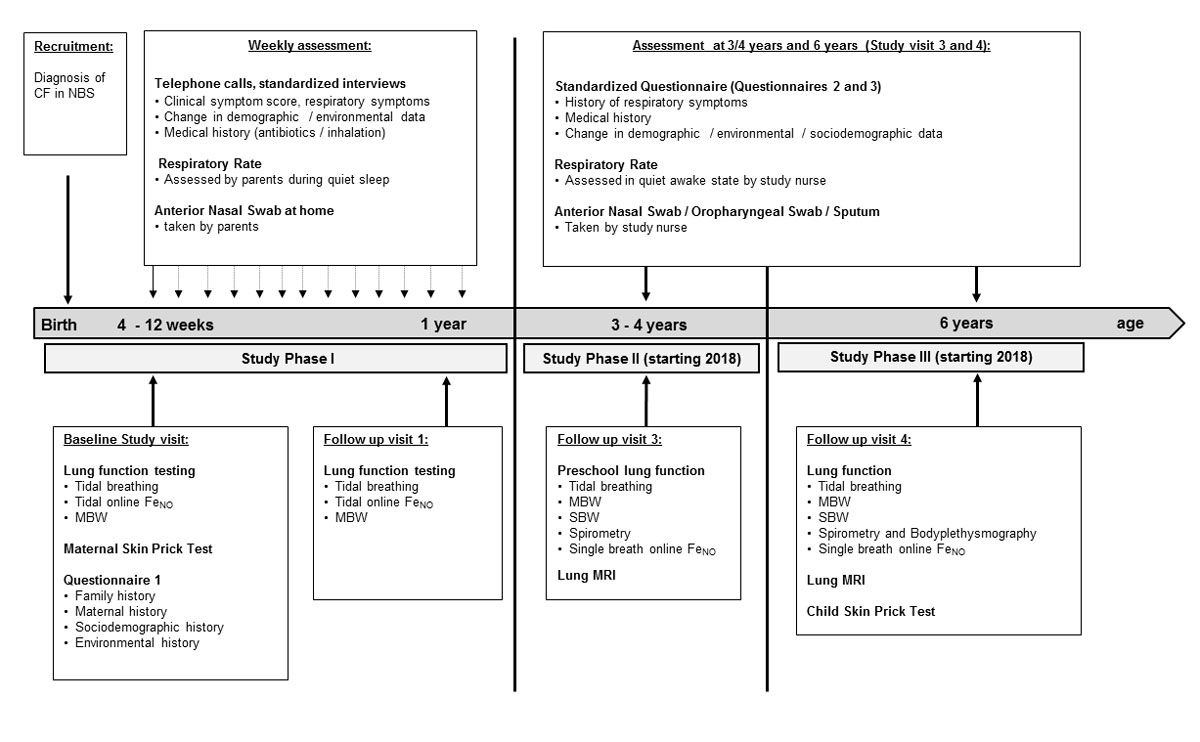
Figure 1 Design of the SCILD cohort study – flow chart of data collection.
NBS = new-born screening; FeNO = exhaled nitric oxide; MBW = multiple breath washout, SBW = single breath washout; MRI = magnet resonance imaging
DOI: https://doi.org/10.4414/smw.2018.14618
Cystic fibrosis is the most common lethal inherited disease affecting Caucasian populations, with a prevalence of approximately 1:3300 [1]. It is a multisystem disorder that affects exocrine glands including the lung, liver, pancreas and intestine [2]. Respiratory morbidity is the leading cause of death, and despite improved survival rates the current median survival age of patients with cystic fibrosis is approximately 40 years [3].
The establishment of new-born screening for cystic fibrosis allows diagnosis before the onset of clinical signs [4, 5]. In Switzerland, new-born screening for cystic fibrosis was implemented nationwide in 2011 [6]. As not all patients with cystic fibrosis display overt symptoms during the first years of life, new-born screening allows early monitoring and treatment of preclinical disease, and a better understanding of the early disease pathogenesis. It is now well recognised that in the first months of life in individuals with cystic fibrosis, pulmonary inflammation, infection and abnormal lung function are present prior to clinically diagnosed respiratory illness [7–13]. Therefore, understanding the pathophysiology of early cystic fibrosis lung disease is essential to delay its onset and progression, implement early therapeutic interventions and further improve outcomes.
The Swiss Cystic Fibrosis Infant Lung Development Cohort (SCILD) was established in 2011 after implementation of the Swiss new-born screening for cystic fibrosis and aims to examine the initiating events of cystic fibrosis lung disease during infancy and their influence on the trajectory of disease progression throughout early childhood. The SCILD cohort was set up in a comparable way to the Bern Basel Infant Lung Development (BILD) cohort study [14], a prospective birth cohort study of unselected healthy infants, which was established in 1999. The BILD and SCILD cohort studies both aim to investigate the physiological properties of the respiratory system, and the environmental and genetic risk factors that influence lung development in individuals from infancy throughout childhood. The SCILD cohort surveillance protocol has been only slightly modified from the BILD cohort, and healthy infants from the BILD cohort serve as controls for longitudinal measurements performed throughout the study. The following paragraphs provide details on the study design and data collection, which have not been published previously, and give a short summary of current publications from the SCILD cohort. The reader will thus gain a comprehensive and detailed overview of the study.
Infants have been recruited since 2011 following diagnosis with cystic fibrosis by new-born screening in the cystic fibrosis centres of Aarau, Basel, Bellinzona, Bern, Geneva, Lausanne, Lucerne, St Gallen and Zurich, therefore covering all of Switzerland. After diagnosis and recruitment at the treating centre, infant lung function testing is proposed to all parents of newly diagnosed infants with cystic fibrosis (Task Force for cystic fibrosis new-born screening on behalf of the Swiss Working Group for Cystic Fibrosis). The University Children’s Hospital Bern is the only centre in Switzerland currently performing infant lung function measurements in infants with cystic fibrosis. Study enrolment occurs on the day of lung function testing at the age of around 10 weeks, irrespective of prior clinical symptoms. The study is performed in Bern and was approved by the Ethics Committee Bern, Switzerland. Written informed consent is obtained from the parents. Exclusion criteria include the need for respiratory support for more than 3 days after birth, severe comorbidities or known diseases other than cystic fibrosis, maternal drug abuse, other known severe maternal diseases and severe problems of communication.
The study design of the SCILD cohort is divided into three phases as shown in figure 1. Details on data assessment at each study time point are displayed in table 1.
Figure 1 Design of the SCILD cohort study – flow chart of data collection.
NBS = new-born screening; FeNO = exhaled nitric oxide; MBW = multiple breath washout, SBW = single breath washout; MRI = magnet resonance imaging
Table 1 Details of data collection from the different sources in the SCILD study.
| Sources | Details of data collection |
|---|---|
| 1. Standardised questionnaires [14–17] | |
| Questionnaire 1 | • Pets and exact type of pet at home, during/after pregnancy • Type of heating, type of stove, chimney, open fire-place • Sleeping environment child (mattress, encasement, sheepskin rug use) • Damage due to damp at home, assessment of exposure to mould • Maternal active or passive smoking, ETS smoke exposure, number of cigarettes, smoking cessation and first year of life • Maternal intake of coffee/tee, caffeine containing soft drinks, vitamin supplement intake, fruit intake • Maternal treatment with antibiotics, steroid treatment during pregnancy • Respiratory infections, gastrointestinal infections, urinary tract infections, other infections during pregnancy • Paternal active smoking, number of cigarettes, smoking cessation during pregnancy and first year of life |
| Questionnaires 2–5 | • Infections, colds (especially upper and lower respiratory tract, ear, nose, throat), number and duration • Exacerbations of disease (number, severity)* • Number, reason and course of hospitalisations† • Medication, especially antibiotics since last study visit, alternative medications and remedies used • Quality of life [17]: limits in physical activity/ ability to participate in social activities / mental health (depression, listlessness) • Atopic disease: atopic rhinitis or conjunctivitis, asthma (severity), atopic dermatitis • Behavioural problems, enuresis (primary, secondary, diurnal, nocturnal) • Level and impairment of indoor and outdoor activity, type of activity • Pets and exact type of pet at home • Farming exposure, animal type • Traffic exposure at home (trucks) • Type of heating, type of stove, chimney, open fire-place • Living condition (type of dwelling, rural/urban, number of rooms, number of people in household) • Diet (fruit, vegetables, sweets, chocolate, fast food, snacks, unpasteurized milk) • Maternal and paternal active or passive smoking, ETS exposure, number of cigarettes, smoking cessation until first major follow-up at age 3 and 6 years |
| 2. Lung function measurements | |
| Infant lung function | 1. Tidal breathing measurements [18, 19]: • Ultrasonic flowmeter (Spiroson®, EcoMedics AG, Duernten, Switzerland) with infant face mask • Main outcome parameters: tidal volume, minute ventilation, respiratory rate 2. Measurement of FENO [20]: • Rapid-response chemoluminescence analyser (CLD 88, EcoMedics AG, Duernten, Switzerland), with a range of 0–100 ppb, infant face mask • Breath-by-breath measurement • Main outcome parameter: mean FENO 3. Multiple breath washouts (SF6) [21]: • Ultrasonic flowmeter (see above), infant face mask • Main outcome parameters: lung volume (FRC) and ventilation inhomogeneity (LCI) |
| Preschool lung function | 1. Tidal breathing measurements: • Quiet tidal breathing • Ultrasonic flowmeter with mouth-piece, filter and nasal clamp • Main outcome parameters: minute ventilation (tidal volume multiplied by respiratory rate) and expiratory flow 2. Measurement of FENO [20]: • Rapid-response chemoluminescence analyser (CLD 88, EcoMedics AG, Duernten, Switzerland), with a range of 0–100 ppb • Breath-by-breath measurement and single-breath manoeuvre, with mouth-piece, no nasal clamp • Main outcome parameter: mean FENO 3. Multiple breath washouts (N2): • Ultrasonic flowmeter (see above), mouthpiece, filter, nasal clamp • Main outcome parameters: lung volume (FRC) and ventilation inhomogeneity (LCI) 4. Single breath washouts (helium, SF6) • Ultrasonic flowmeter (see above), mouthpiece, filter, nasal clamp • Main outcome parameters: Sacin, Scond 5. Spirometry (body plethysmography): • MasterScreen Body, Jaeger, Germany, mouthpiece, filter, nasal clamp • Main outcome parameters: lung volume (FRC, intrathoracic gas volume), forced expiratory flows and volumes |
| 3. Telephone interviews | • Respiratory symptoms (including cough and wheeze during day and night, difficulty breathing, reduced activity) • Standardised score to group symptoms into four levels according to severity and with high sensitivity for lower respiratory tract infections • Main outcome parameter: weeks with severe respiratory symptoms (defined as total number of weeks with day/night score of ≥3 • Any changes in environment such as moving, pets, smoking habits, childcare, breastfeeding, nutrition • Medication, e.g., exact time point, kind and duration of antibiotics |
| 3. Nasal swabs | • Microbiota analysis: microbiota assessment with PCR amplification of the 16S ribosomal RNA (rRNA) gene and 454 amplicon sequencing as described previously [22, 23] • Virological analysis: real-time (7 duplex) PCR will be used to used detect respiratory viruses as previously described [24] |
| 4. Respiratory rate measurements | • Assessed over 60 seconds by hand on infant’s chest in quiet sleep state • Assessed over 60 seconds by a study nurse in awake children at study visit 3 and 4 |
| 6. Skin prick test | • Dog dander, cat dander, Dermatophagoides pteronyssinus, mixed tree pollens, mixed grass pollens, Alternaria tenuis, positive control (histamine), negative control (NaCl) (Allergomed, Switzerland) |
| 7. Magnetic resonance imaging | • Starting in 2018 • Matrix pencil decomposition, a new functional MRI method • Morphological MRI • Main outcome: ventilation and perfusion indices |
ETS = environmental tobacco smoke; FENO = exhaled nitric oxide; FRC = functional residual capacity; LCI = lung clearance index, MRI = magnetic resonance imaging; PCR = polymerase chain-reaction * Pulmonary exacerbation is defined as any increase in respiratory symptoms from baseline that require a doctoral visit and additional treatment and/or decline in lung function measurement. † Specific reasons for hospitalisations are documented including respiratory, gastrointestinal problems or any other.
The first study phase begins with recruitment after infants have been diagnosed with cystic fibrosis following new-born screening and continues throughout the first year of life. Informed written consent is collected at the baseline study visit when the infants are around 10 weeks of age. Throughout phase 1, a study nurse contacts parents weekly by telephone. Phase 1 concludes following the first follow-up visit, which is scheduled for when the child reaches 1 year of age.
The second study phase starts after the first follow-up visit at age one and continues until the second follow up visit at the age of 3 to 4 years for all infants born after 2014. Respiratory and environmental history during phase 2 will be assessed retrospectively at the second follow up visit.
The third study phase starts after the second follow up at age 3 to 4 years and continues until the third follow up visit at the age of 6 years. Respiratory and environmental history during Phase 3 will be assessed retrospectively at the third follow-up visit.
Data acquisition is based on the study design of the BILD cohort [14]. Data are derived from the following sources: (1) questionnaires, (2) lung function measurements, (3) telephone interviews, (4) nasal swab samples, 5) respiratory rate measurements, (6) skin prick tests, and (7) functional and morphological magnetic resonance imaging (MRI) (planned for 2018). Details on the specific time points for assessments are displayed in figure 1. Further details of data collection are displayed in table 1.
We attempt to access data from each source in every participant; however, it is not mandatory to participate in all aspects of data collection, such as weekly nasal swab sampling and/or respiratory rate measurements, and infant lung function tests are also optional. The latter did not yet apply to any potential participants. In addition, they do not have to attend every study phase.
At end of 2017, 70 infants with cystic fibrosis had been recruited in the SCILD study. Of these, 56 (80%) have completed the first year of study, and 14 (20%) have not yet reached one year of age and are still completing the first study phase.
Details on demographic data can be found in table 2. To date there have been no infants lost to follow up in phase 1 of the study and all infants completed the first year of the study. The second and third follow-up visits will begin in 2018. Recruitment is ongoing, with approximately 9 to 13 newly diagnosed infants recruited per year (incidence of cystic fibrosis diagnosed by new-born screening approximately 25/year [1]); for details see table 3.
Table 2 Demographic details of the study population.
| Mean | Standard deviation | Total number of measurements | |
|---|---|---|---|
| Gestational age (weeks) | 39.2 | 1.5 | 70 |
| Birth height (cm) | 49 | 1.8 | 70 |
| Birth weight (kg) | 3.2 | 0.4 | 70 |
| Age at infant lung function testing (weeks) | 6 | 2.3 | 68* |
| Weekly telephone calls†
(calls per infant) |
38.4 | 13.7 | 2614 |
| Weekly respiratory rate†
(measurements per infant) |
30.0 | 16.8 | 1915 |
| Weekly nasal swabs†
(per infant) |
28.7 | 17.1 | 1955 |
Results are displayed as total numbers if not stated otherwise. To date, 70 infants with cystic fibrosis have been included in the study, of whom 56 have completed study phase 1. * In two infants lung function measurements were not successful, because no quiet, regular sleep episode could be obtained. † All infants are included, standard deviations are large, as some infants just started the study period and data collection is still ongoing
Table 3 Numbers of recruited participants in the SCILD cohort (2011–2017).
| Year |
Total
(2011–2016) |
|||||||
|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | ||
| Infants with CF detected by NBS* | 56 | 19 | 35 | 20 | 23 | n.a.‡ | 153 | |
| Recruited after positive NBS† | 9 | 12 | 8 | 9 | 8 | 10 | 14 | 70 |
| Completed study phase 1 |
9 | 12 | 8 | 9 | 8 | 10 | – | 56 |
| Drop-outs during study phase 1 | – | – | – | – | – | – | – | – |
| Phase 2 (starts in 2018) |
– | – | – | – | 2018/19 | 2019/20 | 2020/21 | |
| Phase 3 (starts in 2018) |
2018 | 2018 | 2019 | 2020 | 2021 | 2022 | 2023 | |
CF = cystic fibrosis; NBS = new-born screening * Infants that were diagnosed with CF throughout Switzerland † Infants that were recruited in the SCILD study ‡ Numbers for 2017 are not yet available
Publications to date have focused primarily on longitudinal microbial and viral analyses of the nasal swab material and infant lung function measurements over the first year of life [24, 39, 40, 41]. All studies have included healthy infants from the BILD cohort as control group.
The microbiota is known to play an important role in human health, with important influences on early adaptive immunity and innate resistance to infection [42, 43]. Recent studies suggest that diversity of the microbiota is lower in children with cystic fibrosis than in healthy children, and diversity decreases following the onset of chronic Pseudomonas infection [44–46]. However, little is known about the entire composition of the upper respiratory tract microbiota in infants with cystic fibrosis. We analysed the nasal microbiota longitudinally in 30 infants with cystic fibrosis (461 samples) compared with 47 healthy controls (872 samples). We identified compositional differences in the microbiota of infants with cystic fibrosis compared with healthy controls [39]. Furthermore, a disordered microbiota was found following antibiotic administration. Our data indicate that the microbiota is altered in infants with cystic fibrosis and that early antibiotic therapy influences the microbiota. These findings can be used to inform future studies on the effect of antibiotic treatment on the microbiota in infants with cystic fibrosis, and might be of importance in prevention of early disease progression [39].
Acute viral respiratory tract infections in children and adults with cystic fibrosis play a significant role in morbidity and mortality [47]. However, data about viral detection during infancy remains scarce [48]. We assessed 12 different respiratory viruses in 31 infants with cystic fibrosis (665 samples) and 32 healthy infants (712 samples) in a prospective longitudinal study during the first year of life. By weekly monitoring of respiratory symptoms, we could distinguish between asymptomatic and symptomatic viral detection. In our study, viral detection was not more frequent in cystic fibrosis infants than in healthy controls, and infants with cystic fibrosis were less symptomatic when a virus was present. We can only speculate about underlying reasons, but local epithelial properties, immunological mechanisms and even early treatment could play a role in viral detection and symptoms at the time of detection in early cystic fibrosis lung disease [24].
It is unknown at what age lung function impairment may arise in children with cystic fibrosis. We assessed lung function (multiple breath washout and tidal breathing parameters) shortly after birth in 53 infants with cystic fibrosis diagnosed by new-born screening and 57 healthy controls. We reported that more than 40% of eight-week-old infants with cystic fibrosis showed abnormal lung function independent of clinical symptoms and previous therapeutic approaches. Ventilation inhomogeneity or hyperinflation may serve as noninvasive markers for monitoring cystic fibrosis lung disease and specific treatment effects, and could thus be used as outcome parameters for future intervention studies in this age group [40].
The fractional concentration of exhaled nitric oxide (FENO) is a well-known biomarker for airway inflammation and is elevated in a number of inflammatory lung diseases [49]. However, despite chronic severe airway inflammation in patients with cystic fibrosis, FENO levels are decreased [50]. In order to understand whether reduced FENO in cystic fibrosis airways is primarily related to the loss of function of the cystic fibrosis transmembrane regulator (CFTR) or an epiphenomenon of chronic inflammation, we measured FENO in infants with cystic fibrosis and healthy controls at the age of 5 to 12 weeks prior to first respiratory infection. Airway FENO was reduced in young infants with cystic fibrosis, and the effect was more pronounced in infants with two copies of class I and/or II mutations and thus without CFTR function [41]. Hence, low FENO levels in cystic fibrosis airways shortly after birth are likely to be associated with underlying CFTR dysfunction. As reduced levels of FENO have been linked to a number of adverse effects, including effects on the defence mechanisms of the respiratory tract, this finding might open up a new chapter of research in the field of early FENO measurements in patients with cystic fibrosis.
The SCILD cohort includes infants with cystic fibrosis from early infancy until childhood recruited throughout Switzerland. Therefore, the course of the disease can be prospectively followed throughout early childhood. The dataset of the SCILD cohort combines lung function data, collection of environmental and sociodemographic factors, documentation of respiratory symptoms, and microbiological analyses. Lung function surveillance at several time points (after birth, at 1, 3 to 4, and 6 years of age) allows data collection during important phases of lung development. Weekly questionnaires and surveillance during the first year of life allow close monitoring during both asymptomatic and symptomatic episodes of the disease. This study design has resulted in over 2500 data points about respiratory health being collected to date and almost 2000 nasal samples being collected. This design not only allows tracking of the cystic fibrosis lung disease independent of clinical status, but also surveillance of early disease prior to severe clinical symptoms.
The SCILD cohort is the only prospective birth cohort investigating cystic fibrosis disease in Switzerland and the third ever established in the world, in addition to the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) and the London Cystic Fibrosis Collaboration (LCFC) [51]. The latter cohort, however, recruits from centres in London only. Whereas all three cohorts focus on lung function measurements and aim to find diagnostic tools to monitor early lung impairment [52–54], novel aspects of the SCILD study are the close monitoring by weekly nasal sampling and phone calls. With the newly developed MRI imaging technique, there is no need for radiation or sedation [37, 55]. Furthermore, results are always compared with a contemporary healthy control group, the BILD cohort, for whom data are collected at the same study centre with a similar study design and setting. This comparability throughout the study period is an important feature of our study population.
The incidence of cystic fibrosis in Switzerland is approximately 1:3300 live births [1], thus the number of infants diagnosed with cystic fibrosis per year is relatively low in Switzerland (incidence approximately 25/year [1]). In previous years, logistical and disease-specific factors (e.g., prolonged hospital stay of infants due to meconium ileus, parents who are not able to travel to Bern for the measurements because of long travel distances, delayed diagnosis of cystic fibrosis after the age of 15 weeks) have led to a lower than expected number of recruited patients, which is a weakness of the study. Specific questions (e.g., stratifying by the different causative mutations) might not be possible until additional subjects are recruited, as numbers are too low for certain subgroup analyses. However, continued recruitment of newly diagnosed infants with cystic fibrosis and healthy controls throughout the planned follow-up period will ensure numbers sufficient to address the aim to investigate the physiological properties of the respiratory system and detect risk factors that influence lung development.
We appreciate the contribution of S. Lüscher, S. Schmid, G. Wirz, M. Graf, L. Beul-Beguin and K. Röthlisberger (Division of Respiratory Medicine, Department of Pediatrics, Inselspital and University of Bern, Bern, Switzerland) to the data collection.
The SCILD cohort study is funded by project grants from the Swiss National Science Foundation (SNF), the Swiss Society for Cystic Fibrosis (CFCH), the Botnar Foundation and Ambizione. The funders have no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The University Children`s Hospital in Bern, Switzerland, is the coordinating study centre and provides all infrastructure.
PL has received personal fees from Gilead, Novartis, Polyphor, Roche, Schwabe, Vertex, Vifor and Zambon.
1Jurca M, de Jong C, Kuehni C. Evaluationsbericht 2016 CF-Neugeborenen Screening. Institut für Sozial und Präventivmedizin, Uni Bern. 2017.
2 O’Sullivan BP , Freedman SD . Cystic fibrosis. Lancet. 2009;373(9678):1891–904. doi:.https://doi.org/10.1016/S0140-6736(09)60327-5
3 Ratjen F , Döring G . Cystic fibrosis. Lancet. 2003;361(9358):681–9. doi:.https://doi.org/10.1016/S0140-6736(03)12567-6
4 Comeau AM , Accurso FJ , White TB , Campbell PW, 3rd , Hoffman G , Parad RB , et al.; Cystic Fibrosis Foundation. Guidelines for implementation of cystic fibrosis newborn screening programs: Cystic Fibrosis Foundation workshop report. Pediatrics. 2007;119(2):e495–518. doi:.https://doi.org/10.1542/peds.2006-1993
5 Balfour-Lynn IM . Newborn screening for cystic fibrosis: evidence for benefit. Arch Dis Child. 2008;93(1):7–10. doi:.https://doi.org/10.1136/adc.2007.115832
6 Rueegg CS , Kuehni CE , Gallati S , Baumgartner M , Torresani T , Barben J ; Swiss CF Screening Task Force. One-year evaluation of a neonatal screening program for cystic fibrosis in Switzerland. Dtsch Arztebl Int. 2013;110(20):356–63.
7 Armstrong DS , Grimwood K , Carzino R , Carlin JB , Olinsky A , Phelan PD . Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ. 1995;310(6994):1571–2. doi:.https://doi.org/10.1136/bmj.310.6994.1571
8 Khan TZ , Wagener JS , Bost T , Martinez J , Accurso FJ , Riches DW . Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151(4):1075–82.
9 Linnane BM , Hall GL , Nolan G , Brennan S , Stick SM , Sly PD , et al.; AREST-CF. Lung function in infants with cystic fibrosis diagnosed by newborn screening. Am J Respir Crit Care Med. 2008;178(12):1238–44. doi:.https://doi.org/10.1164/rccm.200804-551OC
10 Ranganathan SC , Dezateux C , Bush A , Carr SB , Castle RA , Madge S , et al.; London Collaborative Cystic Fibrosis Group. Airway function in infants newly diagnosed with cystic fibrosis. Lancet. 2001;358(9297):1964–5. doi:.https://doi.org/10.1016/S0140-6736(01)06970-7
11 Stick SM , Brennan S , Murray C , Douglas T , von Ungern-Sternberg BS , Garratt LW , et al.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF). Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155(5):623–8.e1. doi:.https://doi.org/10.1016/j.jpeds.2009.05.005
12 Ramsey BW , Banks-Schlegel S , Accurso FJ , Boucher RC , Cutting GR , Engelhardt JF , et al. Future directions in early cystic fibrosis lung disease research: an NHLBI workshop report. Am J Respir Crit Care Med. 2012;185(8):887–92. doi:.https://doi.org/10.1164/rccm.201111-2068WS
13 Sly PD , Brennan S , Gangell C , de Klerk N , Murray C , Mott L , et al.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF). Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180(2):146–52. doi:.https://doi.org/10.1164/rccm.200901-0069OC
14 Fuchs O , Latzin P , Kuehni CE , Frey U . Cohort profile: the Bern infant lung development cohort. Int J Epidemiol. 2012;41(2):366–76. doi:.https://doi.org/10.1093/ije/dyq239
15 Kuehni CE , Brooke AM , Strippoli MP , Spycher BD , Davis A , Silverman M . Cohort profile: the Leicester respiratory cohorts. Int J Epidemiol. 2007;36(5):977–85. doi:.https://doi.org/10.1093/ije/dym090
16 Strippoli MP , Silverman M , Michel G , Kuehni CE . A parent-completed respiratory questionnaire for 1-year-old children: repeatability. Arch Dis Child. 2007;92(10):861–5. doi:.https://doi.org/10.1136/adc.2007.117978
17 Ronit A , Gelpi M , Argentiero J , Mathiesen I , Nielsen SD , Pressler T , et al. Electronic applications for the CFQ-R scoring. Respir Res. 2017;18(1):108. doi:.https://doi.org/10.1186/s12931-017-0592-z
18 Latzin P , Roosli M , Huss A , Kuehni CE , Frey U . Air pollution during pregnancy and lung function in newborns: a birth cohort study. Eur Respir J. 2009;33(3):594–603.
19 Latzin P , Roth S , Thamrin C , Hutten GJ , Pramana I , Kuehni CE , et al. Lung volume, breathing pattern and ventilation inhomogeneity in preterm and term infants. PLoS One. 2009;4(2):e4635. doi:.https://doi.org/10.1371/journal.pone.0004635
20 Latzin P , Kuehni CE , Baldwin DN , Roiha HL , Casaulta C , Frey U . Elevated exhaled nitric oxide in newborns of atopic mothers precedes respiratory symptoms. Am J Respir Crit Care Med. 2006;174(12):1292–8. doi:.https://doi.org/10.1164/rccm.200606-782OC
21 Latzin P , Sauteur L , Thamrin C , Schibler A , Baldwin D , Hutten GJ , et al. Optimized temperature and deadspace correction improve analysis of multiple breath washout measurements by ultrasonic flowmeter in infants. Pediatr Pulmonol. 2007;42(10):888–97. doi:.https://doi.org/10.1002/ppul.20674
22 Hilty M , Qi W , Brugger SD , Frei L , Agyeman P , Frey PM , et al. Nasopharyngeal microbiota in infants with acute otitis media. J Infect Dis. 2012;205(7):1048–55. doi:.https://doi.org/10.1093/infdis/jis024
23 Mika M , Mack I , Korten I , Qi W , Aebi S , Frey U , et al. Dynamics of the nasal microbiota in infancy: a prospective cohort study. J Allergy Clin Immunol. 2015;135(4):905–12.e11. doi:.https://doi.org/10.1016/j.jaci.2014.12.1909
24 Korten I , Kieninger E , Klenja S , Mack I , Schläpfer N , Barbani MT , et al.; SCILD and BILD study groups. Respiratory viruses in healthy infants and infants with cystic fibrosis: a prospective cohort study. Thorax. 2018;73(1):13–20. doi:.https://doi.org/10.1136/thoraxjnl-2016-209553
25 Quittner AL , Buu A , Messer MA , Modi AC , Watrous M . Development and validation of The Cystic Fibrosis Questionnaire in the United States: a health-related quality-of-life measure for cystic fibrosis. Chest. 2005;128(4):2347–54. doi:.https://doi.org/10.1378/chest.128.4.2347
26 American Thoracic Society, European Respiratory Society. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am J Respir Crit Care Med. 2005;171(8):912–30. doi:.https://doi.org/10.1164/rccm.200406-710ST
27 Frey U , Stocks J , Sly P , Bates J . Specifications for signal processing and data handling used for infant pulmonary function testing. ERS/ATS Task Force on Standards for Infant Respiratory Function Testing. European Respiratory Society/American Thoracic Society. Eur Respir J. 2000;16(5):1016–22. doi:.https://doi.org/10.1183/09031936.00.16510160
28 Hall GL , Reinmann B , Wildhaber JH , Frey U . Tidal exhaled nitric oxide in healthy, unsedated newborn infants with prenatal tobacco exposure. J Appl Physiol (1985). 2002;92(1):59–66. doi:.https://doi.org/10.1152/jappl.2002.92.1.59
29 Robinson PD , Latzin P , Verbanck S , Hall GL , Horsley A , Gappa M , et al. Consensus statement for inert gas washout measurement using multiple- and single- breath tests. Eur Respir J. 2013;41(3):507–22. doi:.https://doi.org/10.1183/09031936.00069712
30 Miller MR , Hankinson J , Brusasco V , Burgos F , Casaburi R , Coates A , et al.; ATS/ERS Task Force. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38. doi:.https://doi.org/10.1183/09031936.05.00034805
31 Silverman M , Wang M , Hunter G , Taub N . Episodic viral wheeze in preschool children: effect of topical nasal corticosteroid prophylaxis. Thorax. 2003;58(5):431–4. doi:.https://doi.org/10.1136/thorax.58.5.431
32 Bauman G , Bieri O . Matrix pencil decomposition of time-resolved proton MRI for robust and improved assessment of pulmonary ventilation and perfusion. Magn Reson Med. 2017;77(1):336–42. doi:.https://doi.org/10.1002/mrm.26096
33 Bauman G , Lützen U , Ullrich M , Gaass T , Dinkel J , Elke G , et al. Pulmonary functional imaging: qualitative comparison of Fourier decomposition MR imaging with SPECT/CT in porcine lung. Radiology. 2011;260(2):551–9. doi:.https://doi.org/10.1148/radiol.11102313
34 Bauman G , Puderbach M , Deimling M , Jellus V , Chefd’hotel C , Dinkel J , et al. Non-contrast-enhanced perfusion and ventilation assessment of the human lung by means of fourier decomposition in proton MRI. Magn Reson Med. 2009;62(3):656–64. doi:.https://doi.org/10.1002/mrm.22031
35 Bauman G , Puderbach M , Heimann T , Kopp-Schneider A , Fritzsching E , Mall MA , et al. Validation of Fourier decomposition MRI with dynamic contrast-enhanced MRI using visual and automated scoring of pulmonary perfusion in young cystic fibrosis patients. Eur J Radiol. 2013;82(12):2371–7. doi:.https://doi.org/10.1016/j.ejrad.2013.08.018
36 Bauman G , Pusterla O , Bieri O . Ultra-fast Steady-State Free Precession Pulse Sequence for Fourier Decomposition Pulmonary MRI. Magn Reson Med. 2016;75(4):1647–53. doi:.https://doi.org/10.1002/mrm.25697
37 Nyilas S , Bauman G , Sommer G , Stranzinger E , Pusterla O , Frey U , et al. Novel magnetic resonance technique for functional imaging of cystic fibrosis lung disease. Eur Respir J. 2017;50(6):1701464. doi:.https://doi.org/10.1183/13993003.01464-2017
38 Eichinger M , Optazaite DE , Kopp-Schneider A , Hintze C , Biederer J , Niemann A , et al. Morphologic and functional scoring of cystic fibrosis lung disease using MRI. Eur J Radiol. 2012;81(6):1321–9. doi:.https://doi.org/10.1016/j.ejrad.2011.02.045
39 Mika M , Korten I , Qi W , Regamey N , Frey U , Casaulta C , et al.; SCILD study group. The nasal microbiota in infants with cystic fibrosis in the first year of life: a prospective cohort study. Lancet Respir Med. 2016;4(8):627–35. doi:.https://doi.org/10.1016/S2213-2600(16)30081-9
40 Kieninger E , Yammine S , Korten I , Anagnostopoulou P , Singer F , Frey U , et al.; and the SCILD; and BILD study groups. Elevated lung clearance index in infants with cystic fibrosis shortly after birth. Eur Respir J. 2017;50(5):1700580. doi:.https://doi.org/10.1183/13993003.00580-2017
41 Korten I , Liechti M , Singer F , Hafen G , Rochat I , Anagnostopoulou P , et al.; SCILD and BILD study group. Lower exhaled nitric oxide in infants with Cystic Fibrosis compared to healthy controls. J Cyst Fibros. 2018;17(1):105–8. doi:.https://doi.org/10.1016/j.jcf.2017.05.005
42 Hooper LV , Littman DR , Macpherson AJ . Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–73. doi:.https://doi.org/10.1126/science.1223490
43 Huttenhower C , Gevers D , Knight R , Abubucker S , Badger JH , Chinwalla AT , et al.; Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14. doi:.https://doi.org/10.1038/nature11234
44 Cox MJ , Allgaier M , Taylor B , Baek MS , Huang YJ , Daly RA , et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One. 2010;5(6):e11044. doi:.https://doi.org/10.1371/journal.pone.0011044
45 Filkins LM , Hampton TH , Gifford AH , Gross MJ , Hogan DA , Sogin ML , et al. Prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol. 2012;194(17):4709–17. doi:.https://doi.org/10.1128/JB.00566-12
46 Zhao J , Schloss PD , Kalikin LM , Carmody LA , Foster BK , Petrosino JF , et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci USA. 2012;109(15):5809–14. doi:.https://doi.org/10.1073/pnas.1120577109
47 Olesen HV , Nielsen LP , Schiotz PO . Viral and atypical bacterial infections in the outpatient pediatric cystic fibrosis clinic. Pediatr Pulmonol. 2006;41(12):1197–204. doi:.https://doi.org/10.1002/ppul.20517
48 Byrnes CA , Vidmar S , Cheney JL , Carlin JB , Armstrong DS , Cooper PJ , et al.; ACFBAL Study Investigators. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax. 2013;68(7):643–51. doi:.https://doi.org/10.1136/thoraxjnl-2012-202342
49 Barnes PJ , Dweik RA , Gelb AF , Gibson PG , George SC , Grasemann H , et al. Exhaled nitric oxide in pulmonary diseases: a comprehensive review. Chest. 2010;138(3):682–92. doi:.https://doi.org/10.1378/chest.09-2090
50 Balfour-Lynn IM , Laverty A , Dinwiddie R . Reduced upper airway nitric oxide in cystic fibrosis. Arch Dis Child. 1996;75(4):319–22. doi:.https://doi.org/10.1136/adc.75.4.319
51 Bush A , Sly PD . Evolution of cystic fibrosis lung function in the early years. Curr Opin Pulm Med. 2015;21(6):602–8. doi:.https://doi.org/10.1097/MCP.0000000000000209
52 Simpson SJ , Ranganathan S , Park J , Turkovic L , Robins-Browne RM , Skoric B , et al.; AREST CF. Progressive ventilation inhomogeneity in infants with cystic fibrosis after pulmonary infection. Eur Respir J. 2015;46(6):1680–90. doi:.https://doi.org/10.1183/13993003.00622-2015
53 Davies G , Stocks J , Thia LP , Hoo AF , Bush A , Aurora P , et al.; London Cystic Fibrosis Collaboration (LCFC). Pulmonary function deficits in newborn screened infants with cystic fibrosis managed with standard UK care are mild and transient. Eur Respir J. 2017;50(5):1700326. doi:.https://doi.org/10.1183/13993003.00326-2017
54 Hoo AF , Thia LP , Nguyen TT , Bush A , Chudleigh J , Lum S , et al.; London Cystic Fibrosis Collaboration. Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax. 2012;67(10):874–81. doi:.https://doi.org/10.1136/thoraxjnl-2012-201747
55 Ramsey KA , Rosenow T , Turkovic L , Skoric B , Banton G , Adams AM , et al. Lung Clearance Index and Structural Lung Disease on Computed Tomography in Early Cystic Fibrosis. Am J Respir Crit Care Med. 2016;193(1):60–7.
Current study group: Jürg Barben, MD, St. Gallen; Carmen Casaulta, MD, Bern; Andrea Jung, MD, Zurich; Elisabeth Kieninger, MD, PhD, Bern; Insa Korten, MD, Bern; Philipp Latzin, MD, PhD, Bern; Alexander Moeller, MD, Zurich; Anne Mornand, MD, Geneva; Dominik Müller-Suter, MD, Aarau; Nicolas Regamey, MD, Lucerne; Isabelle Rochat MD, Lausanne; Florian Singer, MD, PhD, Bern; Renate Spinas, MD, Zurich; Daniel Trachsel, MD, Basel; Sophie Yammine, MD, PhD, Bern; Maura Zanolari, MD, Bellinzona
The SCILD cohort study is funded by project grants from the Swiss National Science Foundation (SNF), the Swiss Society for Cystic Fibrosis (CFCH), the Botnar Foundation and Ambizione. The funders have no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The University Children`s Hospital in Bern, Switzerland, is the coordinating study centre and provides all infrastructure.
PL has received personal fees from Gilead, Novartis, Polyphor, Roche, Schwabe, Vertex, Vifor and Zambon.