Identification of a MAFB mutation in a patient with multicentric carpotarsal osteolysis
DOI: https://doi.org/10.4414/smw.2017.14529
Lei
Zhuanga, Sabine
Adlerb, Daniel
Aeberlib, Peter M.
Villigerb, Beat
Truebab
aDepartment for BioMedical Research, University of Bern, Switzerland
bDepartment of Rheumatology, University Hospital, Bern, Switzerland
Summary
Multicentric carpotarsal osteolysis (MCTO) is an autosomal dominant disease of the skeleton characterised by progressive destruction of carpal and tarsal bones. Recently, it has been demonstrated that this disease is caused by heterozygous mutations in the gene for the transcriptional repressor MAFB. We analysed genomic DNA and RNA from leucocytes of a female patient diagnosed with MCTO. We identified the mutation c.161C>T in the genomic sequence and in the expressed messenger RNA for MAFB. This is the second report of the c.161C>T mutation in a MCTO patient. Since the parents do not possess this mutation, the daughter must have acquired a de novo mutation. At the level of the gene, this mutation is found at a CpG dinucleotide sequence, suggesting that DNA methylation was involved in the occurrence of the DNA aberration. At the level of the protein, the mutation exchanges a serine with a leucine residue at a position on MAFB that can become phosphorylated in the wild-type protein. MAFB negatively regulates the RANKL-dependent differentiation of monocytes into osteoclasts. It is likely that the mutation will affect the phosphorylation status of the protein and its biological activity. When the activity of the transcriptional repressor is reduced, osteoclastogenesis will be increased, which might explain the carpotarsal bone destruction observed in the patient.
Introduction
Multicentric carpotarsal osteolysis (MCTO, MIM 166300) is a rare disease of the skeleton characterised by aggressive destruction and disappearance of the carpal and tarsal bones, with other bones, including the metacarpals, being barely affected. Often, the patients show slight craniofacial abnormalities such as micrognathia and a triangle face, and frequently they develop a progressive nephropathy, which may lead to renal failure. The disease can be inherited in an autosomal dominant fashion, although most of the cases appear to represent sporadic events that are caused by de novo mutations [1–3].
In 2012, Zankl et al. [1] carefully analysed 11 simplex cases with MCTO as well as two families with autosomal dominant MCTO. By next-generation sequencing, the authors identified ten different missense mutations in the gene for the transcription factor MAFB. All these mutations were found in a short region of 51 base-pairs, corresponding to the amino terminal transactivation domain of the transcription factor. The results were subsequently confirmed by Mehawej et al. [2], who studied eight MCTO cases from six families, and by Mumm et al. [3], who studied nine MCTO patients from eight families.
MAFB is a member of the MAF family of basic leucine zipper transcription factors [4, 5]. This family includes seven members, four large MAF proteins (MAFA, MAFB, c-MAF and NRL) and three small MAF proteins (MAFG, MAFF, MAFK) that lack the transactivation domain. The MAF proteins belong to the activator protein-1 (AP1) superfamily of transcription factors, which are known to form homo- and heterodimers via an extended leucine zipper [6]. The basic domain that precedes this leucine zipper enables the interaction of the protein with the MAF recognition element (MARE) on target genes. MAFB can form dimers with many other AP1 members (MAFA, MAFB, c-MAF, FOS, FOSB, FRA1, FRA2, JUN, BACH1) and as a consequence it is involved in the development of many organs and tissues. Experiments with mice and chickens have demonstrated that MAFB is involved in the segmentation of the hindbrain [7], in the differentiation of pancreatic β-cells [8], in the survival of podocytes in the kidney [9] and in the differentiation of monocytes [10].
In the haematopoietic system, MAFB is specifically expressed in the myeloid lineage that includes monocytes, macrophages and osteoclasts. It appears to induce the differentiation of multipotent progenitor cells into monocytes and macrophages [10], but to negatively regulate the differentiation into osteoclasts [11]. The negative regulation is accomplished by inhibiting RANKL-mediated osteoclastogenesis at the transcriptional level via binding to transcription factors FOS and MITF. Mutations in MAFB, as found in MCTO patients, might therefore release the block of osteoclast differentiation.
Materials and methods
Isolation of genomic DNA
DNA was purified from freshly drawn blood with use of the Wizard genomic DNA purification kit from Promega (Madison, WI, USA). Written consent was obtained from all probands to use the samples for genetic testing and to publish the results in accordance with the guidelines of the Swiss Society of Medical Genetics. Red blood cells were removed by specific cell lysis. White blood cells were collected by centrifugation and lysed in Nuclei Lysis Solution. RNA was digested with RNase and cellular proteins were removed by salt precipitation. Finally, the genomic DNA was precipitated with isopropanol and washed with ethanol.
RNA isolation
Total RNA was extracted from blood by means of the QIAamp RNA Blood Mini kit from Qiagen (Hilden, Germany). Samples of fresh blood (2 ml) were collected in the presence of the anticoagulant EDTA and immediately processed. Erythrocytes were lysed in hypotonic buffer. Leucocytes were collected by centrifugation, lysed with a QIAshredder and dissolved in RLT buffer. The RNA was adsorbed to a silica membrane, washed and eluted with TE buffer. The purified RNA was transcribed into first-strand cDNA using random hexamer primers and Improm-II Reverse Transcriptase (Promega, Madison, WI, USA).
PCR amplification and DNA sequencing
Fragments of interest were amplified via the polymerase chain-reaction (PCR) through 35 cycles (20" at 98°C, 15" at 65°C, 15–50'' at 72°C) with KAPA high fidelity DNA polymerase (KAPA Biosystems, Boston, USA). The following primer pairs were used: ACGTGAAGAAGGAGCCACTG / TGTGTCTTCTGTTCGGTCGG (product 154 bp), AGAGCAAGAGAGCTAGAGAGC / ATGGCCTTCCTGACTTCTCG (product 1647 bp). The fragments were separated on 1% agarose gels. Resolved fragments were recovered from the gels with the GeneElute extraction kit (Sigma, St. Louis, MO, USA). DNA sequences were determined by Sanger cycle sequencing on an ABI 3730 platform and analysed with the program package of MacVector Inc. (Apex, NC, USA).
Parental DNA testing
To unequivocally prove a parental relationship between father, mother and daughter, DNA profiling was done with a set of informative microsatellite markers. Genomic DNA of father, mother and daughter was used to amplify 15 highly polymorphic short tandem repeats (STRs) with the PowerPlex® 16 HS System (Promega, Madison, WI, USA). The STRs included D3S1358, TH01, D21S11, D18S51, D5S818, D13S317, D7S820, D16S539, CSF1PO, vWA, D8S1179, TPOX, FGA, PentaD and PentaE. The PCR fragments were resolved by capillary electrophoresis on a 3730xl DNA analyser (ABI Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) and the resulting data were analysed with the GeneMarker® HID software from SoftGenetics (State College, PA, USA).
Results
The female patient, who is currently 20 years old, developed pain of her right wrist at the age of 4 years. She received intra-articular injections of glucocorticoids, followed by weekly doses of methotrexate. Because of progressive joint erosions, the tumour necrosis factor blocker etanercept was added at the age of 7 years. Bone destruction progressed despite therapy and a histological examination showed activated osteoclasts and signs of fibrosis. Concomitant proteinuria prompted a kidney biopsy at the age of 13 years, which revealed low-grade focal glomerulosclerosis that finally led to the diagnosis of carpotarsal osteolysis with nephropathy (MCTON). Blood levels of calcium, phosphate, vitamin D3 and parathyroid hormone were normal. A tarsal bone biopsy at the age of 18 showed enhanced bone resorption and fibrosis. A single dose of denosumab (60 mg) was able to reduce the inflammation as demonstrated with magnetic resonance imaging (MRI) 9 months later.
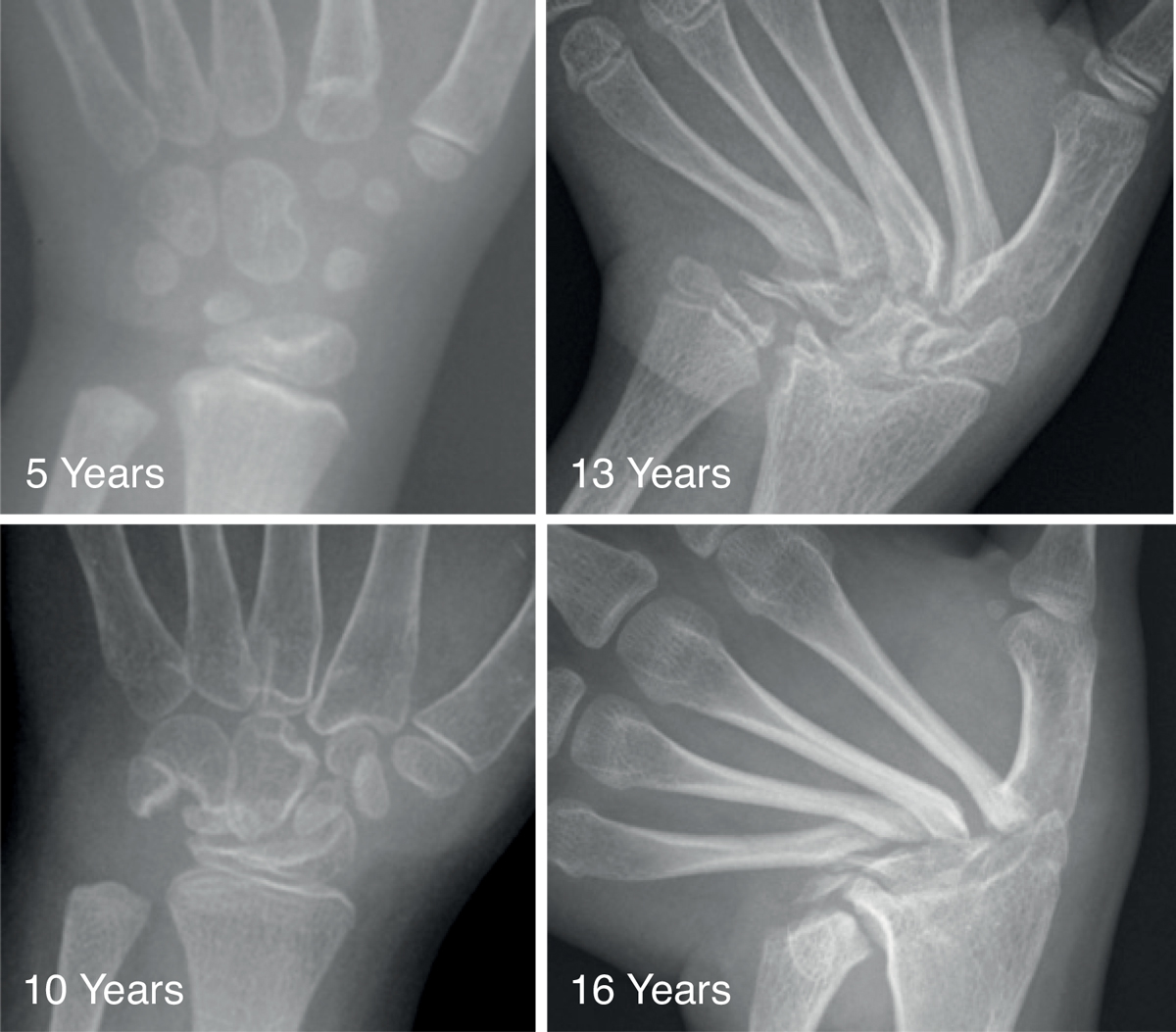
The course of the carpal bone destruction is presented in fig. 1. At the age of 5, the index patient possessed all carpal bones, but some alterations in location and orientation were evident. At the age of 10, the carpal bones showed erosions and partial destruction. At 13 years, the carpal bones and the epiphyses of the ulna and radius were almost completely resorbed. At 16 years, the proximal ends of the metacarpals showed progressive thinning and sclerosis, with ulnar deviation in the left hand. The tarsal bones revealed multilocular erosions at the age of 9 years. In contrast to the carpal bones, they did not become resorbed, but the erosions led to osteoarthritis (not shown).
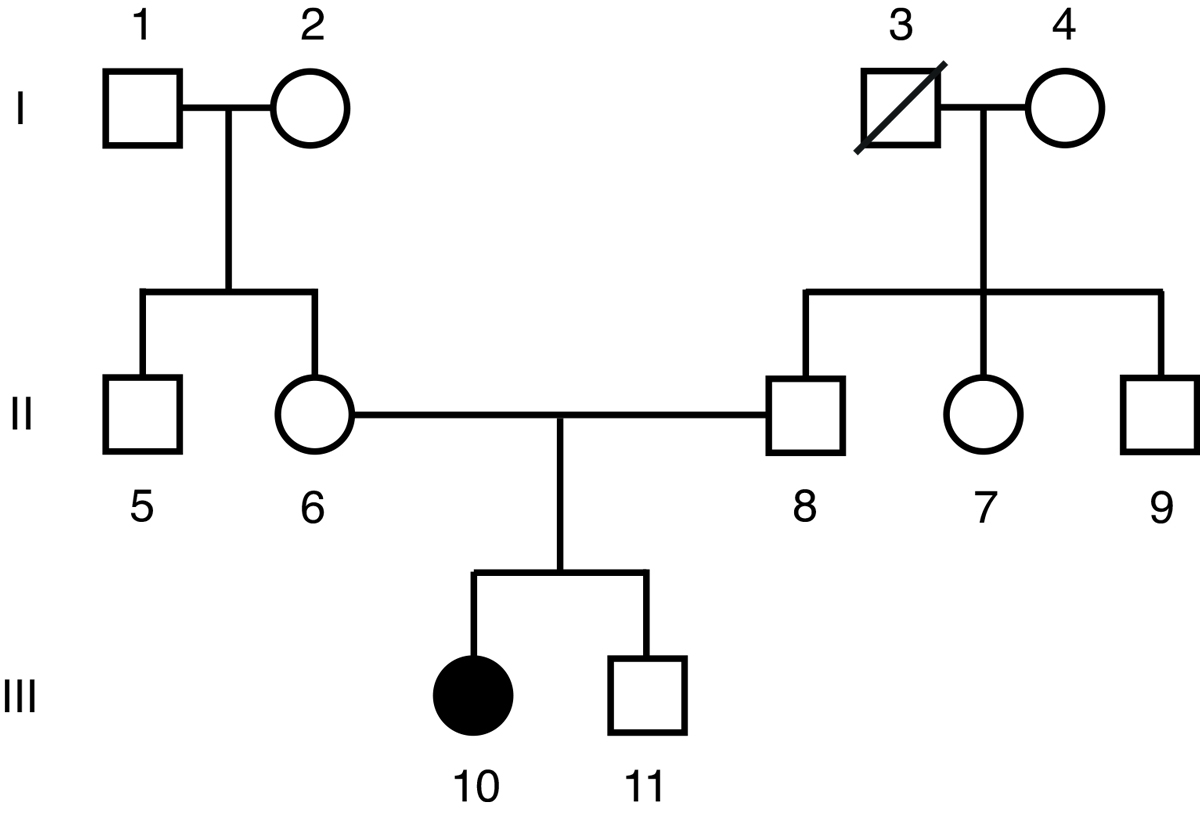
A pedigree of the family of the index patient is presented in fig. 2. Neither the parents nor the brother showed any signs of the disease. A careful analysis of 15 different microsatellite markers proved that the parents were in fact the biological parents of the index patient (probability of parental offspring >99.9999%). Our MCTO patient must therefore have acquired a de novo mutation.
Genomic DNA and total RNA were isolated from leucocytes of freshly drawn blood. Sequencing of the single exon of MAFB on both strands of the genomic DNA revealed the heterozygous mutation c.161C>T (fig. 3). At the protein level, this mutation led to the substitution of serine-54 in MAFB by leucine (p.S54L). Neither father nor mother carried this mutation, confirming the presence of a de novo mutation (fig. 3). Quantification of the areas under the chromatogram of the index patient revealed 49% C and 51% T (n = 3), indicating complete penetrance of the heterozygous disease in leucocytes. Sequencing of the MAFB mRNA after reverse transcription into cDNA proved the same c.161C>T mutation. Quantification of the areas under the chromatogram revealed a similar proportion of C and T (47% C, 53% T), suggesting that the two alleles of the MAFB gene were transcribed into mRNA with similar efficiency (not shown).
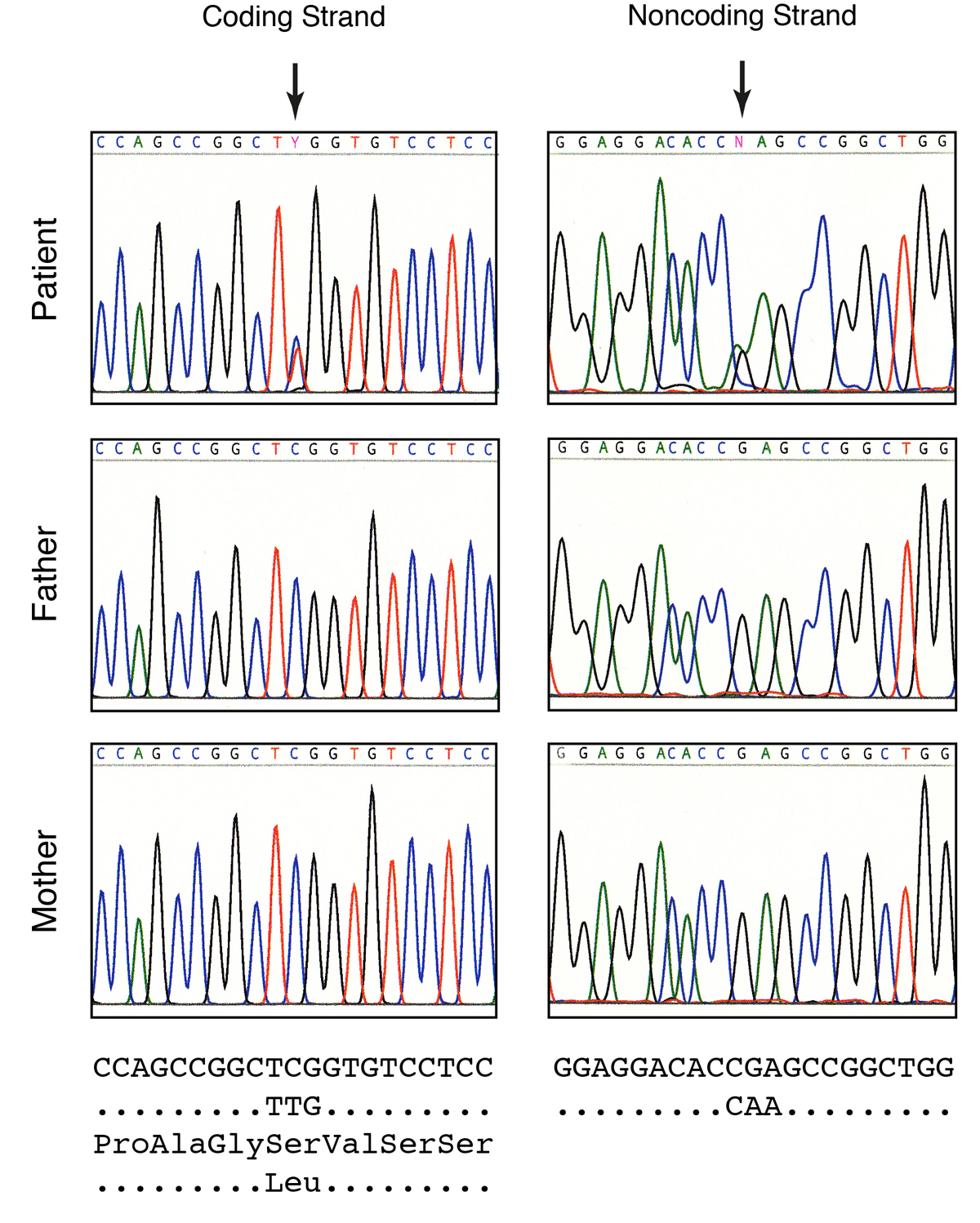
Figure 3
Sequencing of the MAFB gene. Parts of the original sequencing chromatograms are shown together with the genomic DNA sequence and the predicted amino acid sequence. The patient reveals a C>T transition that is found on the coding as well as on the noncoding strand. The heterozygous mutation substitutes a serine by a leucine residue. Note that neither father nor mother contains this mutation.
Discussion
The genetic cause of MCTO was discovered 5 years ago when Zankl et al. [1] reported that patients diagnosed with the disease contain mutations in the gene for the transcriptional repressor MAFB. So far, 15 different missense mutations that affect the codons for 10 different amino acids have been reported [1–3]. All these mutations cluster in the transactivation domain of MAFB, which is thought to interact with other proteins of the transcription machinery. Our patient has a heterozygous mutation at nucleotide-161 (c.161C>T), which changes serine-54 into leucine (p.Ser54Leu). This is the second report of the c.161C>T mutation in a MCTO patient; the same mutation was found by Zankl et al. [1] in two families with autosomal dominant MCTO. Furthermore, Mehawej et al. [2] described a different mutation at the same position (c.161C>G), which changes serine-54 into tryptophan. Of note, all 15 mutations described so far in MCTO patients are situated within a short region of MAFB (residues 54-71) that can become phosphorylated in the wild-type protein (fig. 4). This region encompasses eight serine and two threonine residues. Experiments with a phospho-specific antibody showed that at least two of these residues, T58 and T62, could be phosphorylated by glycogen synthase kinase-3 (GSK3) [12]. Phosphorylation has been studied in more detail with the homologous protein MAFA. MAFA is phosphorylated on residues S49, T53, T57, S61 and S65 in a sequential fashion [13]. These residues correspond to S54, T58, T62, S66 and S70 in MAFB (fig. 4). Interestingly, phosphorylation of MAFA increases its biological activity, but at the same time induces its degradation [14]. Since the sequences of MAFA and MAFB are highly conserved in this particular region, it is likely that phosphorylation of S54 in MAFB, which would correspond to the mutated site of our patient, will also affect the activity of the protein.
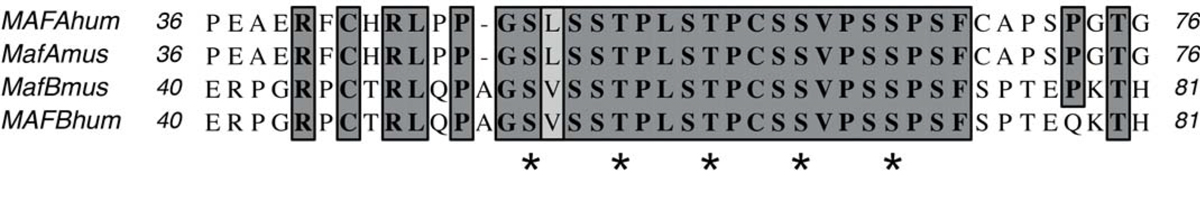
Figure 4
Alignment of the amino acid sequences of MAFA and MAFB from mice and humans at the region that is mutated in MCTO patients. Identical residues are boxed. Serine and threonine residues, which can become phosphorylated by GSK3, are indicated by asterisks. The mutation of the index patient corresponds to serine-54, which represents the first of the five phosphorylation sites.
MAFB is known as an important modulator of osteoclastogenesis. It induces development of monocytes from progenitor cells [10], but at the same time inhibits their terminal differentiation into active osteoclasts [11]. Osteoclastogenesis is controlled by the RANK/RANKL signalling pathway [15]. RANKL-deficient mice show severe osteopetrosis with a complete lack of osteoclasts [16]. RANKL overexpressing mice, in contrast, have an osteoporotic phenotype with loss of trabecular bone [17]. MAFB appears to affect RANK/RANKL signalling by interfering at the genomic level with the expression of transcription factors NFATc1 and OSCAR. There is experimental evidence that MAFB binds to the transcriptional activators FOS and MITF and prevents them from binding to their target genes, which would include NFTAc1 and OSCAR [11]. When MAFB activity is missing, FOS can interact with NFATc1 and enhance transcription of NFATc1 in a positive feedback loop, whereas MITF can combine with NFTAc1 and drive transcription of OSCAR. In combination, the two proteins will induce the programme of osteoclast differentiation and ultimately lead to the breakdown of bone.
Our patient must have acquired a de novo mutation since the parents do not possess the mutation. This is in contrast to the probands of the two families investigated by Zankl et al., who obviously inherited the c.161C>T mutation from one of their parents. This publication is therefore the first description of a de novo mutation at this particular position of the MAFB gene. It appears that this region represents a hotspot for mutation. The codon for serine 54 (TCG) contains a CpG dinucleotide, which can become methylated at the 5′ position of cytosine. DNA methylation is the only major modification of the genomic DNA in humans [18]. Typically, methylated regions of the DNA are inactive and tightly packed into condensed heterochromatin, whereas unmethylated regions are transcriptionally active. Five-methylcytosine can spontaneously convert to thymine [18, 19]. During evolution, the human genome has gradually lost most of its CpG dinucleotide sequences and those CpG dinucleotides that persisted are mostly restricted to CpG islands. Such islands are typically found at the 5′ regions of genes (promoters), which interact with regulatory factors that shield the DNA from methylation. In fact, our CpG site is located in a CpG island that includes the 5′ half of the MAFB gene (Fig. 5). This gene might stay free of methylation during development when it is actively transcribed. However, during spermatogenesis up to 90% of the CpG dinucleotides become methylated in order to keep mature sperm cells in a transcriptionally inactive state [20]. After modification, 5-methylcytosine can mutate with low frequency to thymine and when this aberration is not repaired, the sperm cells incorporate a mutation that can be passed on to the offspring. It is of interest to note that 5 out of the 15 MAFB mutations described today are C>T transitions occurring at CpG dinucleotides. In fact, approximately 37% of all point mutations responsible for genetic human diseases are transitional mutations found at CpG dinucleotides [21].
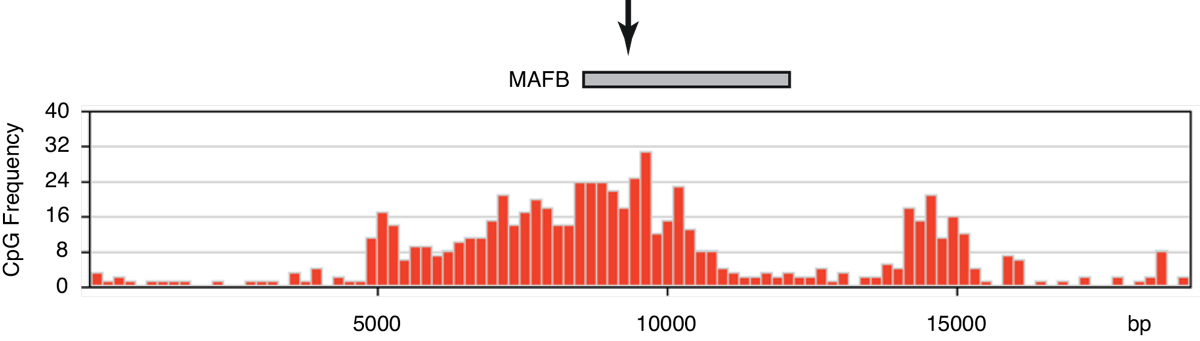
Figure 5
The mutation occurs in a typical CpG island. The frequency of CpG dinucleotides within a window of 200 is depicted along the genomic sequence of human chromosome 20 at the region of the MAFB gene. A grey box shows the single exon of the MAFB gene. An arrow indicates the site of mutation. This site is located in a region with relatively high CpG content (25 CpG/200 nt), whereas the regions further upstream and downstream have a very low CpG content (0–3 CpG/200 nt).
Why is the osteolysis in MCTO patients restricted to the carpal and tarsal bones? What makes these bones so special that they are completely resorbed while other bones, including the metacarpals, are barely affected? Carpal bones differ in at least two aspects from tubular bones, although they too grow by endochondral ossification. Carpal bones are spherical bones that grow on all sides, whereas tubular bones are bipolar and grow only between the epiphysis and the diaphysis. The term acrophysis has therefore been introduced for the growth plates of carpal and tarsal bones [22]. Like the metaphysis of long bones, the acrophysis contains cartilage cells, which are lined up in defined layers (resting cells, zone of division, zone of columniation, hypertrophic zone and zone of calcification). Acrophyses are not only found in carpal and tarsal bones but also in the patella. In our patient, the patella is not affected. Another difference between carpal bones and long bones is the formation of multiple joints. Tubular bones contain two regions that form joints with adjacent bones, but carpal bones are involved in a large number of joints. The human wrist is made up of eight bones that enable articulation with the metacarpal bones of the digits, with the radius and the ulna of the forearm and among themselves. Most of these bones (except the pisium) have six surfaces. Three to four of these surfaces (depending on whether they are in contact with contiguous bones) are articular and consequently covered by hyaline cartilage. Little is known about the molecular mechanisms that regulate the formation of such multiple joints. It is possible that these mechanisms are responsible for the specific location of the lesions observed in MCTO.
It remains to be demonstrated how all these findings would translate into a better treatment of MCTO patients. At the moment, the only feasible treatment appears to be the inhibition of terminal differentiation of monocytes into osteoclasts. This can be achieved by the administration of neutralising antibodies against RANKL such as denosumab.
References
1
Zankl
A
,
Duncan
EL
,
Leo
PJ
,
Clark
GR
,
Glazov
EA
,
Addor
MC
, et al.
Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am J Hum Genet. 2012;90(3):494–501. doi:. Correction in: Am J Hum Genet. 2014;94(4):643.doi: https://doi.org/10.1016/j.ajhg.2012.01.003
2
Mehawej
C
,
Courcet
JB
,
Baujat
G
,
Mouy
R
,
Gérard
M
,
Landru
I
, et al.
The identification of MAFB mutations in eight patients with multicentric carpo-tarsal osteolysis supports genetic homogeneity but clinical variability. Am J Med Genet A. 2013;161(12):3023–9. doi:.https://doi.org/10.1002/ajmg.a.36151
3
Mumm
S
,
Huskey
M
,
Duan
S
,
Wenkert
D
,
Madson
KL
,
Gottesman
GS
, et al.
Multicentric carpotarsal osteolysis syndrome is caused by only a few domain-specific mutations in MAFB, a negative regulator of RANKL-induced osteoclastogenesis. Am J Med Genet A. 2014;164(9):2287–93. doi:.https://doi.org/10.1002/ajmg.a.36641
4
Tsuchiya
M
,
Misaka
R
,
Nitta
K
,
Tsuchiya
K
. Transcriptional factors, Mafs and their biological roles. World J Diabetes. 2015;6(1):175–83. doi:.https://doi.org/10.4239/wjd.v6.i1.175
5
Eychène
A
,
Rocques
N
,
Pouponnot
C
. A new MAFia in cancer. Nat Rev Cancer. 2008;8(9):683–93. doi:.https://doi.org/10.1038/nrc2460
6
Shaulian
E
,
Karin
M
. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131–6. doi:.https://doi.org/10.1038/ncb0502-e131
7
Giudicelli
F
,
Gilardi-Hebenstreit
P
,
Mechta-Grigoriou
F
,
Poquet
C
,
Charnay
P
. Novel activities of Mafb underlie its dual role in hindbrain segmentation and regional specification. Dev Biol. 2003;253(1):150–62. doi:.https://doi.org/10.1006/dbio.2002.0864
8
Matsuoka
TA
,
Zhao
L
,
Artner
I
,
Jarrett
HW
,
Friedman
D
,
Means
A
, et al.
Members of the large Maf transcription family regulate insulin gene transcription in islet beta cells. Mol Cell Biol. 2003;23(17):6049–62. doi:.https://doi.org/10.1128/MCB.23.17.6049-6062.2003
9
Sadl
V
,
Jin
F
,
Yu
J
,
Cui
S
,
Holmyard
D
,
Quaggin
S
, et al.
The mouse Kreisler (Krml1/MafB) segmentation gene is required for differentiation of glomerular visceral epithelial cells. Dev Biol. 2002;249(1):16–29. doi:.https://doi.org/10.1006/dbio.2002.0751
10
Kelly
LM
,
Englmeier
U
,
Lafon
I
,
Sieweke
MH
,
Graf
T
. MafB is an inducer of monocytic differentiation. EMBO J. 2000;19(9):1987–97. doi:.https://doi.org/10.1093/emboj/19.9.1987
11
Kim
K
,
Kim
JH
,
Lee
J
,
Jin
HM
,
Kook
H
,
Kim
KK
, et al.
MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109(8):3253–9. doi:.https://doi.org/10.1182/blood-2006-09-048249
12
Herath
NI
,
Rocques
N
,
Garancher
A
,
Eychène
A
,
Pouponnot
C
. GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J. 2014;4(1):e175. doi:.https://doi.org/10.1038/bcj.2013.67
13
Rocques
N
,
Abou Zeid
N
,
Sii-Felice
K
,
Lecoin
L
,
Felder-Schmittbuhl
MP
,
Eychène
A
, et al.
GSK-3-mediated phosphorylation enhances Maf-transforming activity. Mol Cell. 2007;28(4):584–97. doi:.https://doi.org/10.1016/j.molcel.2007.11.009
14
Benkhelifa
S
,
Provot
S
,
Nabais
E
,
Eychène
A
,
Calothy
G
,
Felder-Schmittbuhl
MP
. Phosphorylation of MafA is essential for its transcriptional and biological properties. Mol Cell Biol. 2001;21(14):4441–52. doi:.https://doi.org/10.1128/MCB.21.14.4441-4452.2001
15
Whyte
MP
,
Mumm
S
. Heritable disorders of the RANKL/OPG/RANK signaling pathway. J Musculoskelet Neuronal Interact. 2004;4(3):254–67.
16
Kim
N
,
Odgren
PR
,
Kim
DK
,
Marks
SC, Jr
,
Choi
Y
. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci USA. 2000;97(20):10905–10. doi:.https://doi.org/10.1073/pnas.200294797
17
Rinotas
V
,
Niti
A
,
Dacquin
R
,
Bonnet
N
,
Stolina
M
,
Han
CY
, et al.
Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res. 2014;29(5):1158–69. doi:.https://doi.org/10.1002/jbmr.2112
18
Messerschmidt
DM
,
Knowles
BB
,
Solter
D
. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014;28(8):812–28. doi:.https://doi.org/10.1101/gad.234294.113
19
Mugal
CF
,
Ellegren
H
. Substitution rate variation at human CpG sites correlates with non-CpG divergence, methylation level and GC content. Genome Biol. 2011;12(6):R58. doi:.https://doi.org/10.1186/gb-2011-12-6-r58
20
Popp
C
,
Dean
W
,
Feng
S
,
Cokus
SJ
,
Andrews
S
,
Pellegrini
M
, et al.
Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–5. doi:.https://doi.org/10.1038/nature08829
21
Cooper
DN
,
Youssoufian
H
. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78(2):151–5. doi:.https://doi.org/10.1007/BF00278187
22
Oestreich
AE
. The acrophysis: a unifying concept for enchondral bone growth and its disorders. I. Normal growth. Skeletal Radiol. 2003;32(3):121–7. doi:.https://doi.org/10.1007/s00256-002-0604-y