJ
,
Podolsky
DK
. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–34. doi:.https://doi.org/10.1038/nature06005
16
Knights
D
,
Lassen
KG
,
Xavier
RJ
. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62(10):1505–10. doi:.https://doi.org/10.1136/gutjnl-2012-303954
17
Moran
CJ
. Very early onset inflammatory bowel disease. Semin Pediatr Surg. 2017;26(6):356–9. doi:.https://doi.org/10.1053/j.sempedsurg.2017.10.004
18
Cleynen
I
,
Vermeire
S
. The genetic architecture of inflammatory bowel disease: past, present and future. Curr Opin Gastroenterol. 2015;31(6):456–63. doi:.https://doi.org/10.1097/MOG.0000000000000215
19
Cleynen
I
,
Boucher
G
,
Jostins
L
,
Schumm
LP
,
Zeissig
S
,
Ahmad
T
, et al.; International Inflammatory Bowel Disease Genetics Consortium. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387(10014):156–67. doi:.https://doi.org/10.1016/S0140-6736(15)00465-1
20
Vind
I
,
Jespersgaard
C
,
Hougs
L
,
Riis
L
,
Dinesen
L
,
Andersen
PS
, et al.
Genetic and environmental factors in monozygotic twins with Crohn’s disease and their first-degree relatives: a case report. Digestion. 2005;71(4):262–5. doi:.https://doi.org/10.1159/000087053
21
Tysk
C
,
Lindberg
E
,
Järnerot
G
,
Flodérus-Myrhed
B
. Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut. 1988;29(7):990–6. doi:.https://doi.org/10.1136/gut.29.7.990
22
Waddington
CH
. The epigenotype. Int J Epidemiol. 2012;41(1):10–3. Published online December 20, 2011. doi:.https://doi.org/10.1093/ije/dyr184
23
Jenke
AC
,
Zilbauer
M
. Epigenetics in inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28(6):577–84. doi:.https://doi.org/10.1097/MOG.0b013e328357336b
24
Portela
A
,
Esteller
M
. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68. doi:.https://doi.org/10.1038/nbt.1685
25
Ushijima
T
,
Watanabe
N
,
Okochi
E
,
Kaneda
A
,
Sugimura
T
,
Miyamoto
K
. Fidelity of the methylation pattern and its variation in the genome. Genome Res. 2003;13(5):868–74. doi:.https://doi.org/10.1101/gr.969603
26
Faulk
C
,
Dolinoy
DC
. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6(7):791–7. doi:.https://doi.org/10.4161/epi.6.7.16209
27
Chen
T
,
Li
E
. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. doi:.https://doi.org/10.1007/3-540-31390-7_6
28
Ventham
NT
,
Kennedy
NA
,
Nimmo
ER
,
Satsangi
J
. Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology. 2013;145(2):293–308. Published online June 08, 2013. doi:.https://doi.org/10.1053/j.gastro.2013.05.050
29
Fogel
O
,
Richard-Miceli
C
,
Tost
J
. Epigenetic Changes in Chronic Inflammatory Diseases. Adv Protein Chem Struct Biol. 2017;106:139–89. Published online October 18, 2016. doi:.https://doi.org/10.1016/bs.apcsb.2016.09.003
30
Däbritz
J
,
Menheniott
TR
. Linking immunity, epigenetics, and cancer in inflammatory bowel disease. Inflamm Bowel Dis. 2014;20(9):1638–54. doi:.https://doi.org/10.1097/MIB.0000000000000063
31
Bestor
TH
. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–402. doi:.https://doi.org/10.1093/hmg/9.16.2395
32
Reik
W
,
Dean
W
,
Walter
J
. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089–93. doi:.https://doi.org/10.1126/science.1063443
33
Hughes
T
,
Webb
R
,
Fei
Y
,
Wren
JD
,
Sawalha
AH
. DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks. Genes Immun. 2010;11(7):554–60. Published online May 13, 2010. doi:.https://doi.org/10.1038/gene.2010.24
34
Kohli
RM
,
Zhang
Y
. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–9. doi:.https://doi.org/10.1038/nature12750
35
Zhang
T
,
Cooper
S
,
Brockdorff
N
. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–81. Published online October 15, 2015. doi:.https://doi.org/10.15252/embr.201540945
36
Henikoff
S
,
Shilatifard
A
. Histone modification: cause or cog?
Trends Genet. 2011;27(10):389–96. Published online July 20, 2011. doi:.https://doi.org/10.1016/j.tig.2011.06.006
37
Zentner
GE
,
Henikoff
S
. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–66. doi:.https://doi.org/10.1038/nsmb.2470
38
Fuks
F
,
Hurd
PJ
,
Deplus
R
,
Kouzarides
T
. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31(9):2305–12. doi:.https://doi.org/10.1093/nar/gkg332
39
Glória
L
,
Cravo
M
,
Pinto
A
,
de Sousa
LS
,
Chaves
P
,
Leitão
CN
, et al.
DNA hypomethylation and proliferative activity are increased in the rectal mucosa of patients with long-standing ulcerative colitis. Cancer. 1996;78(11):2300–6. doi:.https://doi.org/10.1002/(SICI)1097-0142(19961201)78:11<2300::AID-CNCR5>3.0.CO;2-Q
40
Issa
JP
,
Ahuja
N
,
Toyota
M
,
Bronner
MP
,
Brentnall
TA
. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61(9):3573–7.
41
Azarschab
P
,
Porschen
R
,
Gregor
M
,
Blin
N
,
Holzmann
K
. Epigenetic control of the E-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosomes Cancer. 2002;35(2):121–6. doi:.https://doi.org/10.1002/gcc.10101
42
Hsieh
CJ
,
Klump
B
,
Holzmann
K
,
Borchard
F
,
Gregor
M
,
Porschen
R
. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58(17):3942–5.
43
Sato
F
,
Shibata
D
,
Harpaz
N
,
Xu
Y
,
Yin
J
,
Mori
Y
, et al.
Aberrant methylation of the HPP1 gene in ulcerative colitis-associated colorectal carcinoma. Cancer Res. 2002;62(23):6820–2.
44
Sato
F
,
Harpaz
N
,
Shibata
D
,
Xu
Y
,
Yin
J
,
Mori
Y
, et al.
Hypermethylation of the p14(ARF) gene in ulcerative colitis-associated colorectal carcinogenesis. Cancer Res. 2002;62(4):1148–51.
45
Wheeler
JM
,
Kim
HC
,
Efstathiou
JA
,
Ilyas
M
,
Mortensen
NJ
,
Bodmer
WF
. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001;48(3):367–71. doi:.https://doi.org/10.1136/gut.48.3.367
46
Fleisher
AS
,
Esteller
M
,
Harpaz
N
,
Leytin
A
,
Rashid
A
,
Xu
Y
, et al.
Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60(17):4864–8.
47
Franke
A
,
McGovern
DP
,
Barrett
JC
,
Wang
K
,
Radford-Smith
GL
,
Ahmad
T
, et al.
Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–25. doi:.https://doi.org/10.1038/ng.717
48
Saito
S
,
Kato
J
,
Hiraoka
S
,
Horii
J
,
Suzuki
H
,
Higashi
R
, et al.
DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm Bowel Dis. 2011;17(9):1955–65. Published online January 06, 2011. doi:.https://doi.org/10.1002/ibd.21573
49
Nimmo
ER
,
Prendergast
JG
,
Aldhous
MC
,
Kennedy
NA
,
Henderson
P
,
Drummond
HE
, et al.
Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18(5):889–99. Published online October 21, 2011. doi:.https://doi.org/10.1002/ibd.21912
50
Harris
RA
,
Nagy-Szakal
D
,
Pedersen
N
,
Opekun
A
,
Bronsky
J
,
Munkholm
P
, et al.
Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18(12):2334–41. Published online March 29, 2012. doi:.https://doi.org/10.1002/ibd.22956
51
Häsler
R
,
Feng
Z
,
Bäckdahl
L
,
Spehlmann
ME
,
Franke
A
,
Teschendorff
A
, et al.
A functional methylome map of ulcerative colitis. Genome Res. 2012;22(11):2130–7. Published online July 23, 2012. doi:.https://doi.org/10.1101/gr.138347.112
52
Smith
PJ
,
Levine
AP
,
Dunne
J
,
Guilhamon
P
,
Turmaine
M
,
Sewell
GW
, et al.
Mucosal transcriptomics implicates under expression of BRINP3 in the pathogenesis of ulcerative colitis. Inflamm Bowel Dis. 2014;20(10):1802–12. doi:.https://doi.org/10.1097/MIB.0000000000000169
53
Cooke
J
,
Zhang
H
,
Greger
L
,
Silva
AL
,
Massey
D
,
Dawson
C
, et al.
Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(11):2128–37. Published online March 14, 2012. doi:.https://doi.org/10.1002/ibd.22942
54
Anderson
CA
,
Boucher
G
,
Lees
CW
,
Franke
A
,
D’Amato
M
,
Taylor
KD
, et al.
Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–52. Published online February 06, 2011. doi:.https://doi.org/10.1038/ng.764
55
Tsaprouni
LG
,
Ito
K
,
Powell
JJ
,
Adcock
IM
,
Punchard
N
. Differential patterns of histone acetylation in inflammatory bowel diseases. J Inflamm (Lond). 2011;8(1):1. doi:.https://doi.org/10.1186/1476-9255-8-1
56
Glauben
R
,
Batra
A
,
Fedke
I
,
Zeitz
M
,
Lehr
HA
,
Leoni
F
, et al.
Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176(8):5015–22. doi:.https://doi.org/10.4049/jimmunol.176.8.5015
57
de Zoeten
EF
,
Wang
L
,
Sai
H
,
Dillmann
WH
,
Hancock
WW
. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. 2010;138(2):583–94. Published online October 29, 2009. doi:.https://doi.org/10.1053/j.gastro.2009.10.037
58
Glauben
R
,
Siegmund
B
. Inhibition of histone deacetylases in inflammatory bowel diseases. Mol Med. 2011;17(5-6):426–33. doi:.https://doi.org/10.2119/molmed.2011.00069
59
Arpaia
N
,
Campbell
C
,
Fan
X
,
Dikiy
S
,
van der Veeken
J
,
deRoos
P
, et al.
Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–5. Published online November 13, 2013. doi:.https://doi.org/10.1038/nature12726
60
Furusawa
Y
,
Obata
Y
,
Fukuda
S
,
Endo
TA
,
Nakato
G
,
Takahashi
D
, et al.
Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. doi:. Correction in: Nature. 2014;506:254. doi:https://doi.org/10.1038/nature13041https://doi.org/10.1038/nature12721
61
Louis
P
,
Flint
HJ
. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. Published online February 13, 2009. doi:.https://doi.org/10.1111/j.1574-6968.2009.01514.x
62
Sokol
H
,
Seksik
P
,
Furet
JP
,
Firmesse
O
,
Nion-Larmurier
I
,
Beaugerie
L
, et al.
Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–9. doi:.https://doi.org/10.1002/ibd.20903
63
Berni Canani
R
,
Di Costanzo
M
,
Leone
L
. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics. 2012;4(1):4. doi:.https://doi.org/10.1186/1868-7083-4-4
64
Fofanova
TY
,
Petrosino
JF
,
Kellermayer
R
. Microbiome-Epigenome Interactions and the Environmental Origins of Inflammatory Bowel Diseases. J Pediatr Gastroenterol Nutr. 2016;62(2):208–19. doi:.https://doi.org/10.1097/MPG.0000000000000950
65
Chang
PV
,
Hao
L
,
Offermanns
S
,
Medzhitov
R
. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. 2014;111(6):2247–52. Published online January 03, 2014. doi:.https://doi.org/10.1073/pnas.1322269111
66
Couturier-Maillard
A
,
Secher
T
,
Rehman
A
,
Normand
S
,
De Arcangelis
A
,
Haesler
R
, et al.
NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–11. Published online January 02, 2013. doi:.https://doi.org/10.1172/JCI62236