Excipients: not so inert? When the excipient plays the role of an active substance, as exemplified by systemic lupus
DOI: https://doi.org/10.4414/smw.2018.14631
aCNRS/Strasbourg University Unit Biotechnology and Cell Signalling; team Neuroimmunology and Peptide Therapeutics / Laboratory of Excellence Medalis,
Strasbourg, France
bUniversity of Strasbourg Institute for Advanced Study (USIAS), Strasbourg, France
Summary
It is well recognised that the historical timeline required for developing a drug, beginning with target identification and validation, is long and often tedious. It requires a large set of competences in various areas of molecular and cellular biology, chemistry, pharmacology, imaging, and model animal experimentation. Once the active molecule appears to be ready for human testing in controlled clinical trials, then the question arises of how to formulate it to render it stable, adequately packaged, according to the chosen route of administration, and bioavailable to reach its target in the affected organs. Historically, excipients have been considered inert and devoid of medicinal effect or influence. In fact, excipients are seldom neutral and some of them have been found to play a significant role, for example by initiating or participating in chemical and physical interactions with the active substance, leading in certain cases to compromise its therapeutic activity. It is difficult today to appreciate the number of potential drugs that have been discarded as a result of limited efficacy due to inappropriate excipients. This matter is presented here, with the peptide P140 (Lupuzor™) as example. Two formulations of P140, differing in the excipients used (mannitol or trehalose), have been evaluated in patients affected by systemic lupus erythematosus in two successive phase IIb clinical trials. P140 was shown to reduce excessive autophagy activity discovered in some lupus immune cell subsets. One of the two excipients, namely trehalose, has been claimed to exert an intrinsic stimulating activity on autophagy process, which was found therefore to counteract the beneficial peptide effects.
Introduction
In France, the recent controversy about Levothyrox (levothyroxine or L-thyroxine) raised awareness of unavoidable components of medicines, the excipients. These components, also called fillers, package bioactive molecules to render them administrable to patients. In the case of Levothyrox tablets, lactose was replaced by mannitol, and citric acid was added as preservative. Levothyrox is principally given to patients with primary (thyroidal), secondary (pituitary) or tertiary (hypothalamic) congenital or acquired hypothyroidism. Widespread reports of side effects apparently linked to the change in the Levothyrox formulation forced the producer to return to the first version of its drug.
These unfortunate events, and others that had been previously reported, deserve serious attention in the scientific community. Harmless components, such as gelatine, carrageenan and many others (including dimethyl sulphoxide at very low dose [1]), are often used as additives to food, cosmetics and also, in some cases, to vaccines. In sensitive individuals, such components, which are appropriately said to be “safe” by the health regulatory agencies, can nevertheless induce unwanted deleterious effects that can affect the general status of patients and healthy (vaccinated) individuals.
Beyond the situation of Levothyrox and examples of inappropriate usages of excipients that have been reported previously in the specialised literature [2] and sometimes widely related in the media, the focus of this short review mainly concerns research made upstream, and the design of preclinical and clinical trials.
When preclinical studies are conducted in animal models, active molecules are generally administered in neutral media, such as NaCl 9 g/l or dimethyl sulphoxide 0.1–3% (v/v), depending on the route of administration and the solubility of the bioactive compound. Successful candidate molecules that finally enter into clinical trials are then formulated to obtain the final medicinal product and administered to healthy volunteers and patients. A number of excipients have been validated by the US Food and Drug Administration (FDA), the European Medicines Agency, the European/US Pharmacopoeia, and other drug agencies. Pharmaceutical excipients are defined in USP General Chapter <1078> Good Manufacturing Practices for Bulk Pharmaceutical Excipients as follows [3]: “Pharmaceutical excipients are substances other than the active pharmaceutical ingredient that have been appropriately evaluated for safety and are intentionally included in a drug delivery system. For example, excipients can aid in the processing of the drug delivery system during its manufacture; protect, support, or enhance stability, bioavailability, or patient acceptability; assist in product identification; and enhance other attributes of the overall safety, effectiveness, or delivery of the drug during storage or use.” A similar definition of excipients is given by the International Pharmaceutical Excipients Council [4]. I refer interested readers to specialised publications reporting exhaustive information on excipients commonly used nowadays and emphasising the advantages and limitations, even some major drawbacks and dangers, displayed by some of them [5–8].
A key decision is selection of the excipients most appropriate for the type of molecule, its inherent pharmacological properties (solubility, stability, pharmacodynamics) and route of administration. As conducting clinical trials is expensive and time-consuming, it is generally impossible to evaluate a variety of excipients in humans. The decisions that are taken are therefore the result of empirical considerations and past experience.
As mentioned above, it is assumed that excipients are inactive ingredients that are added to the active drug to protect the latter from oxidation and too fast degradation at the site of administration, favour its pharmacokinetics, and facilitate its physiological absorption and bioavailability. In some cases, excipients acting as sweeteners can also improve the taste of the active drug. Others are incorporated to colour the final medicinal product to allow its differentiation from other pharmaceuticals and consequently avoid some medication mistakes. These so-called functional properties (e.g., antioxidative preservative, diluent, lubricant, or glidant) clearly require that the excipient interacts, at least transiently, with the bioactive compound. It is undeniable that, through this interaction, the additive can potentially alter the intrinsic properties of the active drug, and even abolish and/or counterbalance its therapeutic activity. To ensure that this is not the case, in addition to the excipient- or vehicle-controlled studies that we routinely include in the preclinical phases, we should also systematically include additional arms in which structurally similar inactive molecules formulated with the same conditioning medium are tested in parallel.
The choice of excipients is therefore central in the drug development process. It is not a step “secondary” to the production of new medicines, and their selection has to be carefully planned at a very early stage, upstream of the research into the beneficial drug effects. A bad choice of excipient can counteract drug development by modifying or masking its activity. Some so-called excipients also display bioactivity themselves, alone or in association with the active component, and these activities can lead to unwanted effects in patients. This is the case, for instance, with castor oil, which is well-accepted as a vehicle, solvent and plasticiser, but which is also prescribed as a laxative. The disaccharide trehalose, is also a well-known excipient, used primarily as a freeze-drying agent. It also exhibit other properties [9] and is used, for example, in ophthalmic solutions to ensuring the protection, hydration and lubrication of the ocular surface (Thealoz Duo®, laboratoires Thea, Clermont-Ferrand, France [10, 11]), or as an agent to improve bone metabolism and prevent osteoporosis [12]. Based on the finding that trehalose stabilises aggregation-prone proteins, this sugar has also been evaluated in proteinopathies such as Huntington’s chorea and Alzheimer’s disease [13–16]. In the case of the P140/Lupuzor™ clinical trial, however, trehalose used as an excipient instead of mannitol had negative influence that could have had serious consequences, but fortunately led only to an unsuccessful trial [17]. At the scientific level, we took advantage of this bad experience to reinforce our knowledge of the mode of action of P140 peptide [18, 19].
A brief history of P140 peptide and its preclinical evaluation in mouse models
A peptide corresponding to the sequence 131–151 of the spliceosomal U1-snRNP 70K protein has been found to contain an epitope recognised by T cells in lupus [20, 21]. On exposure to this peptide, autoreactive CD4+ T cells collected from lupus-prone MRL/lpr mice underwent proliferation and secreted interleukin-2. These mutant mice, derived from original crosses among strains LG, AKR, C3H, and C57BL/6 at the North American Jackson Laboratory, show systemic autoimmunity, massive lymphadenopathy associated with proliferation of aberrant T cells, arthritis, and immune complex glomerulonephritis. They are a model for systemic lupus erythematosus (SLE)-like autoimmune syndromes. Subsequent studies with peptide 131–151 in this mouse model of lupus identified an analogue of the nominal sequence 131–151 in which the serine at position 140 is phosphorylated (hence called P140) [22]. In the widely available MRL/lpr mouse model, lupus-like disease correlates with proteinuria (an indicator of renal failure) and high serum levels of anti-double-stranded DNA antibody (anti-dsDNA Ab), both of which were found to be significantly attenuated by P140 peptide [22], demonstrating the ability of the phosphorylated form of 131–151 to reduce disease progression and severity, and mortality in a severe lupus-prone mouse model (fig. 1A). P140 also lowered the hypercellularity (fig. 1B) that is typically observed in the peripheral blood of MRL/lpr mice [18, 23].
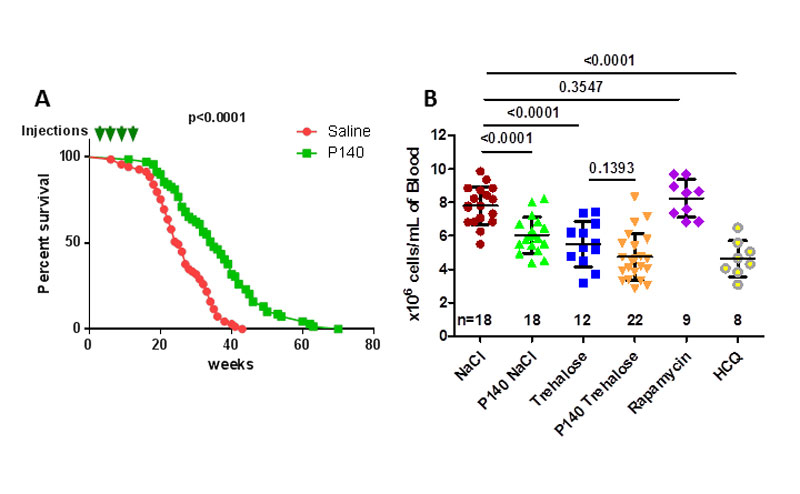
Figure 1
Therapeutic effects of P140 treatment in MRL/lpr mice. A. Long-term effect of P140 peptide on the survival of MRL/lpr mice (69 female mice per group corresponding to eight different experiments performed over 4 years). Pre-autoimmune MRL/lpr mice were treated with either saline or P140 (four doses of 100 µg, given intravenously at 4, 6, 8 and 12 weeks of age). The arrows represent injection timepoints. The median lifespan was 25 weeks in the untreated group vs 35 weeks in the P140-treated group (p <0.0001 using both the log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test). B. Effect of autophagy enhancers and blockers on peripheral hypercellularity in MRL/lpr mice. Female MRL/lpr (11–13 weeks old) each received a single intravenous administration of 100 µg P140 in either saline or 10% trehalose, or 100 µg rapamycin or 100 µg hydroxychloroquine (HCQ). The control groups received saline or trehalose only. The number of leukocytes/ml was evaluated by counting cells 5 days later. Each symbol represents one individual mouse (n, number of mice/group). The results are expressed as the means ± standard deviation. The horizontal bars represent the respective average cell count values. Statistical significance was assessed using the Student’s t-test.
Determination of the mode of action of P140 in lupus individuals has been an area of active investigation in our laboratory for several years, with the MRL/lpr mouse used as a model system. Experiments designed to identify the cellular receptor for P140 demonstrated binding of the peptide to the constitutively expressed chaperone protein HSPA8/HSC70 on the surface of cells [24]. The expression of HSPA8 and of major histocompatibility class-II (MHCII) molecules that are abnormally elevated in splenic B cells from MRL/lpr mice compared with those isolated from CBA/J control mice was decreased on upon treatment with P140 [23]. Since we knew from the existing literature that HSPA8 plays a central role in autophagy and therefore influences the loading of endogenous peptides onto MHCII molecules in the so-called late endosomal MIIC compartment, the effect of P140 on autophagy was examined. We found that autophagy markers accumulated in MRL/lpr B cells treated with P140, suggesting that the peptide decreased the macroautophagic flux that we found to be abnormally elevated in this mouse [23, 25, 26]. Later on, it was demonstrated that the primary target of P140 is chaperone-mediated autophagy (CMA), with implication of macroautophagy processes [27]. CMA was found to be hyperactivated in MRL/lpr lupus mice and significantly attenuated after P140 in vivo treatment. As a working hypothesis, these results, and notably the downregulation of excessive CMA processes, are interpreted to indicate that the mode of action of P140 involves a decrease in autoantigen processing in MRL/lpr antigen-presenting B cells, resulting in a decrease of MHCII expression followed by a reduction of autoreactive T-cell priming and signalling (fig. 2). Past studies, which were not fully understood when our publications came out, have effectively shown that, compared with CD4+ T cells from untreated mice, CD4+ T cells from P140-treated MRL/lpr mice reacted poorly with peptides containing self-T cell epitopes [28]. Consequently, this effect on T helper cells leads to much weaker activation of B cells, which no longer maturate into plasma cells that secrete deleterious antibodies ([29, 30] Schall, Muller et al. unpublished; fig. 2). Step by step, this mode of action of P140 peptide has been confirmed using B cells from normal individuals and patients with SLE [29].
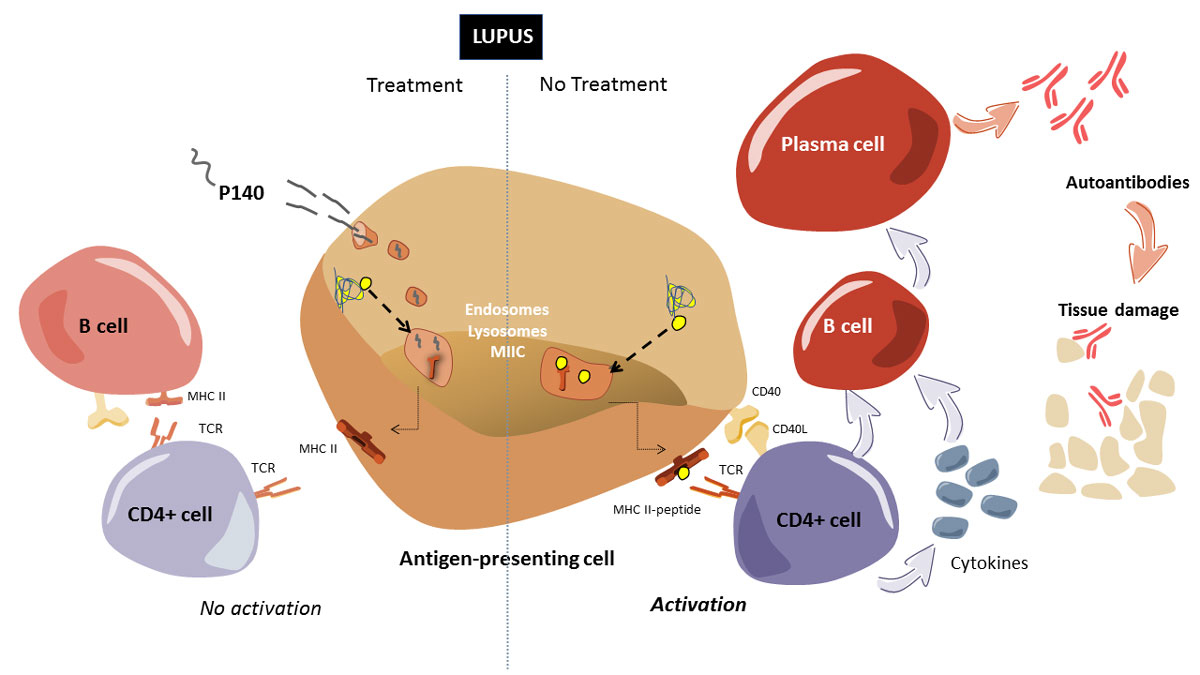
Figure 2
Schematic representation of the mode of action of P140.
Right side: In antigen presenting cells (APCs), lysosomal compartments (lysosomes and autophagosomes) derive to MIIC. In this compartment, antigenic peptides resulting from endogenous processing systems (e.g., autophagy, proteasome; dotted arrow) and exogenous pathways of antigen processing, are loaded onto MHCII molecules, which are then presented to the plasma membrane (thin arrows) and able in this context to recruit/activate CD4+ T cells via their receptor (TCR). This step requires co-stimulatory molecules expressed by T cells such as CD28, which interacts with CD80 (B7.1) and CD86 (B7.2) on the membrane of the APC, or CD40 ligand, which is primarily expressed on activated T cells. In lupus, which is characterised by dysfunction of T cells, polyclonal B-cell activation and failures occurring in several immune compartments, the cascade of events is hyperactivated (differentiation of T cells, autoreactive B cells, maturation into plasma cells and production of autoantibodies). Pro-inflammatory cytokines and chemokines are produced by these hyperactivated lymphocytes. Autoantibodies, alone or linked to self-antigens (forming thus circulating immune complexes) can induce inflammatory reactions after their deposition in targeted tissues. Left side: In the presence of P140 peptide, the whole (hyperactivated) process is slowed. Shortly after its intravenous administration to MRL/lpr mice, P140 enters MRL/lpr B-lymphocytes acting as APCs via a clathrin-dependent endo-lysosomal pathway and accumulates at the lysosomal lumen where it can interact with lysosomal HSPA8 and hamper its chaperone function in CMA (experimentally shown in vitro). This correlates with the observation that P140 decreases the overexpression of LAMP2A (a rate limiting factor in CMA) in MRL/lpr B cells in vivo and of MHCII molecules. Loss of HSPA8 chaperoning function and destabilisation of LAMP2A induced by P140 may thus interfere with the endogenous (auto)antigen processing and loading to MHCII molecules, which are then destabilised, leading to lower (or no more) activation of autoreactive T cells and B cells, and consequently to an improvement of autoimmune condition observed in mice and patients with lupus.
A remarkable observation is that P140-treatment has no effect in MRL/lpr mice on T- or B-cell reactivity to non-self (e.g., viral) peptides. Compared with MRL/lpr mice that received saline, P140-treated MRL/lpr mice normally respond to a viral challenge and mount specific CD4+ T cell and antibody responses of equal magnitude [28]. MRL/lpr mice that are pre-treated or not with P140 peptide develop equally potent IgG antibody responses to a foreign immunogen, ovalbumin (Schall, Muller, et al., unpublished). Based on all these data, we concluded that P140 displays immunomodulatory (but not immunosuppressive) effects on antigen-presenting cells, T and B cells, resulting in fine in the observed therapeutic effects summarised in figures 1 and 2
.
Clinical investigations of P140/Lupuzor™ in lupus patients
Following our very promising results in MRL/lpr mice that develop a severe lupus disease and the regulatory safety investigations that are required before administrating any product to human, an open phase IIa clinical study was performed in 2006 by the young company ImmuPharma (Mulhouse/London). It included 20 patients with lupus in two clinical centres in Bulgaria. This preliminary trial allowed us to determine that a 200-µg dose of P140 peptide conditioned in 5.4% mannitol (Lupuzor™) and given subcutaneously to patients reduced the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score and levels of anti-dsDNA Ab [30]. The peptide was a white to off-white, amorphous powder and was supplied in single-dose glass vials as a lyophilised product for reconstitution. A phase IIb clinical study was then designed and conducted by ImmuPharma [17, 31]. This phase IIb trial was a double-blind, randomised, placebo-controlled study in patients and the purpose was to assess the safety, tolerability, biological activity and efficacy of Lupuzor™ in combination with standard of care therapy (SOC). Patients who met ≥4 of the American College of Rheumatology criteria, had a score of ≥6 on the SLE Disease Activity Index-2000 (SLEDAI-2K), and did not have an A score on the British Isles Lupus Assessment Group (BILAG)-2004 scale were eligible. They were randomly assigned to receive subcutaneous injections of Lupuzor™ (200 μg) every 4 weeks (n = 49; group 1) or every 2 weeks (n = 52; group 2) or placebo (n = 49; group 3) in addition to SOC. For efficacy analysis at week 12, results were available for 147 patients (49, 51 and 47 patients in groups 1, 2 and 3, respectively). The target population (136 patients in the intention-to-treat [ITT] population) comprised patients having a clinical SLEDAI score of at least 6 points at week 0. The clinical SLEDAI score is the SLEDAI-2K score omitting low complement increased DNA binding components. In the total ITT population, 53.1% (26 patients; p = 0.048), 45.1% (23 patients; p = 0.185), and 36.2% (17 patients), respectively, achieved a response at week 12 according to the SLE Response Index (SRI) combined score response, which was the primary efficacy endpoint. In the target population, the results were more impressive: 61.9% (p <0.025), 47.9% (not significant) and 38.6%, respectively, achieved a SRI response at week 12. The most common adverse event was injection-site erythema, which was generally mild. An interim analysis including 114 randomised patients out of the target population was also performed; this demonstrated significantly better efficacy (according to SLEDAI score) of Lupuzor™ 200 µg/month compared with placebo (67.6% vs. 41.5%; p<0.025) at week 12 and (84.2% vs. 45.8%; p<0.025) at week 24. From this pivotal study it was concluded that Lupuzor™ administered three times, once a month, together with standard therapy was safe and well-tolerated, with no significant drug-related adverse events recorded, and was effective in lupus patients [31].
Following this successful phase IIb clinical study, the efficacy of a P140 peptide-based strategy in lupus patients was evaluated in a second phase IIb trial by the biopharmaceutical company Cephalon (Frazer, Pennsylvania). In this study, 10% trehalose was used as excipient (Forigerimod, CEP-33457) instead of mannitol. The clinical data that were reported indicated a negative impact of this excipient on the efficacy of the peptide [17, 19]. In the open label follow-up of 11 patients given Forigerimod following the CEP-33457 phase IIb clinical trial, it was observed that, unlike Lupuzor™, there was no apparent response for several months, the peptide in trehalose even tending to be less effective than placebo. Approximately 6 months later clear-cut improvements in response measures were observed, suggesting that at this stage the negative influence of trehalose was overcome [19]. A phase III clinical study (with a Special Protocol Assessment and “Fast Track” designation from the FDA) is currently being conducted by Immupharma based on Lupuzor™ (P140 in mannitol) in Europe, US and Mauritius.
Trehalose, an activator of autophagy, severely interferes with the beneficial effect of P140 peptide.
We have shown that P140 treatment rescues lupus mice from both macroautophagy and CMA hyperactivation [23, 27, 32]. In parallel, we also have pertinent experimental information from the literature that trehalose acts as an enhancer of autophagy [14, 33–36]. We therefore did a number of control experiments to better define the consequences of using trehalose together with P140 [18, 19]. When compared with the efficacy of P140 in 9 g/l NaCl or 5.4% mannitol, P140 in 10% trehalose was much less efficient at reducing peripheral cell counts (fig. 1B). The effect of the blocker rapamycin and activator hydroxychloroquine (fig. 3) tested in the same conditions is shown for comparison in figure 1B (although it presents some risk, the antimalarial hydroxychloroquine is still widely used for treating lupus patients [37, 38]). To observe a significant effect of P140 in trehalose, the dose of the peptide had to be at least doubled in mice [18]. We noticed a great variability of response in the group of mice that received the P140 in trehalose and, more dramatically, that some mice of this group displayed very low blood cell counts, a feature that was never observed when P140 was conditioned with mannitol or saline [18]. This severe peripheral depletion of blood cells might reflect extensive death of pre-activated cells. P140 in trehalose was also less efficient than P140 in mannitol or saline at reducing human leukocyte antigen (HLA) DR class II molecule expression on the surface of human B cells [19]. HLA-DR is a MHCII cell surface receptor encoded by HLA complex on 6p21.31. The level of circulating plasmocytes expressing CD45R/B220-CD138, as measured with flow cytometry analysis, remained low in all groups of mice with, however, unequal results in mice that received the P140 trehalose formulation. Even more impressive was the finding that trehalose administrated alone, without any peptide, tended to accelerate the course of lupus disease in young MRL/lpr mice (affecting both levels of proteinuria and survival) when compared to saline only, a harmful effect that disappeared after the cessation of trehalose administration [19].

Figure 3 Simplified scheme showing the process of macroautophagy and a partial list of known enhancers and inhibitors that can affect autophagic activity.
Abbreviations not defined in the text: ATG = autophagy related; 3-MA = 3-methyladenine; PE = phosphatidylethanolamine; MAP1LC3B = microtubule-associated proteins 1A/1B light chain 3B; PI3K = phosphoinositide 3-kinase; SMER = small-molecule enhancers; UBL = ubiquitin-like; VPs = vacuolar protein sorting
These data very clearly indicate that trehalose is not recommended and should be systematically avoided in lupus conditions. The disaccharide, like rapamycin, is a potent autophagy enhancer that increases autophagic flux in a variety of mammalian cells (fig. 3). However, in contrast to rapamycin, which acts by the inhibiting mammalian target of rapamycin (mTOR) complex 1, trehalose enhances autophagy via an mTOR-independent pathway [16, 39–44] (fig. 3). The mode of action of trehalose, which remained elusive until recently, has been elucidated in part, which supports some beneficial effects of this molecule in prevention or treatment of diseases related to autophagic deficiency [44–46]. Important new insight into the biological activity of trehalose was generated from studies performed in the context of nonalcoholic fatty liver disease. DeBosh et al. [44, 45] showed in this setting that trehalose reduces the activity of glucose transporters GLUT 1, 2, 3 and 8 (also known as solute carrier 2A or SLC2A family) at the plasma membrane, leading to the activation of adenosine 5′-monophosphate-activated protein kinase (AMPK) and the phosphorylation at Ser317 of autophagy-inducer kinase unc-52-like kinase-1 (ULK1; fig. 3). The GLUT family encompasses fourteen 12-transmembrane domain-containing proteins that mediate uptake of hexoses, such as fructose and glucose, across cellular membranes. GLUT2 and GLUT8, which transport fructose and glucose, are the most abundant hepatic GLUTs. Although deciphering the mode of action of trehalose in other settings is certainly required, as its effects might differ in another cell context [47], nowadays, there is a strong converging evidence to suggest that exogenous sugars, such as trehalose, can impair cellular energetics and induce autophagy [44].
Conclusion
The disaccharide trehalose, a glucose-glucose disaccharide linked by an α,α-1,1-glycoside bond, is not a neutral compound. It is a bioactive molecule able, in particular, to enhance the autophagy flux in certain pathological situations. Used alone or in association with other autophagy enhancers, trehalose might represent a good option to treat conditions where the autophagy status is deficient (as in nonalcoholic fatty liver disease and amyotrophic lateral sclerosis, for example). Conversely, however, it can have unfavourable effects when the autophagy status is abnormally elevated (as in lupus and Crohn’s disease, for example [48]). Close attention should be paid therefore to the selection of excipients used with bioactive compounds.
Our data indicate that P140, a CMA blocker, has an opposite effect to trehalose, reinforcing further our assumptions about its mode of action. P140 diminishes the excessive autophagic flux in the spleen of murine models of lupus [18, 23, 49]. It very significantly improves the clinical course of the disease in sick MRL/lpr mice, with less pronounced lupus-related renal damage and reduced cellular infiltrates in the salivary glands, a feature found in Sjögren’s syndrome [22, 23, 49]. Conversely it aggravates the disease in animal models of amyotrophic lateral sclerosis [48, 50] and showed no effect in models of nonalcoholic steatohepatitis (unpublished).
To conclude, our attention is more generally drawn to the possibility that a number of clinical trials might have failed due to the improper use of additives. Some of these substances, which are added to bioactive compounds to maintain or improve their stability, texture, bioavailability, or just as excipients to fill the sealed container, can considerably affect the metabolic pathways that are supposed to be targeted by the bioactive drug. Since autophagy activity appears to be increased in certain organs, tissues or cells, while abnormally decreased in others in the same individual [49], high doses (or repeated daily doses) of excipients that display multiple effects on autophagy should be used with particular caution. More experimental work is certainly necessary for unravelling the intricacies of this question. This will be time consuming, but the result can be of the greatest value to have fewer misunderstandings and false information.
Acknowledgements
I thank Hélène Jeltsch-David, Nicolas Schall and Mykolas Bendorius for valuable help with the figures. I would also like to acknowledge the support of the TRANSAUTOPHAGY COST Action, CA15138, and the Club francophone de l’autophagie.
References
1
Galvao
J
,
Davis
B
,
Tilley
M
,
Normando
E
,
Duchen
MR
,
Cordeiro
MF
. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014;28(3):1317–30. doi:.https://doi.org/10.1096/fj.13-235440
2
Bochner
F
,
Hooper
WD
,
Tyrer
JH
,
Eadie
MJ
. Factors involved in an outbreak of phenytoin intoxication. J Neurol Sci. 1972;16(4):481–7. doi:.https://doi.org/10.1016/0022-510X(72)90053-6
3USP General Information Chapter <1078> Good manufacturing practices for bulk pharmaceutical excipients. USP 35-NF 30 (United States Pharmacopeial Convention) USP, Rockville, MD, USA, 2011.
4
http://ipec-europe.org/UPLOADS/IPECCompositionGuidefinal.pdf
5
Chaudhari
SP
,
Patil
PS
. Pharmaceutical excipients: a review. Int J Adv Pharm Biol Chem.
2012;1:21–34.
6
Chakraborty
A
,
Dhar
P
. A review on potential of proteins as an excipient for developing a nano-carrier delivery system. Crit Rev Ther Drug Carrier Syst. 2017;34(5):453–88. doi:.https://doi.org/10.1615/CritRevTherDrugCarrierSyst.2017018612
7
Irwin
JJ
,
Pottel
J
,
Zou
L
,
Wen
H
,
Zuk
S
,
Zhang
X
, et al.
A molecular basis for innovation in drug excipients. Clin Pharmacol Ther. 2017;101(3):320–3. doi:.https://doi.org/10.1002/cpt.458
8
Darji
MA
,
Lalge
RM
,
Marathe
SP
,
Mulay
TD
,
Fatima
T
,
Alshammari
A
, et al.
Excipient Stability in Oral Solid Dosage Forms: A Review. AAPS PharmSciTech. 2018;19(1):12–26. doi:.https://doi.org/10.1208/s12249-017-0864-4
9
Ohtake
S
,
Wang
YJ
. Trehalose: current use and future applications. J Pharm Sci. 2011;100(6):2020–53. doi:.https://doi.org/10.1002/jps.22458
10
Luyckx
J
,
Baudouin
C
. Trehalose: an intriguing disaccharide with potential for medical application in ophthalmology. Clin Ophthalmol. 2011;5:577–81.
11
Pinto-Bonilla
JC
,
Del Olmo-Jimeno
A
,
Llovet-Osuna
F
,
Hernández-Galilea
E
. A randomized crossover study comparing trehalose/hyaluronate eyedrops and standard treatment: patient satisfaction in the treatment of dry eye syndrome. Ther Clin Risk Manag. 2015;11:595–603.
12
Zhao
J
,
Wang
S
,
Bao
J
,
Sun
X
,
Zhang
X
,
Zhang
X
, et al.
Trehalose maintains bioactivity and promotes sustained release of BMP-2 from lyophilized CDHA scaffolds for enhanced osteogenesis in vitro and in vivo. PLoS One. 2013;8(1):e54645. doi:.https://doi.org/10.1371/journal.pone.0054645
13
Izmitli
A
,
Schebor
C
,
McGovern
MP
,
Reddy
AS
,
Abbott
NL
,
de Pablo
JJ
. Effect of trehalose on the interaction of Alzheimer’s Aβ-peptide and anionic lipid monolayers. Biochim Biophys Acta. 2011;1808(1):26–33. doi:.https://doi.org/10.1016/j.bbamem.2010.09.024
14
Sarkar
S
,
Davies
JE
,
Huang
Z
,
Tunnacliffe
A
,
Rubinsztein
DC
. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282(8):5641–52. doi:.https://doi.org/10.1074/jbc.M609532200
15
Jain
NK
,
Roy
I
. Effect of trehalose on protein structure. Protein Sci. 2009;18(1):24–36.
16
Vidal
RL
,
Matus
S
,
Bargsted
L
,
Hetz
C
. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. 2014;35(11):583–91. doi:.https://doi.org/10.1016/j.tips.2014.09.002
17
Zimmer
R
,
Wallace
DJ
,
Muller
S
. Randomized, double-blind, placebo-controlled studies of P140 peptide in mannitol (Lupuzor) and trehalose (Forigerimod) in patients with SLE. Arthritis Rheum. 2012;64(Suppl 10):2620.
18
Schall
N
,
Page
N
,
Macri
C
,
Chaloin
O
,
Briand
JP
,
Muller
S
. Peptide-based approaches to treat lupus and other autoimmune diseases. J Autoimmun. 2012;39(3):143–53. doi:.https://doi.org/10.1016/j.jaut.2012.05.016
19
Muller
S
,
Wallace
DJ
. The importance of implementing proper selection of excipients in lupus clinical trials. Lupus. 2014;23(7):609–14. doi:.https://doi.org/10.1177/0961203314525249
20
Monneaux
F
,
Briand
JP
,
Muller
S
. B and T cell immune response to small nuclear ribonucleoprotein particles in lupus mice: autoreactive CD4(+) T cells recognize a T cell epitope located within the RNP80 motif of the 70K protein. Eur J Immunol. 2000;30(8):2191–200. doi:.https://doi.org/10.1002/1521-4141(2000)30:8<2191::AID-IMMU2191>3.0.CO;2-R
21
Monneaux
F
,
Hoebeke
J
,
Sordet
C
,
Nonn
C
,
Briand
JP
,
Maillère
B
, et al.
Selective modulation of CD4+ T cells from lupus patients by a promiscuous, protective peptide analog. J Immunol. 2005;175(9):5839–47. doi:.https://doi.org/10.4049/jimmunol.175.9.5839
22
Monneaux
F
,
Lozano
JM
,
Patarroyo
ME
,
Briand
JP
,
Muller
S
. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MR/lpr mice. Eur J Immunol. 2003;33(2):287–96. doi:.https://doi.org/10.1002/immu.200310002
23
Page
N
,
Gros
F
,
Schall
N
,
Décossas
M
,
Bagnard
D
,
Briand
JP
, et al.
HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann Rheum Dis. 2011;70(5):837–43. doi:.https://doi.org/10.1136/ard.2010.139832
24
Page
N
,
Schall
N
,
Strub
JM
,
Quinternet
M
,
Chaloin
O
,
Décossas
M
, et al.
The spliceosomal phosphopeptide P140 controls the lupus disease by interacting with the HSC70 protein and via a mechanism mediated by gammadelta T cells. PLoS One. 2009;4(4):e5273. doi:.https://doi.org/10.1371/journal.pone.0005273
25
Page
N
,
Gros
F
,
Schall
N
,
Briand
JP
,
Muller
S
. A therapeutic peptide in lupus alters autophagic processes and stability of MHCII molecules in MRL/lpr B cells. Autophagy. 2011;7(5):539–40. doi:.https://doi.org/10.4161/auto.7.5.14845
26
Gros
F
,
Arnold
J
,
Page
N
,
Décossas
M
,
Korganow
A-S
,
Martin
T
, et al.
Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8(7):1113–23. doi:.https://doi.org/10.4161/auto.20275
27
Macri
C
,
Wang
F
,
Tasset
I
,
Schall
N
,
Page
N
,
Briand
JP
, et al.
Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy. 2015;11(3):472–86. doi:.https://doi.org/10.1080/15548627.2015.1017179
28
Monneaux
F
,
Parietti
V
,
Briand
JP
,
Muller
S
. Importance of spliceosomal RNP1 motif for intermolecular T-B cell spreading and tolerance restoration in lupus. Arthritis Res Ther. 2007;9(5):R111. doi:.https://doi.org/10.1186/ar2317
29
Wilhelm
M
,
Wang
F
,
Schall
N
,
Kleinmann
J-F
,
Faludi
M
,
Nashi
EP
, et al.
Lupus regulator peptide P140 represses B-cell antigen differentiation by reducing HLA class II overexpression. Arthritis Rheum. 2018. [Epub ahead of print.] doi:.https://doi.org/10.1002/art.40470
30
Muller
S
,
Monneaux
F
,
Schall
N
,
Rashkov
RK
,
Oparanov
BA
,
Wiesel
P
, et al.
Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis Rheum. 2008;58(12):3873–83. doi:.https://doi.org/10.1002/art.24027
31
Zimmer
R
,
Scherbarth
HR
,
Rillo
OL
,
Gomez-Reino
JJ
,
Muller
S
. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann Rheum Dis. 2013;72(11):1830–5. doi:.https://doi.org/10.1136/annrheumdis-2012-202460
32
Wang
F
,
Muller
S
. Manipulating autophagic processes in autoimmune diseases: a special focus on modulating chaperone-mediated autophagy, an emerging therapeutic target. Front Immunol. 2015;6:252. doi:.https://doi.org/10.3389/fimmu.2015.00252
33
Aguib
Y
,
Heiseke
A
,
Gilch
S
,
Riemer
C
,
Baier
M
,
Ertmer
A
, et al.
Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5(3):361–9. doi:.https://doi.org/10.4161/auto.5.3.7662
34
Renna
M
,
Jimenez-Sanchez
M
,
Sarkar
S
,
Rubinsztein
DC
. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010;285(15):11061–7. doi:.https://doi.org/10.1074/jbc.R109.072181
35
Castillo
K
,
Nassif
M
,
Valenzuela
V
,
Rojas
F
,
Matus
S
,
Mercado
G
, et al.
Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9(9):1308–20. doi:.https://doi.org/10.4161/auto.25188
36
Fernandez-Estevez
MA
,
Casarejos
MJ
,
López Sendon
J
,
Garcia Caldentey
J
,
Ruiz
C
,
Gomez
A
, et al.
Trehalose reverses cell malfunction in fibroblasts from normal and Huntington’s disease patients caused by proteosome inhibition. PLoS One. 2014;9(2):e90202. doi:.https://doi.org/10.1371/journal.pone.0090202
37
Sumpter
MD
,
Tatro
LS
,
Stoecker
WV
,
Rader
RK
. Evidence for risk of cardiomyopathy with hydroxychloroquine. Lupus. 2012;21(14):1594–6. doi:.https://doi.org/10.1177/0961203312462757
38
Tselios
K
,
Gladman
DD
,
Su
J
,
Urowitz
MB
. Antimalarials as a risk factor for elevated muscle enzymes in systemic lupus erythematosus. Lupus. 2016;25(5):532–5. doi:.https://doi.org/10.1177/0961203315617845
39
Sarkar
S
,
Rubinsztein
DC
. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4(9):895–901. doi:.https://doi.org/10.1039/b804606a
40
Mizushima
N
,
Yoshimori
T
,
Levine
B
. Methods in mammalian autophagy research. Cell. 2010;140(3):313–26. doi:.https://doi.org/10.1016/j.cell.2010.01.028
41
Fleming
A
,
Noda
T
,
Yoshimori
T
,
Rubinsztein
DC
. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7(1):9–17. doi:.https://doi.org/10.1038/nchembio.500
42
Gros
F
,
Muller
S
. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br J Pharmacol. 2014;171(19):4337–59. doi:.https://doi.org/10.1111/bph.12792
43
Levine
B
,
Packer
M
,
Codogno
P
. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. doi:.https://doi.org/10.1172/JCI73938
44
DeBosch
BJ
,
Heitmeier
MR
,
Mayer
AL
,
Higgins
CB
,
Crowley
JR
,
Kraft
TE
, et al.
Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9(416):ra21. doi:.https://doi.org/10.1126/scisignal.aac5472
45
Mayer
AL
,
Higgins
CB
,
Heitmeier
MR
,
Kraft
TE
,
Qian
X
,
Crowley
JR
, et al.
SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep. 2016;6(1):38586. doi:.https://doi.org/10.1038/srep38586
46
Mardones
P
,
Rubinsztein
DC
,
Hetz
C
. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci Signal. 2016;9(416):fs2. doi:.https://doi.org/10.1126/scisignal.aaf1937
47
Yoon
YS
,
Cho
ED
,
Jung Ahn
W
,
Won Lee
K
,
Lee
SJ
,
Lee
HJ
. Is trehalose an autophagic inducer? Unraveling the roles of non-reducing disaccharides on autophagic flux and alpha-synuclein aggregation. Cell Death Dis. 2017;8(10):e3091. doi:.https://doi.org/10.1038/cddis.2017.501
48
Muller
S
,
Brun
S
,
René
F
,
de Sèze
J
,
Loeffler
JP
,
Jeltsch-David
H
. Autophagy in neuroinflammatory diseases. Autoimmun Rev. 2017;16(8):856–74. doi:.https://doi.org/10.1016/j.autrev.2017.05.015
49
Li
B
,
Wang
F
,
Schall
N
,
Muller
S
. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J Autoimmun. 2018;90:132–45. doi:.https://doi.org/10.1016/j.jaut.2018.02.005
50
Baek
KH
,
Park
J
,
Shin
I
. Autophagy-regulating small molecules and their therapeutic applications. Chem Soc Rev. 2012;41(8):3245–63. doi:.https://doi.org/10.1039/c2cs15328a