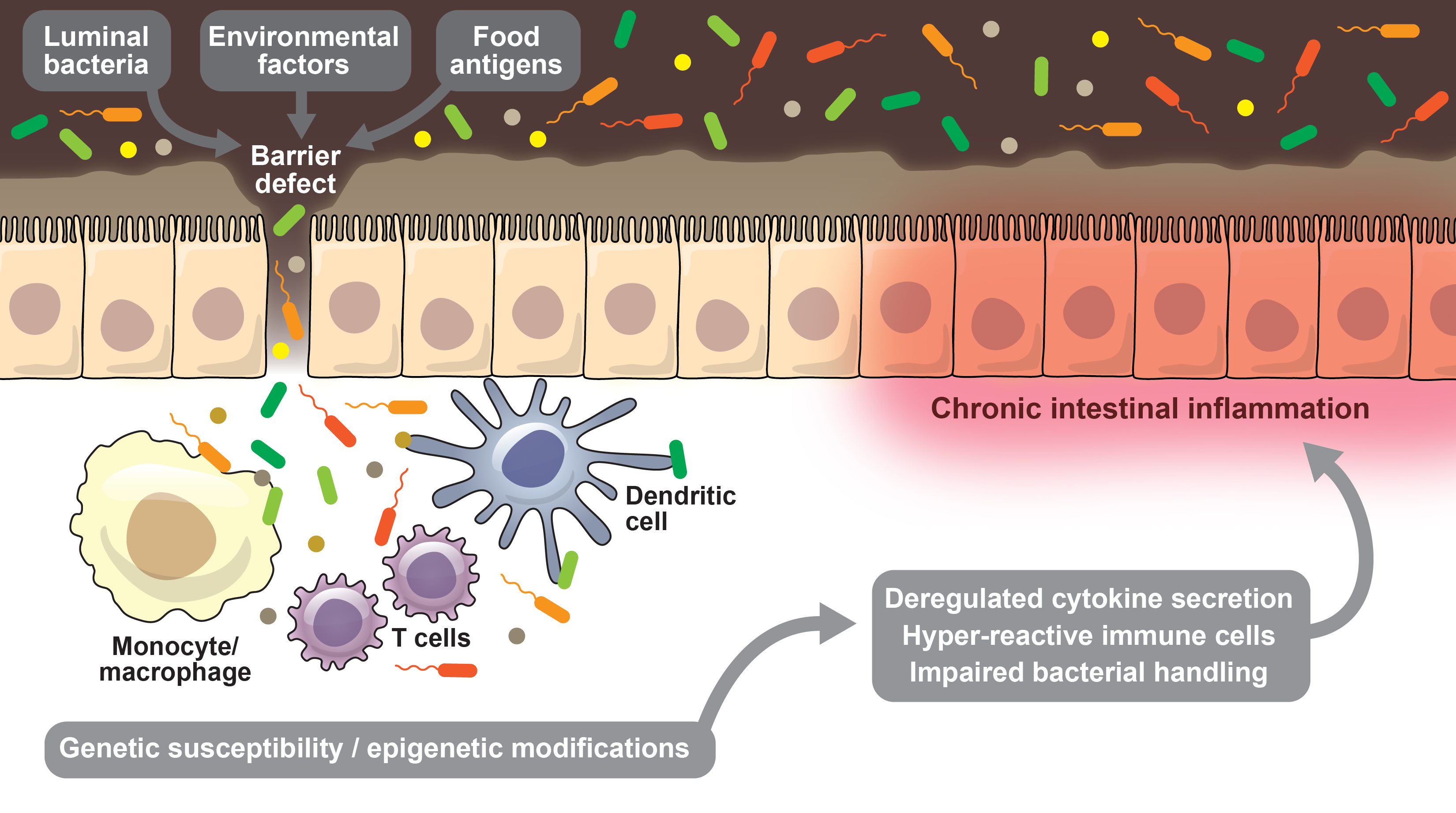
New insights into the pathophysiology of inflammatory bowel disease: microbiota, epigenetics and common signalling pathways
DOI: https://doi.org/10.4414/smw.2018.14599
Gerhard
Rogler, Luc
Biedermann, Michael
Scharl
Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Switzerland
Summary
The exact pathophysiology of inflammatory bowel disease (IBD) is still unknown. However, over the years important insights allowed the development of novel therapeutic approaches that are at the threshold of introduction into clinical practice, or at least in clinical trials. After being first described by Burrill B. Crohn, Crohn’s disease, one of the two major forms of IBD, was perceived as an infectious disease. When the concept of autoimmune diseases was formulated, Crohn’s disease and ulcerative colitis were thought to be members of this disease group. T cells certainly contribute to the chronification of the intestinal inflammation and targeting T cell migration has been introduced some years ago as a successful therapeutic approach in IBD. Despite the development of successful therapy based on this pathophysiological concept, IBD is no longer seen as a typical autoimmune disease. After the millennium, genome wide association studies on genetic variants and risk factors in these polygenetic diseases have told us a lot about pathogenetic pathways. However, genetic susceptibility explains only up to one third of the cases. Environmental factors also must play a role. Those environmental factors may “transfer” their disease-promoting potential into pathophysiological pathways with the intestinal microbiota as mediator. Hence, the intestinal microbiota has gained much attention as an important factor in disease development. Microbial factors, as well as other direct environmental influences, have been shown to affect epigenetic signatures, intestinal epithelial cells and the innate immune system, providing another important concept on how these diseases originate and can cause repeated flares at the same gut segments even after years of remission and after intermediate complete mucosal healing.
Current pathophysiological concepts of IBD not only help us to better understand these diseases and develop new therapies. They also illustrate the evolution of basic scientific concepts over time and that sometimes partially or even largely abandoned concepts persistently influence out current thinking/clinical practice.
Introduction
Inflammatory bowel diseases (IBDs) are chronic, relapsing inflammatory disorders of the intestinal tract usually starting in young adults and potentially causing a life-long burden and reduction of quality of life for the patients. There are approximately 12,000–15,000 adult cases in Switzerland. Data from the Swiss IBD cohort study (SIBDCS) suggest that (consistent with the majority of global epidemiological investigations [1]) the overall incidence is increasing [2]. Worldwide, over 2.5 million people of European ancestry are affected [3–7]. Increasingly, IBD is diagnosed in countries where it was almost absent 20 years ago, such as in Asia [8].
Crohn’s disease may occur anywhere in the gut from the mouth to the anus. It is an often segmental but transmural inflammation of the gut wall. Inflammation in Crohn’s disease may trigger fibrosis [9, 10] or fistula formation [11, 12]. Currently, despite recent advances in medical treatment options, up to 80% of the patients must undergo at least one surgical removal of an intestinal segment [13, 14]. In contrast, ulcerative colitis affects only the large bowel, always starting with highest activity in the rectum. It may be associated with high numbers of bloody bowel movements per day and has a huge impact on quality of life – up to 10 to 15% of patients will ultimately need colectomy [15, 16]. However, numbers seem to be decreasing.
Due to the associated severe morbidity and – at least for many patients with more severe disease – unsatisfactory treatment options available, research on IBD pathogenesis and factors triggering disease flares is currently intense. In addition, IBD has become a “prototype disease group” for chronic autoinflammatory disorders with a polygenic background and important multifaceted, environmental triggers.
There is clear evidence that environmental factors must contribute to both disease pathogenesis and disease flares [17–20]. The genetic risk factors have not changed over hundreds or thousands of years, but the disease incidence and prevalence is still increasing as outlined above. Almost absent as a disease until 100 years ago, IBD now affects millions, including non-Caucasians, in whom the disease had been exceedingly rare – clearly indicating environmental influences. These environmental factors may either directly affect the gut or may mediate their effects via the intestinal microbiota [18]. Those effects may not only be immediate and direct. Epigenetic changes, for instance, may impact the intestinal ecosystem months and years after the environmental influence that caused the epigenetic alterations was present. But classical environmental factors, such as exposure to antibiotics with regards to microbial composition, may also exert an influence that is much more sustained than originally expected [21, 22].
Thus, IBD is a disease group par excellence from which to learn about the important interactions between genetics/genetic risk factors, environmental influences and “in-vironmental” factors, for example, the intestinal microbiota [18]. Despite the numerous genetic factors identified, it turns out that a limited number of main pathways are affected. Some of these important pathways will be discussed in the following paragraphs.
Pathophysiological insights obtained from IBD research are now being translated into other chronic inflammatory diseases such as arthritis, asthma or diabetes. This increases the value of results obtained in the IBD field. However, there are multiple gaps in our knowledge of the factors influencing disease progression and development of complications, which are responsible for a large part of the disease burden along the course of the diseases.
What to learn from history: historical facts and their role for current pathophysiological concepts in IBD
Crohn’s disease as a disease entity was described by Burrill B. Crohn, Leon Ginzburg and Gordon D. Oppenheimer in New York in 1932 as regional ileitis and later named after Crohn (an impressive example of how a surname’s place in the alphabet may affect future recognition – at that time, alphabetic order determined positioning of co-authors) [23]. However, Crohn and colleagues were not the first to report this disease pattern: previous reports by other authors had been ignored or neglected in the medical-scientific community. This was described in detail by Crohn himself in the 1940s [24–26]. There were several reasons why Crohn's disease was named after Burrill B. Crohn on the basis of the above-mentioned publications, and the disease aroused medical-scientific interest. Some are still relevant for the disease concepts today.
In the 1920s, it became very popular to group patients with similar symptoms into disease entities and try to categorise symptoms. The concept of disease, as we know it today, evolved at the beginning of the 20th century. It became common to systematically record clinical symptoms that were similar and to define them as a syndrome or new disease. It was a transformation of the concept of disease, in which the enormous influence of the causal concepts of microbiology played a major role. The 1920s and 1930s were the heyday of microbiology. Many new diseases were based on the finding of a causative bacteria. Therefore, Burrill B. Crohn was initially convinced that regional ileitis or Crohn’s disease was caused by a microbiological agent, Mycobacterium avium subsp. paratuberculosis (MAP). Crohn’s first manuscripts came to this conclusion by an analogy. He found that the so-called Johne’s disease in cattle has many similarities with the human ileitis terminalis in terms of anatomical changes and histological findings. Based on these similarities and analogies, he concluded that both diseases – Johne’s disease in cattle and regional ileitis in humans – must have the same trigger. In the context of the “microbiological revolution” mentioned above, it indeed was shown that MAP causes John’s disease in cattle.
Despite unsuccessful attempts to detect Mycobacterium avium in patients with Crohn’s disease, this hypothesis was maintained over the years and there is still a community of gastroenterologists that strongly believes in this pathophysiological hypothesis [27, 28]. The tissue of the terminal ileum of affected patients with Crohn’s disease was homogenised and inoculated into guinea pigs or rabbits. Although this led to signs of disease of the respective species in other diseases with a microbiological, that is, bacterial, origin, there were no signs of disease in the animals for Crohn’s disease. The experiment thus contradicted the infection hypothesis. Nevertheless, antibiotics were tested as a therapy for Crohn’s disease and ulcerative colitis based on this hypothesis. And some of them proved to have some benefit for the patients.
The other reason why Crohn’s description of the disease was picked up was its prevalence. At that time, its incidence and prevalence started to rise, so that many doctors had seen patients with similar symptoms. This indicates that indeed there was a change of (environmental) conditions that triggered disease onset in an increasing number of patients. MAP has been around for centuries. It is hard to believe that it would suddenly cause an “epidemic”. On the other hand, the genetic signature of the population has not changed within decades making a “gene” as the cause of those “new” diseases rather unlikely.
The “immunological concept” of IBD pathology
The concept of autoimmune diseases was introduced into medicine 25 or 30 years ago. Immunology was one of the most successful biomedical sciences at that time [29–31]. According to the basic hypothesis, excessive or uncontrolled activation of the adaptive immune system (e.g., T cells and B cells) can lead to uncontrolled and chronic inflammation. Numerous publications deal with T cell subpopulations and adaptive immunity in chronic inflammatory bowel diseases [32–34]. The role of T cells has been studied extensively in animal models. The absence of regulatory T cells (FoxP3 positive cells) leads to the onset of colitis [35–37]. This led to concepts that suggest the adoptive transfer of regulatory T cells in patients with severe and refractory IBD [35]. In addition, it is well known that colitis can be induced via T cell costimulatory molecules [38, 39]. The activation of T cells is used in cancer therapy today (e.g., anti-CTLA4 antibodies, ipilimumab or programmed cell death protein 1 [PD1] and programmed death-ligand 1 [PDL-1] antibodies such as nivolumab and pembrolizumab) [40]. An important side effect of such therapies is the induction of “immune-mediated colitis” in melanoma or adenocarcinoma patients [41–43]. To some extent, this is proof of the concept that adaptive immune cells contribute to the pathogenesis of IBD. Therapeutic principles that are established in IBD (such as systemic steroids and anti-tumour necrosis factor antibodies such as infliximab) are now successfully used to treat this immune-mediated colitis [44–47]. Interestingly, whether or not a patient will develop immune-mediated colitis as a side effect of tumour therapy seems to be dependent on the composition of the intestinal microbiota [48]. This points to an important role of the microbiota composition in the local activation of mucosal T cells – an aspect of IBD pathophysiology that will be discussed in more detail later.
T cells usually do not proliferate in the mucosa. They invade from the circulation to contribute to mucosal inflammation. Insights into the pathophysiological role of T cells, generated the idea to inhibit their migration to the site of tissue inflammation. Vedolizumab specifically inhibits the interaction between alpha4/beta7 integrin on lymphocytes and mucosal vascular addressin cell adhesion molecule 1 (MAdCAM), which is almost exclusively expressed on endothelial cells in the gut. This approach has now been successfully introduced into clinical routine [49–53].
IBD is no longer seen as a classic autoimmune disease because most likely the antigens against which the adaptive immune system is primed originate from the intestinal microbiota. Furthermore, “autoimmune disease” implies an over-reactive, hyper-stimulated immune system, whereas rather the opposite appears to be a primary factor in the pathogenesis of IBD. Several of the hitherto established IBD risk single nucleotide polymorphisms code for genes involved in microbial defence (see below). The latter, in conjunction with the increase of microbes in the deeper layer of the mucus directly above the mucosal layer or even having penetrated the mucosal barrier in patients with IBD, indicates that activation in inflammatory pathways rather represents a secondary response due to initially impaired defence mechanisms. Nevertheless, the autoimmune disease concept has generated important insights that led to improvements of therapy.
Genetic factors contributing to IBD pathogenesis
In older twin pair studies, 40 to 50% concordance of Crohn’s disease has been observed [54, 55]. The fact that even monozygotic twins have a concordance of less than 50% again indicates the important role of environmental factors in IBD pathogenesis. On the other hand, it clearly shows that genetic risk factors have an important role in disease onset. With the technique of genome-wide association studies (GWAS), those genetic risk factors could be detected. The first genetic risk factor identified to contribute to Crohn’s disease were variants in the nucleotide-binding oligomerisation domain protein 2 (NOD2) DNA in 2001 [56–59]. NOD2 is a pattern recognition receptor mainly expressed in cells of the innate immune system as well as in intestinal epithelial cells, binding muramyl dipeptide, which is a component of the bacterial cell wall. NOD2 variants that are associated with increased susceptibility to develop Crohn’s disease show impaired or deficient recognition of these bacterial wall products, again pointing to the importance of the microbiota for the pathogenesis of IBD. In the meantime, more than 250 genetic risk factor have been identified [60, 61]. There are many shared genetic risk factors with other autoinflammatory diseases, such as lupus erythematosus, rheumatoid arthritis, psoriasis or type I diabetes [60]. There is also a large overlap between ulcerative colitis and Crohn’s disease with respect to genetic risk factors. This tells us that, based on the same genetic risk profile of an individual, different diseases may finally develop – again pointing to an additional role of environmental factors. The risk increases (odds ratios) associated with many of those genetic variants are only 1.2, meaning that the chance to get the disease is only 20% higher than normal having such a risk factors. In addition, it usually means that many healthy people or at least persons who are not affected by the specific diseases carry such risk factors. This is the reason why determining genetic risk factors cannot be used for diagnosis or even disease risk estimation. We learned a great deal about the pathophysiological pathways involved from genetic findings. However, the whole effort so far has not translated into clinical developments.
Nevertheless, the concept of risk genes has spread to the population. 23andme is the story of a great commercial success (“Find out what your DNA says about you and your family”) on a very dubious ethical and scientific basis.
A crucial role for environmental factors in the pathogenesis of IBD
There is abundant evidence that in IBD, as in other chronic inflammatory diseases, environmental factors play an important role, for both disease onset and disease course (onset and duration of disease flares) [17–19, 62–64]. The risk of developing IBD and the subsequent disease behaviour in children of immigrants from low-incidence areas (Asia) coming to high-incidence areas (western Europe) is similar to that of the indigenous western population [65–67]. Living in an “urban environment” is a confirmed risk factor for IBD [68]. An increased prevalence of IBD in urban environments has been documented in Switzerland also [17]. It is obvious that research into these environmental factors causing or influencing IBD is important as, in contrast to the genetic risk factors, they can be changed or at least modulated. The prevention of environmentally triggered disease flares would be most relevant for IBD patients.
Unfortunately, reliable data on distinct environmental factors are limited. Only a few environmental factors influencing IBD disease course are unequivocally relevant [17]. Probably the best investigated environmental factor influencing IBD disease courses is smoking. Active smoking worsens the course of Crohn’s disease [69–73]. In Switzerland, twice as many patients with Crohn’s disease are active smokers compared with ulcerative colitis [74]. In striking contrast to the general population, significantly more women than men with Crohn’s disease smoke (42.8 vs 35.8%, p = 0.025) [74]. Despite the well-established negative effects on the disease course, smoking rates in Crohn’s disease are alarmingly high, especially in female patients. Interestingly, smoking affects the colonic microbiota, providing a further link between the environment and the “in-vironment” [75, 76].
Other environmental factors that have been associated with clinical presentation or risk of inflammatory flares, as well as increased incidence, are diet and food additives [77–81]. Diet also may mediate its effects via the microbiota composition. Oral contraceptives and nonsteroidal anti-inflammatory drugs are the two main classes of frequently taken drugs that have been attributed to have potential to cause flares of the disease [17, 82–86].
Specific food additives such as titanium dioxide [87] or emulsifiers [77] also may contribute to the disease course as they impair the intestinal barrier function and allow bacteria to get into contact with intestinal epithelial cells that are normally protected by the mucosal mucus layer.
The important role of the intestinal microbiota
The important role of the intestinal microbiota for the pathogenesis of IBD has been mentioned several times in other paragraphs. Evidence for a crucial role of the microbiota in IBD pathogenesis comes from several fields. In animal models of colitis, inflammation is prevented by germ-free conditions [88, 89]. Surgical diversion of the faecal stream is followed by improvement of inflammation in many patients with recurrence after restoration of the intestinal faecal stream [90, 91]. Several probiotics (such as Escherichia coli Nissle 1917) have shown efficacy in the treatment or the prophylaxis of flares in ulcerative colitis [92–94].
In addition, in recent years many authors confirmed an alteration of the intestinal microbiota in patients with IBD [95–101]. The changes found are generally referred to as “dysbiosis” or “reduced diversity”. It is unclear whether a reduction of specific bacterial strains such as Faecalibacterium prausnici is just an epiphenomenon or has indeed pathophysiological relevance. Unfortunately, most studies so far have analysed the microbiome with 16s-RNA gene sequencing. This allows only a rather crude estimation of the real changes occurring in the patients. In addition, the alterations are usually detected in patients with inflammation that itself may contribute to the observed changes.
The technique that has allowed the study of the intestinal microbiota composition is called high-throughput sequencing or pyrosequencing. It allows the analysis of 100,000 or 500,000 different DNA sequences of bacteria of the intestinal flora within a few hours. In order to process the obtained information, the latest computer technology is necessary.
The pathophysiological concepts, for Crohn’s disease at least, have now somehow returned to the point of origin. Now it is not a single bacterium that causes the disease as an infectious agent, but a dysbalance of the entire intestinal flora, which leads to an activation of the immune system. It appears that interesting disease concepts are never completely abandoned, but return in modifications.
How epigenetic changes are involved in IBD pathogenesis
An important question has always puzzled investigators working on the pathogenesis of IBD: why is it possible that in Crohn’s disease the intestinal mucosa can be completely normal and healed for years and then the inflammation reappears at exactly the same location? Why can Crohn’s disease reoccur at a site where the involved area has been resected? How can a “disease memory” in the mucosa be explained? How can it happen that environmental influences act on the intestinal mucosa and the disease onsets years after this influence is gone (in the case of smoking)?
The concept of epigenetic imprinting might give an answer to all these open questions and an explanation why there is a “disease memory” in certain areas of the gut wall. In fact, recent data provide significant evidence that epigenetic alterations also in healthy persons occur during lifetime. In patients with IBD, specific methylation patterns have been described [102–105]. Nimmo and co-workers reported a methylation profile that was found to be characteristic for Crohn’s disease of the terminal ileum [104]. In their analysis they found 1117 methylation sites to be differentially methylated, of which 50 showed significantly altered methylation in cases compared with controls [104]. There were distinct pathways that had an enrichment of differences in methylation that had been associated with the pathogenesis of IBD before, including genes relevant in the adaptive and innate immune system such as MAPK13, FASLG, PRF1, S100A13, RIPK3 and IL-21R [104]. Interestingly, the authors also found a significant, 8.6-fold enrichment of methylation changes near GWAS loci of genetic risk factors of IBD [104]. Furthermore, certain complications such as fibrosis and strictures seem to be associated with a specific methylation pattern and subsequently with epigenetic modifications [102].
Which common pathways may be important?
GWAS analyses and epigenetic profiling gave us insights into the genetic/epigenetic risk factors relevant in the pathogenesis of IBD. Many of them play a role in the recognition of and the response to the intestinal microbiota. In these defence pathways bacterial sensing, intestinal barrier regulating proteins and autophagy associated proteins, as well as cell stress and hypoxia stress response genes, play an important role [106–111]. Autophagy is an intracellular clearance system that leads to the degradation of intracellular debris such as misfolded proteins or invading bacteria [112]. Autophagy plays an important role in host defences against many bacteria, viruses and parasites [112] and for the maintenance of intestinal homeostasis. For many of the genetic variants and differentially methylated genes, however, the function and their role in IBD pathogenesis has not clearly been elucidated.
Interestingly some of the intracellular pathways in which the risk genes are involved are interconnected. Variants in the protein tyrosine phosphatase, non-receptor type 22 (PTPN22), for example, have been shown to be a risk factor for many autoinflammatory diseases such as IBD, rheumatoid arthritis, systemic lupus erythematosus or psoriasis. We recently demonstrated that this enzyme regulates NOD2-induced cytokine release, thereby modulating the response to the microbiota, as well as autophagy [113]. Further, PTPN22 controls the phosphorylation of NALP3, a protein of the inflammasome complex involved in interleukin-1beta and interleukin-18 secretion and subsequently activation of the inflammasome [114, 115]. NALP3 itself has been identified to be a risk gene for Crohn’s disease. Another protein tyrosine phosphatase, PTPN2, also regulates the NLRP3 inflammasome (Cell reports, in press).
Another interesting field in which pathways converge is the regulation of tissue pH. Tissue pH is altered by inflammation as well as during hypoxia. A G-protein coupled pH sensing receptor of the GPR4 family, TDAG8, also has been identified to be a risk variant for Crohn’s disease in GWAS analyses. Hypoxia and inflammation are linked on many levels and influence each other [116]. Hypoxia also decreases the local pH in the mucosal tissue [117]. We recently reported that deletions of GPR4 or OGR1 protect from DSS induced colitis or ameliorates colitis in IL-10/pH receptor double knockout mice [118–120]. These pH receptors also are involved in the regulation of the intestinal barrier when activated [119]. Again we find a connection of the different pathophysiological relevant pathways in the mucosa such as stress response, hypoxia and autophagy [121].
Recently, we have seen first examples, that profound mechanistic and basic science investigations may not only improve our understanding on how and why IBD develops in a previous healthy human gut but also may directly support therapeutic decision making. For instance, an in-depth analysis of microbial composition and functional properties at baseline and during the administration of vedolizumab treatment in patients with IBD in conjunction with clinical data using sophisticated mathematical modelling revealed, that the functional microbial profile (including an increase in butyrate producing microbes in responding Crohn’s disease patients) could be associated with therapeutic response.
Elucidating these critical pathways (summarised in fig. 1) and their role in IBD pathophysiology will help to develop new therapeutic targets and treatments in the future.
Search strategy and selection criteria
References for this review were identified through searches of PubMed with the search terms “Crohn’s disease, ulcerative colitis, pathophysiology”, from 1938 until September, 2017. Articles were also identified through searches of the authors’ own files. Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this review.
Author contributions
All authors participated sufficiently, intellectually or practically, in the work to take public responsibility for the content of the article, including the conception, design, data interpretation and writing of the manuscript. The final version of the manuscript was approved by all authors.
References
1
Molodecky
NA
,
Soon
IS
,
Rabi
DM
,
Ghali
WA
,
Ferris
M
,
Chernoff
G
, et al.
Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54.e42, quiz e30. doi:.https://doi.org/10.1053/j.gastro.2011.10.001
2
Braegger
CP
,
Ballabeni
P
,
Rogler
D
,
Vavricka
SR
,
Friedt
M
,
Pittet
V
; Swiss IBD Cohort Study Group. Epidemiology of inflammatory bowel disease: Is there a shift towards onset at a younger age?
J Pediatr Gastroenterol Nutr. 2011;53(2):141–4. doi:.https://doi.org/10.1097/MPG.0b013e318218be35
3
Ray
K
. IBD: The changing epidemiology of IBD. Nat Rev Gastroenterol Hepatol. 2017;14(12):690.
4
Ananthakrishnan
AN
. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205–17. doi:.https://doi.org/10.1038/nrgastro.2015.34
5
Ng
SC
. Epidemiology of inflammatory bowel disease: focus on Asia. Best Pract Res Clin Gastroenterol. 2014;28(3):363–72. doi:.https://doi.org/10.1016/j.bpg.2014.04.003
6
Ng
SC
,
Tang
W
,
Ching
JY
,
Wong
M
,
Chow
CM
,
Hui
AJ
, et al.; Asia–Pacific Crohn’s and Colitis Epidemiologic Study (ACCESS) Study Group. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology. 2013;145(1):158–165.e2. doi:.https://doi.org/10.1053/j.gastro.2013.04.007
7
Cosnes
J
,
Gower-Rousseau
C
,
Seksik
P
,
Cortot
A
. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785–94.e4. doi:.https://doi.org/10.1053/j.gastro.2011.01.055
8
Ng
SC
,
Leung
WK
,
Shi
HY
,
Li
MK
,
Leung
CM
,
Ng
CK
, et al.
Epidemiology of Inflammatory Bowel Disease from 1981 to 2014: Results from a Territory-Wide Population-Based Registry in Hong Kong. Inflamm Bowel Dis. 2016;22(8):1954–60. doi:.https://doi.org/10.1097/MIB.0000000000000846
9
Lawrance
IC
,
Rogler
G
,
Bamias
G
,
Breynaert
C
,
Florholmen
J
,
Pellino
G
, et al.
Cellular and Molecular Mediators of Intestinal Fibrosis. J Crohn’s Colitis. 2017;11(12):1491–503
. [doi:.].https://doi.org/10.1016/j.crohns.2014.09.008
10
Latella
G
,
Rogler
G
,
Bamias
G
,
Breynaert
C
,
Florholmen
J
,
Pellino
G
, et al.
Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohn’s Colitis. 2014;8(10):1147–65. doi:.https://doi.org/10.1016/j.crohns.2014.03.008
11
Scharl
M
,
Bruckner
RS
,
Rogler
G
. The two sides of the coin: Similarities and differences in the pathomechanisms of fistulas and stricture formations in irritable bowel disease. United European Gastroenterol J. 2016;4(4):506–14. doi:.https://doi.org/10.1177/2050640616635957
12
Scharl
M
,
Rogler
G
. Pathophysiology of fistula formation in Crohn’s disease. World J Gastrointest Pathophysiol. 2014;5(3):205–12. doi:.https://doi.org/10.4291/wjgp.v5.i3.205
13
Safroneeva
E
,
Vavricka
SR
,
Fournier
N
,
Pittet
V
,
Peyrin-Biroulet
L
,
Straumann
A
, et al.; Swiss IBD Cohort Study Group. Impact of the early use of immunomodulators or TNF antagonists on bowel damage and surgery in Crohn’s disease. Aliment Pharmacol Ther. 2015;42(8):977–89. doi:.https://doi.org/10.1111/apt.13363
14
Pittet
V
,
Rogler
G
,
Michetti
P
,
Fournier
N
,
Vader
JP
,
Schoepfer
A
, et al.; Swiss Inflammatory Bowel Disease Cohort Study Group. Penetrating or stricturing diseases are the major determinants of time to first and repeat resection surgery in Crohn’s disease. Digestion. 2013;87(3):212–21. doi:.https://doi.org/10.1159/000350954
15
Targownik
LE
,
Singh
H
,
Nugent
Z
,
Bernstein
CN
. The epidemiology of colectomy in ulcerative colitis: results from a population-based cohort. Am J Gastroenterol. 2012;107(8):1228–35. doi:.. Corrected in: Am J Gastroenterol. 2013;108:157. https://doi.org/10.1038/ajg.2012.127
16
Kaplan
GG
,
Seow
CH
,
Ghosh
S
,
Molodecky
N
,
Rezaie
A
,
Moran
GW
, et al.
Decreasing colectomy rates for ulcerative colitis: a population-based time trend study. Am J Gastroenterol. 2012;107(12):1879–87. doi:.https://doi.org/10.1038/ajg.2012.333
17
Rogler
G
,
Zeitz
J
,
Biedermann
L
. The Search for Causative Environmental Factors in Inflammatory Bowel Disease. Dig Dis. 2016;34(Suppl 1):48–55. doi:.https://doi.org/10.1159/000447283
18
Rogler
G
,
Vavricka
S
. Exposome in IBD: recent insights in environmental factors that influence the onset and course of IBD. Inflamm Bowel Dis. 2015;21(2):400–8. doi:.https://doi.org/10.1097/MIB.0000000000000229
19
Ananthakrishnan
AN
. Environmental risk factors for inflammatory bowel diseases: a review. Dig Dis Sci. 2015;60(2):290–8. doi:.https://doi.org/10.1007/s10620-014-3350-9
20
O’Toole
A
,
Korzenik
J
. Environmental triggers for IBD. Curr Gastroenterol Rep. 2014;16(7):396. doi:.https://doi.org/10.1007/s11894-014-0396-y
21
Dethlefsen
L
,
Relman
DA
. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4554–61. doi:.https://doi.org/10.1073/pnas.1000087107
22
Pérez-Cobas
AE
,
Gosalbes
MJ
,
Friedrichs
A
,
Knecht
H
,
Artacho
A
,
Eismann
K
, et al.
Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62(11):1591–601. doi:.https://doi.org/10.1136/gutjnl-2012-303184
23
Crohn
BB
,
Ginzburg
L
,
Oppenheimer
GD
. Regional ileitis: a pathologic and clinical entity. 1932. Mt Sinai J Med. 2000;67(3):263–8.
24
Crohn
BB
. Inflammatory diseases of the small intestine. J Omaha Midwest Clin Soc. 1946;7(3):77–83.
25
Crohn
BB
. Ileojejunitis. N Y State J Med. 1949;49(15):1808–11.
26
Crohn
BB
,
Ginzburg
L
,
Oppenheimer
GD
. Regional ileitis; a pathologic and clinical entity. Am J Med. 1952;13(5):583–90. doi:.https://doi.org/10.1016/0002-9343(52)90025-9
27
Gitlin
L
,
Borody
TJ
,
Chamberlin
W
,
Campbell
J
. Mycobacterium avium ss paratuberculosis-associated diseases: piecing the Crohn’s puzzle together. J Clin Gastroenterol. 2012;46(8):649–55. doi:.https://doi.org/10.1097/MCG.0b013e31825f2bce
28
Sartor
RB
. Does Mycobacterium avium subspecies paratuberculosis cause Crohn’s disease?
Gut. 2005;54(7):896–8. doi:.https://doi.org/10.1136/gut.2004.055889
29
Bach
JF
. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–20. doi:.https://doi.org/10.1056/NEJMra020100
30
Sneller
MC
,
Wang
J
,
Dale
JK
,
Strober
W
,
Middelton
LA
,
Choi
Y
, et al.
Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89(4):1341–8.
31
Keren
DF
,
Goeken
JA
. Autoimmune reactivity in inflammatory bowel disease. Clin Lab Med. 1997;17(3):465–81.
32
Kaistha
A
,
Levine
J
. Inflammatory bowel disease: the classic gastrointestinal autoimmune disease. Curr Probl Pediatr Adolesc Health Care. 2014;44(11):328–34. doi:.https://doi.org/10.1016/j.cppeds.2014.10.003
33
Ravi
K
,
Chari
ST
,
Vege
SS
,
Sandborn
WJ
,
Smyrk
TC
,
Loftus
EV, Jr
. Inflammatory bowel disease in the setting of autoimmune pancreatitis. Inflamm Bowel Dis. 2009;15(9):1326–30. doi:.https://doi.org/10.1002/ibd.20898
34
Wen
Z
,
Fiocchi
C
. Inflammatory bowel disease: autoimmune or immune-mediated pathogenesis?
Clin Dev Immunol. 2004;11(3-4):195–204. doi:.https://doi.org/10.1080/17402520400004201
35
Geem
D
,
Harusato
A
,
Flannigan
K
,
Denning
TL
. Harnessing regulatory T cells for the treatment of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(6):1409–18.
36
Mayne
CG
,
Williams
CB
. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2013;19(8):1772–88. doi:.https://doi.org/10.1097/MIB.0b013e318281f5a3
37
Groux
H
,
Powrie
F
. Regulatory T cells and inflammatory bowel disease. Immunol Today. 1999;20(10):442–5. doi:.https://doi.org/10.1016/S0167-5699(99)01510-8
38
Liu
H
,
Hu
B
,
Xu
D
,
Liew
FY
. CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-beta, and CTLA4. J Immunol. 2003;171(10):5012–7. doi:.https://doi.org/10.4049/jimmunol.171.10.5012
39
Davenport
CM
,
McAdams
HA
,
Kou
J
,
Mascioli
K
,
Eichman
C
,
Healy
L
, et al.
Inhibition of pro-inflammatory cytokine generation by CTLA4-Ig in the skin and colon of mice adoptively transplanted with CD45RBhi CD4+ T cells correlates with suppression of psoriasis and colitis. Int Immunopharmacol. 2002;2(5):653–72. doi:.https://doi.org/10.1016/S1567-5769(01)00201-6
40
Minor
DR
,
Chin
K
,
Kashani-Sabet
M
. Infliximab in the treatment of anti-CTLA4 antibody (ipilimumab) induced immune-related colitis. Cancer Biother Radiopharm. 2009;24(3):321–5. doi:.https://doi.org/10.1089/cbr.2008.0607
41
Shah
R
,
Witt
D
,
Asif
T
,
Mir
FF
. Ipilimumab as a Cause of Severe Pan-Colitis and Colonic Perforation. Cureus. 2017;9(4):e1182.
42
O’Connor
A
,
Marples
M
,
Mulatero
C
,
Hamlin
J
,
Ford
AC
. Ipilimumab-induced colitis: experience from a tertiary referral center. Therap Adv Gastroenterol. 2016;9(4):457–62. doi:.https://doi.org/10.1177/1756283X16646709
43
Verschuren
EC
,
van den Eertwegh
AJ
,
Wonders
J
,
Slangen
RM
,
van Delft
F
,
van Bodegraven
A
, et al.
Clinical, Endoscopic, and Histologic Characteristics of Ipilimumab-Associated Colitis. Clin Gastroenterol Hepatol. 2016;14(6):836–42. doi:.https://doi.org/10.1016/j.cgh.2015.12.028
44
Fukumoto
T
,
Fujiwara
S
,
Tajima
S
,
Tamesada
Y
,
Sakaguchi
M
,
Oka
M
, et al.
Infliximab for severe colitis associated with nivolumab followed by ipilimumab. J Dermatol. 2018;45(1):e1–2. doi:.https://doi.org/10.1111/1346-8138.14034
45
Hillock
NT
,
Heard
S
,
Kichenadasse
G
,
Hill
CL
,
Andrews
J
. Infliximab for ipilimumab-induced colitis: A series of 13 patients. Asia Pac J Clin Oncol. 2017;13(5):e284–90. doi:.https://doi.org/10.1111/ajco.12651
46
Hsieh
AH
,
Ferman
M
,
Brown
MP
,
Andrews
JM
. Vedolizumab: a novel treatment for ipilimumab-induced colitis. BMJ Case Rep. 2016;2016.
47
Beniwal-Patel
P
,
Matkowskyj
K
,
Caldera
F
. Infliximab Therapy for Corticosteroid-Resistant Ipilimumab-Induced Colitis. J Gastrointestin Liver Dis. 2015;24(3):274.
48
Chaput
N
,
Lepage
P
,
Coutzac
C
,
Soularue
E
,
Le Roux
K
,
Monot
C
, et al.
Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–79. doi:.https://doi.org/10.1093/annonc/mdx108
49
Feagan
BG
,
Rutgeerts
P
,
Sands
BE
,
Hanauer
S
,
Colombel
JF
,
Sandborn
WJ
, et al.; GEMINI 1 Study Group. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710. doi:.https://doi.org/10.1056/NEJMoa1215734
50
Stallmach
A
,
Schmidt
C
,
Teich
N
. Vedolizumab for the treatment of ulcerative colitis. Expert Rev Gastroenterol Hepatol. 2016;10(2):165–75. doi:.https://doi.org/10.1586/17474124.2016.1123618
51
Shahidi
N
,
Bressler
B
,
Panaccione
R
. Vedolizumab for the treatment of ulcerative colitis. Expert Opin Biol Ther. 2016;16(1):129–35. doi:.https://doi.org/10.1517/14712598.2016.1121231
52
Lau
MS
,
Tsai
HH
. Review of vedolizumab for the treatment of ulcerative colitis. World J Gastrointest Pharmacol Ther. 2016;7(1):107–11. doi:.https://doi.org/10.4292/wjgpt.v7.i1.107
53
Colombel
JF
,
Sands
BE
,
Rutgeerts
P
, et al.
The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut. 2017;66(5):839–51.
54
Vind
I
,
Jespersgaard
C
,
Hougs
L
,
Riis
L
,
Dinesen
L
,
Andersen
PS
, et al.
Genetic and environmental factors in monozygotic twins with Crohn’s disease and their first-degree relatives: a case report. Digestion. 2005;71(4):262–5. doi:.https://doi.org/10.1159/000087053
55
Tysk
C
,
Lindberg
E
,
Järnerot
G
,
Flodérus-Myrhed
B
. Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut. 1988;29(7):990–6. doi:.https://doi.org/10.1136/gut.29.7.990
56
Hampe
J
,
Cuthbert
A
,
Croucher
PJ
,
Mirza
MM
,
Mascheretti
S
,
Fisher
S
, et al.
Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet. 2001;357(9272):1925–8. doi:.https://doi.org/10.1016/S0140-6736(00)05063-7
57
Ogura
Y
,
Bonen
DK
,
Inohara
N
,
Nicolae
DL
,
Chen
FF
,
Ramos
R
, et al.
A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–6. doi:.https://doi.org/10.1038/35079114
58
Hugot
JP
,
Chamaillard
M
,
Zouali
H
,
Lesage
S
,
Cézard
JP
,
Belaiche
J
, et al.
Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi:.https://doi.org/10.1038/35079107
59
McGovern
DP
,
van Heel
DA
,
Ahmad
T
,
Jewell
DP
. NOD2 (CARD15), the first susceptibility gene for Crohn’s disease. Gut. 2001;49(6):752–4. doi:.https://doi.org/10.1136/gut.49.6.752
60
Lees
CW
,
Barrett
JC
,
Parkes
M
,
Satsangi
J
. New IBD genetics: common pathways with other diseases. Gut. 2011;60(12):1739–53. doi:.https://doi.org/10.1136/gut.2009.199679
61
de Lange
KM
,
Moutsianas
L
,
Lee
JC
,
Lamb
CA
,
Luo
Y
,
Kennedy
NA
, et al.
Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–61. doi:.https://doi.org/10.1038/ng.3760
62
Wang
MH
,
Achkar
JP
. Gene-environment interactions in inflammatory bowel disease pathogenesis. Curr Opin Gastroenterol. 2015;31(4):277–82. doi:.https://doi.org/10.1097/MOG.0000000000000188
63
Abegunde
AT
,
Muhammad
BH
,
Bhatti
O
,
Ali
T
. Environmental risk factors for inflammatory bowel diseases: Evidence based literature review. World J Gastroenterol. 2016;22(27):6296–317. doi:.https://doi.org/10.3748/wjg.v22.i27.6296
64
Cholapranee
A
,
Ananthakrishnan
AN
. Environmental Hygiene and Risk of Inflammatory Bowel Diseases: A Systematic Review and Meta-analysis. Inflamm Bowel Dis. 2016;22(9):2191–9. doi:.https://doi.org/10.1097/MIB.0000000000000852
65
Ng
SC
,
Shi
HY
,
Hamidi
N
,
Underwood
FE
,
Tang
W
,
Benchimol
EI
, et al.
Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–78. doi:.https://doi.org/10.1016/S0140-6736(17)32448-0
66
Loftus
EV, Jr
. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126(6):1504–17. doi:.https://doi.org/10.1053/j.gastro.2004.01.063
67
Zlotogora
J
,
Zimmerman
J
,
Rachmilewitz
D
. Prevalence of inflammatory bowel disease in family members of Jewish Crohn’s disease patients in Israel. Dig Dis Sci. 1991;36(4):471–5. doi:.https://doi.org/10.1007/BF01298876
68
Soon
IS
,
Molodecky
NA
,
Rabi
DM
,
Ghali
WA
,
Barkema
HW
,
Kaplan
GG
. The relationship between urban environment and the inflammatory bowel diseases: a systematic review and meta-analysis. BMC Gastroenterol. 2012;12(1):51. doi:.https://doi.org/10.1186/1471-230X-12-51
69
Cosnes
J
. Smoking and Diet: Impact on Disease Course?
Dig Dis. 2016;34(1-2):72–7. doi:.https://doi.org/10.1159/000442930
70
Cosnes
J
. Smoking, physical activity, nutrition and lifestyle: environmental factors and their impact on IBD. Dig Dis. 2010;28(3):411–7. doi:.https://doi.org/10.1159/000320395
71
Cosnes
J
,
Beaugerie
L
,
Carbonnel
F
,
Gendre
JP
. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120(5):1093–9. doi:.https://doi.org/10.1053/gast.2001.23231
72
Cosnes
J
,
Carbonnel
F
,
Carrat
F
,
Beaugerie
L
,
Cattan
S
,
Gendre
J
. Effects of current and former cigarette smoking on the clinical course of Crohn’s disease. Aliment Pharmacol Ther. 1999;13(11):1403–11. doi:.https://doi.org/10.1046/j.1365-2036.1999.00630.x
73
Cosnes
J
,
Carbonnel
F
,
Beaugerie
L
,
Le Quintrec
Y
,
Gendre
JP
. Effects of cigarette smoking on the long-term course of Crohn’s disease. Gastroenterology. 1996;110(2):424–31. doi:.https://doi.org/10.1053/gast.1996.v110.pm8566589
74
Biedermann
L
,
Fournier
N
,
Misselwitz
B
,
Frei
P
,
Zeitz
J
,
Manser
CN
, et al.; Swiss Inflammatory Bowel Disease Cohort Study Group. High Rates of Smoking Especially in Female Crohn’s Disease Patients and Low Use of Supportive Measures to Achieve Smoking Cessation--Data from the Swiss IBD Cohort Study. J Crohn’s Colitis. 2015;9(10):819–29. doi:.https://doi.org/10.1093/ecco-jcc/jjv113
75
Biedermann
L
,
Brülisauer
K
,
Zeitz
J
,
Frei
P
,
Scharl
M
,
Vavricka
SR
, et al.
Smoking cessation alters intestinal microbiota: insights from quantitative investigations on human fecal samples using FISH. Inflamm Bowel Dis. 2014;20(9):1496–501. doi:.https://doi.org/10.1097/MIB.0000000000000129
76
Biedermann
L
,
Zeitz
J
,
Mwinyi
J
,
Sutter-Minder
E
,
Rehman
A
,
Ott
SJ
, et al.
Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One. 2013;8(3):e59260. doi:.https://doi.org/10.1371/journal.pone.0059260
77
Chassaing
B
,
Koren
O
,
Goodrich
JK
,
Poole
AC
,
Srinivasan
S
,
Ley
RE
, et al.
Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519(7541):92–6. doi:.. Corrected in: Nature. 2016;536:238. https://doi.org/10.1038/nature14232
78
Ananthakrishnan
AN
,
Khalili
H
,
Song
M
,
Higuchi
LM
,
Richter
JM
,
Nimptsch
K
, et al.
High School Diet and Risk of Crohn’s Disease and Ulcerative Colitis. Inflamm Bowel Dis. 2015;21(10):2311–9.
79
Lukens
JR
,
Gurung
P
,
Vogel
P
,
Johnson
GR
,
Carter
RA
,
McGoldrick
DJ
, et al.
Dietary modulation of the microbiome affects autoinflammatory disease. Nature. 2014;516(7530):246–9. doi:.https://doi.org/10.1038/nature13788
80
Hou
JK
,
Lee
D
,
Lewis
J
. Diet and inflammatory bowel disease: review of patient-targeted recommendations. Clin Gastroenterol Hepatol. 2014;12(10):1592–600. doi:.https://doi.org/10.1016/j.cgh.2013.09.063
81
Ananthakrishnan
AN
,
Khalili
H
,
Konijeti
GG
,
Higuchi
LM
,
de Silva
P
,
Fuchs
CS
, et al.
Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut. 2014;63(5):776–84. doi:.https://doi.org/10.1136/gutjnl-2013-305304
82
Dam
AN
,
Berg
AM
,
Farraye
FA
. Environmental influences on the onset and clinical course of Crohn’s disease-part 1: an overview of external risk factors. Gastroenterol Hepatol (N Y). 2013;9(11):711–7.
83
Ananthakrishnan
AN
. Environmental triggers for inflammatory bowel disease. Curr Gastroenterol Rep. 2013;15(1):302. doi:.https://doi.org/10.1007/s11894-012-0302-4
84
Molodecky
NA
,
Kaplan
GG
. Environmental risk factors for inflammatory bowel disease. Gastroenterol Hepatol (N Y). 2010;6(5):339–46.
85
Carbonnel
F
,
Jantchou
P
,
Monnet
E
,
Cosnes
J
. Environmental risk factors in Crohn’s disease and ulcerative colitis: an update. Gastroenterol Clin Biol. 2009;33(Suppl 3):S145–57. doi:.https://doi.org/10.1016/S0399-8320(09)73150-1
86
Halfvarson
J
,
Jess
T
,
Magnuson
A
,
Montgomery
SM
,
Orholm
M
,
Tysk
C
, et al.
Environmental factors in inflammatory bowel disease: a co-twin control study of a Swedish-Danish twin population. Inflamm Bowel Dis. 2006;12(10):925–33. doi:.https://doi.org/10.1097/01.mib.0000228998.29466.ac
87
Ruiz
PA
,
Moron
B
,
Becker
HM
, et al.
Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut. 2017;66(7):1216–24.
88
Rath
HC
. Role of commensal bacteria in chronic experimental colitis: lessons from the HLA-B27 transgenic rat. Pathobiology. 2002;70(3):131–8. doi:.https://doi.org/10.1159/000068144
89
Sadlack
B
,
Merz
H
,
Schorle
H
,
Schimpl
A
,
Feller
AC
,
Horak
I
. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75(2):253–61. doi:.https://doi.org/10.1016/0092-8674(93)80067-O
90
Villanacci
V
,
Falchetti
D
,
Liserre
B
,
Soresina
AR
,
Plebani
A
,
Ekema
G
, et al.
Diversion of the fecal stream resolves ulcerative colitis complicating chronic granulomatous disease in an adult patient. J Clin Gastroenterol. 2007;41(5):491–3. doi:.https://doi.org/10.1097/01.mcg.0000212638.44735.78
91
Winslet
MC
,
Allan
A
,
Poxon
V
,
Youngs
D
,
Keighley
MR
. Faecal diversion for Crohn’s colitis: a model to study the role of the faecal stream in the inflammatory process. Gut. 1994;35(2):236–42. doi:.https://doi.org/10.1136/gut.35.2.236
92
Ukena
SN
,
Singh
A
,
Dringenberg
U
,
Engelhardt
R
,
Seidler
U
,
Hansen
W
, et al.
Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS One. 2007;2(12):e1308. doi:.https://doi.org/10.1371/journal.pone.0001308
93
Kanauchi
O
,
Matsumoto
Y
,
Matsumura
M
,
Fukuoka
M
,
Bamba
T
. The beneficial effects of microflora, especially obligate anaerobes, and their products on the colonic environment in inflammatory bowel disease. Curr Pharm Des. 2005;11(8):1047–53. doi:.https://doi.org/10.2174/1381612053381675
94
Kruis
W
,
Fric
P
,
Pokrotnieks
J
,
Lukás
M
,
Fixa
B
,
Kascák
M
, et al.
Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004;53(11):1617–23. doi:.https://doi.org/10.1136/gut.2003.037747
95
Michail
S
,
Durbin
M
,
Turner
D
, et al.
Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm Bowel Dis. 2012;18(10):1799–808.
96
Watson
AJM
,
Biancheri
P
,
Patterson
A
. The Mucosal Microbiome and Recurrence After Surgery for Crohn’s Disease. Gastroenterology. 2016;150(7):1682–4. doi:.https://doi.org/10.1053/j.gastro.2016.04.026
97
Øyri
SF
,
Műzes
G
,
Sipos
F
. Dysbiotic gut microbiome: A key element of Crohn’s disease. Comp Immunol Microbiol Infect Dis. 2015;43:36–49. doi:.https://doi.org/10.1016/j.cimid.2015.10.005
98
Hall
LJ
,
Walshaw
J
,
Watson
AJ
. Gut microbiome in new-onset Crohn’s disease. Gastroenterology. 2014;147(4):932–4. doi:.https://doi.org/10.1053/j.gastro.2014.08.014
99
Rajca
S
,
Grondin
V
,
Louis
E
,
Vernier-Massouille
G
,
Grimaud
JC
,
Bouhnik
Y
, et al.
Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20(6):978–86.
100
Hofer
U
. Microbiome: bacterial imbalance in Crohn’s disease. Nat Rev Microbiol. 2014;12(5):312. doi:.https://doi.org/10.1038/nrmicro3255
101
Gevers
D
,
Kugathasan
S
,
Denson
LA
,
Vázquez-Baeza
Y
,
Van Treuren
W
,
Ren
B
, et al.
The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–92. doi:.https://doi.org/10.1016/j.chom.2014.02.005
102
Sadler
T
,
Bhasin
JM
,
Xu
Y
,
Barnholz-Sloan
J
,
Chen
Y
,
Ting
AH
, et al.
Genome-wide analysis of DNA methylation and gene expression defines molecular characteristics of Crohn’s disease-associated fibrosis. Clin Epigenetics. 2016;8(1):30. doi:.https://doi.org/10.1186/s13148-016-0193-6
103
Adams
AT
,
Kennedy
NA
,
Hansen
R
,
Ventham
NT
,
OʼLeary
KR
,
Drummond
HE
, et al.
Two-stage genome-wide methylation profiling in childhood-onset Crohn’s Disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm Bowel Dis. 2014;20(10):1784–93. doi:.https://doi.org/10.1097/MIB.0000000000000179
104
Nimmo
ER
,
Prendergast
JG
,
Aldhous
MC
,
Kennedy
NA
,
Henderson
P
,
Drummond
HE
, et al.
Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18(5):889–99. doi:.https://doi.org/10.1002/ibd.21912
105
Lin
Z
,
Hegarty
JP
,
Yu
W
,
Cappel
JA
,
Chen
X
,
Faber
PW
, et al.
Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig Dis Sci. 2012;57(12):3145–53. doi:.https://doi.org/10.1007/s10620-012-2288-z
106
Adolph
TE
,
Niederreiter
L
,
Blumberg
RS
,
Kaser
A
. Endoplasmic reticulum stress and inflammation. Dig Dis. 2012;30(4):341–6. doi:.https://doi.org/10.1159/000338121
107
Fritz
T
,
Niederreiter
L
,
Adolph
T
,
Blumberg
RS
,
Kaser
A
. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut. 2011;60(11):1580–8. doi:.https://doi.org/10.1136/gut.2009.206466
108
Hubbard-Lucey
VM
,
Shono
Y
,
Maurer
K
,
West
ML
,
Singer
NV
,
Ziegler
CG
, et al.
Autophagy gene Atg16L1 prevents lethal T cell alloreactivity mediated by dendritic cells. Immunity. 2014;41(4):579–91. doi:.https://doi.org/10.1016/j.immuni.2014.09.011
109
Cadwell
K
,
Patel
KK
,
Maloney
NS
,
Liu
TC
,
Ng
AC
,
Storer
CE
, et al.
Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141(7):1135–45. doi:.https://doi.org/10.1016/j.cell.2010.05.009
110
Geremia
A
,
Biancheri
P
,
Allan
P
,
Corazza
GR
,
Di Sabatino
A
. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):3–10. doi:.https://doi.org/10.1016/j.autrev.2013.06.004
111
Knights
D
,
Lassen
KG
,
Xavier
RJ
. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62(10):1505–10. doi:.https://doi.org/10.1136/gutjnl-2012-303954
112
Deretic
V
. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240(1):92–104. doi:.https://doi.org/10.1111/j.1600-065X.2010.00995.x
113
Spalinger
MR
,
Lang
S
,
Vavricka
SR
,
Fried
M
,
Rogler
G
,
Scharl
M
. Protein tyrosine phosphatase non-receptor type 22 modulates NOD2-induced cytokine release and autophagy. PLoS One. 2013;8(8):e72384. doi:.https://doi.org/10.1371/journal.pone.0072384
114
Spalinger
MR
,
Kasper
S
,
Gottier
C
,
Lang
S
,
Atrott
K
,
Vavricka
SR
, et al.
NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. 2016;126(5):1783–800. doi:.https://doi.org/10.1172/JCI83669
115
Spalinger
MR
,
Lang
S
,
Gottier
C
,
Dai
X
,
Rawlings
DJ
,
Chan
AC
, et al.
PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy. 2017;13(9):1590–601. doi:.https://doi.org/10.1080/15548627.2017.1341453
116
Eltzschig
HK
,
Bratton
DL
,
Colgan
SP
. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014;13(11):852–69. doi:.https://doi.org/10.1038/nrd4422
117
Palazon
A
,
Goldrath
AW
,
Nizet
V
,
Johnson
RS
. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–28. doi:.https://doi.org/10.1016/j.immuni.2014.09.008
118
de Vallière
C
,
Wang
Y
,
Eloranta
JJ
,
Vidal
S
,
Clay
I
,
Spalinger
MR
, et al.
G Protein-coupled pH-sensing Receptor OGR1 Is a Regulator of Intestinal Inflammation. Inflamm Bowel Dis. 2015;21(6):1269–81.
119
de Vallière
C
,
Vidal
S
,
Clay
I
,
Jurisic
G
,
Tcymbarevich
I
,
Lang
S
, et al.
The pH-sensing receptor OGR1 improves barrier function of epithelial cells and inhibits migration in an acidic environment. Am J Physiol Gastrointest Liver Physiol. 2015;309(6):G475–90. doi:.https://doi.org/10.1152/ajpgi.00408.2014
120
Wang
Y
,
de Vallière
C
,
Imenez Silva
PH
,
Leonardi
I
,
Gruber
S
,
Gerstgrasser
A
, et al.
The proton-activated receptor GPR4 modulates intestinal inflammation. J Crohn’s Colitis. 2017. [Epub ahead of print.] doi:.https://doi.org/10.1093/ecco-jcc/jjx147
121
Cosin-Roger
J
,
Simmen
S
,
Melhem
H
,
Atrott
K
,
Frey-Wagner
I
,
Hausmann
M
, et al.
Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017;8(1):98. doi:.https://doi.org/10.1038/s41467-017-00213-3