Genetic determinants of the epigenome in development and cancer
DOI: https://doi.org/10.4414/smw.2017.14523
Wellcome Trust Centre for Cell Biology, University of Edinburgh, UK
Summary
Although we have detailed maps of epigenetic marks on DNA and chromatin for many cell types and disease states, the origin and significance of these patterns is incompletely understood. Deregulation of the epigenome is a frequent accompaniment to cancer, and it is therefore important that we learn how it contributes to tumour formation. Here it is proposed that the roles of DNA sequence signals as determinants of the epigenome have been underappreciated. Taking as a paradigm the part played by the dinucleotide CpG in regulating gene expression via its effects on the epigenome, it is suggested that factors recognising other short, frequent sequence motifs also recruit chromatin modifying enzymes in response to DNA sequence. A screen for factors of this kind promises to aid our understanding of the mechanisms by which gene activity is globally regulated.
Introduction
Proteins that recognise specific DNA base sequences are uniquely able to target biological activity to a specific “address” in genomic DNA. It follows that development and maintenance of multiple cell types will be largely achieved by varying the availability of sequence-specific factors of this kind, which in turn enable specific genetic programmes by activating or repressing genes. Attractive though this hypothesis may be, there is evidence that the “transcription factors-only” scenario is oversimplified. Specifically, epigenetic marking, laid down during the developmental history of the cell, may be required to “condition” the response of the genome to transcription factors. In support of a role for epigenetics, reprogramming of cell fate exclusively by transcription factors is an inefficient process [1], suggesting that chromatin conditioning may help to buffer the cell against phenotypic change. Moreover, an increasing number of disease states involve mis-regulation of readers, writers or erasers of epigenetic information, emphasising their key role as modulators of gene activity. This is particularly striking in the case of cancer, where global redistribution of DNA methylation is well documented, but not fully understood [2]. These findings highlight our need to understand the forces that define the epigenome. It is normally considered that developmental history, disease or the influence of the environment are primary epigenome determinants (fig. 1) and consequently that disease states may also be triggered in this way. This article considers the alternative possibility that the epigenetic patterning is determined to a significant extent by the underlying genomic DNA sequence.
Patterns of DNA methylation
Methylation of the vertebrate genome is largely biphasic. The vast majority of DNA, including gene bodies, intergenic DNA transposable elements and other repeats, is highly methylated at CpGs, but a small fraction (about 2%) comprises CpG islands (CGIs), which are methylation-free patches surrounding promoters (fig. 2). A long-standing paradox is that CGIs are usually free of DNA methylation, despite containing an abnormally high concentration of the methylate-able sequence CpG [3]. Earlier work from this laboratory and others showed that in mice, sites for the transcription factor Sp1 are required to prevent DNA methylation [4, 5]. More recently the Schübeler laboratory has documented the negative influence of transcription factor-binding sites on local methylation status [6, 7]. Although reduced DNA methylation at promoters and enhancers is partly due to binding of transcription factors, presumably via steric interference with DNA methyltransferase (Dnmt) access [7], this mechanism is unlikely to explain the CGI phenomenon where methylation is absent at many CpG sites over hundreds of base pairs. An alternative explanation is that de novo Dnmts are excluded by chromatin bearing the H3K4me3 mark [8]. Indeed CGIs mark many promoters and frequently coincide with peaks of H3K4me3 and this is therefore likely to contribute to their immunity to DNA methylation. Promoter activity is not required to exclude DNA methylation, however, as artificial CGI-like constructs that are transcriptionally inert nevertheless remain DNA methylation-free [9]. This may be due to the presence of CXXC domains in all H3K4 methyltransferases, which recognise and bind to the CpG dinucleotide regardless of transcription and may recruit these enzymes to CGIs. Further studies of artificial CGIs have uncovered evidence that, in addition to CpG density, base composition per se is important for CGI function, as AT-rich DNA reproducibly succumbs to DNA methylation, whereas GC-rich DNA is methylation resistant. These AT-rich sequences can still recruit H3K4 methylation when DNA methylation is excluded (that is, in Dnmt3a/b double mutant cells), but this is evidently insufficient to render them methylation-free in wild-type cells [9]. Exclusion of DNA methylation is observed with a variety of unrelated synthetic sequences integrated into the embryonic stem cell genome, even if the density of CpGs is kept at a high level typical of CGIs. The transition between effective absence of methylation (<10%) and dense methylation (>85%) is quite sharp, with a midpoint near 55% GC. These results provide unexpected evidence for an instructive effect of base composition per se on the epigenome.
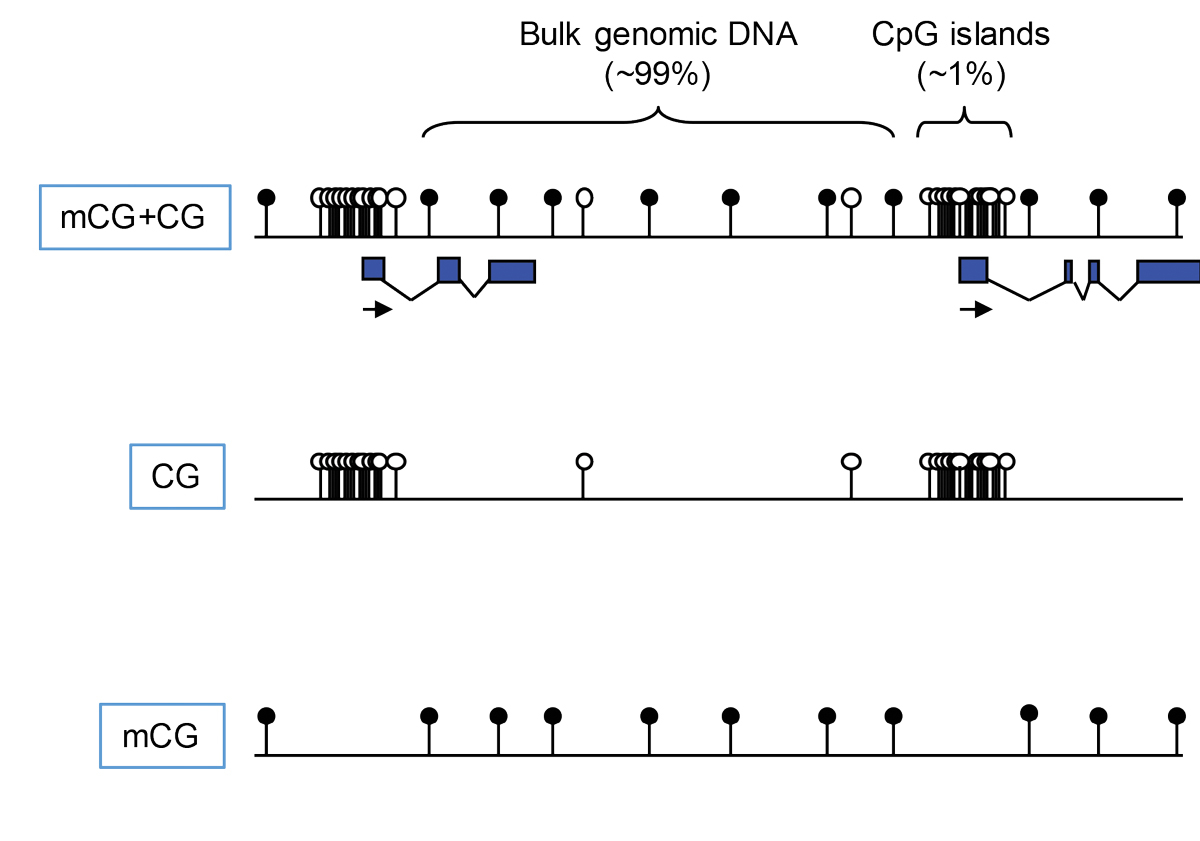
Figure 2 CpG and methyl-CpG have very different distributions across the genome. The dinucleotide CpG occurs in two chemical forms that attract or repel different protein complexes (e.g., via CpG- or mCpG-binding proteins).
Beyond CGIs, there are numerous examples where long range alterations in DNA methylation levels correlate with DNA base composition. This is particularly evident in cancer. For example, opposite shifts in the global distribution of DNA methylation (i.e., AT-rich → GC-rich) are prominent when normal human colon cells are compared with colorectal cancer cells [2]. This also occurs in normal tissues, as our recent analysis of DNA methylation landscapes in the human brain illustrates [10]. Analysis of differentially methylated regions shows that in the cerebellum, sequences losing DNA methylation relative to other brain regions share a GC-rich base composition, whereas regions gaining methylation are relatively GC-poor. These results point to a global shift in DNA methylation, away from GC-rich towards AT-rich sequences. Strikingly, cerebellum resembles ES cells, which also show absence of CGI methylation, but high methylation of the AT-rich bulk genome. Why the cerebellum should exhibit such a different DNA methylome from other parts of the brain is entirely unknown. Hypothetically, global redistribution of DNA methylation may adjust and optimise gene expression levels specific to this brain region, though experimental evidence in support of this explanation is currently lacking.
DNA methylation and cancer – the role of mutation
The role of DNA methylation in cancer has been the subject of intense study, but uncertainty remains regarding its precise causal relevance. The cancer DNA methylome is evidently abnormal in several respects. In particular, CGI promoters are often methylated and associated with silencing of the associated gene. Also, as mentioned above, hypomethylation of large genomic domains is frequent. But whether these effects are a primary or secondary effect of disease and what upstream influences are responsible remain tantalisingly ill-defined. Potentially, de novo methylation may cause a gene silencing event that initiates the transition of a cell to a pre-cancerous state (e.g., by shutting down a tumour suppressor gene). This would be the most direct possible involvement of an epigenetic change in tumorigenesis. Examples of “primary constitutional epimutations” of this kind are rare, however, as most de novo methylation events are accompanied by DNA sequence changes and these mutations are likely to be the primary genetic trigger [11]. For example, the H19 gene is normally mono-allelically expressed owing to imprinting, but de novo methylation of the imprinting control region on the maternal allele often accompanies paediatric Wilm’s tumour. In 98 to 99% of cases, however, methylation is accompanied by a base sequence change, indicating that the epigenetic change is almost always secondary. Similarly, a variety of tumours can be initiated by de novo methylation of the promoter of the MLH1 gene, whose product is required for efficient mismatch repair, but only in 1 to 10% of cases is a primary constitutional epimutation implicated. In the case of MLH2, another mismatch repair gene, all examples of de novo CGI methylation so far associated with tumorigenesis appear to be downstream of an altered DNA sequence. Thus examples where epigenetic changes are clearly the root cause of cancer are few and far between. This is not to argue that DNA methylation plays no role in tumorigenesis, as there are many examples where its secondary involvement appears to reinforce tumour growth or survival. Indeed, DNA methyltransferase inhibitors provide benefit in the clinic, indicating a key supporting role for epigenetic changes that may be further exploited therapeutically [12]. Moreover, DNA methylomes have emerged as useful biomarkers for tumour classification and progression [13].
CpG – a short, frequent motif that influences the epigenome
How might base sequence influence the DNA methylome, or, more broadly, the epigenome as a whole? Here I discuss the possibility that local features of the genomic DNA sequence combine to exert a global influence on chromatin modification and gene activity via DNA binding proteins that recognise short, frequent sequence motifs [14]. The best understood precedent for this assertion comes from studies of the dinucleotide CpG. Despite the limited information content of a two base-pair sequence, this motif has several features that appear to adapt it as a genomic signalling module: (i) it can occur in several chemical states as a result of methylation or hydroxymethylation (plus other oxidation states) of the cytosine moiety on position 5 of the cytosine ring; (ii) it is symmetrical, meaning it is paired with the same sequence on the anti-parallel opposite strand, a property that provides a mechanism for semi-conservatively copying CpG methylation patterns onto the daughter strand at DNA replication; (iii) its frequency is highly variable in the genome, ranging from dense clusters at CGIs, to sparse underrepresentation in most of the genome due to the mutagenic pressure associated with DNA methylation; and (iv) last but not least, we know of proteins that recognise different chemical forms of CpG and appear to mediate effects on genome function [15, 16].
Maps of methylated and unmethylated CpGs often include both forms together, usually as interspersed open and filled circles. Given that the two motifs represent distinct DNA binding signals, however, there is a case for displaying them separately. The resulting maps emphasise their contrasting distributions (see fig. 2). The CpG map greatly emphasises the clustering of unmethylated sites at CGIs, whereas the mCpG map renders CGIs almost invisible. It is evident that two such distinct landscapes will be interpreted very differently by proteins with a specific affinity for one motif. CpG readers include Cfp1, KDM2a, KDM2b and Mll2, all of which possess a CXXC domain that binds exclusively to unmethylated CpG in duplex DNA [17–22]. In contrast, MeCP2, MBD1, MBD2 and UHRF1, among others, require methylated CpG to bind [23–26]. Most of these motif readers influence chromatin modification states by recruiting enzymatic complexes. Cfp1, for example, is part of the Set1 complex that methylates lysine 4 of histone H3 (H3K4me3) and is found at CGIs. The coincidence immediately suggests that Cfp1, by targeting CGIs, may recruit the Set1 complex leading to deposition of H3K4me3 (fig. 3), which is a signature histone mark at CGIs [17, 27, 28]. In strong support of this scenario, artificial CGI-like sequences that are transcriptionally inert acquire H3K4me3 when integrated into the genome [9, 17]. Studies of other proteins with CpG-binding CXXC domains report similar recruitment of chromatin modifying activities [19–21]. It appears that a major function of CGIs is to attract proteins of this kind in order to facilitate regulation of gene expression. Methyl-CpG-binding proteins, on the other hand, avoid unmethylated CGIs and track CpG methylation across the genome. Unlike most (but not all) CpG binding proteins, they generally associate with co-repressor complexes that inhibit gene expression and deacetylate histone tails (see fig. 3). For example, MeCP2 recruits the NCoR/SMRT [29] and Sin3a corepressors [30, 31], while Mbd2 is part of the NuRD co-repressor complex [32].
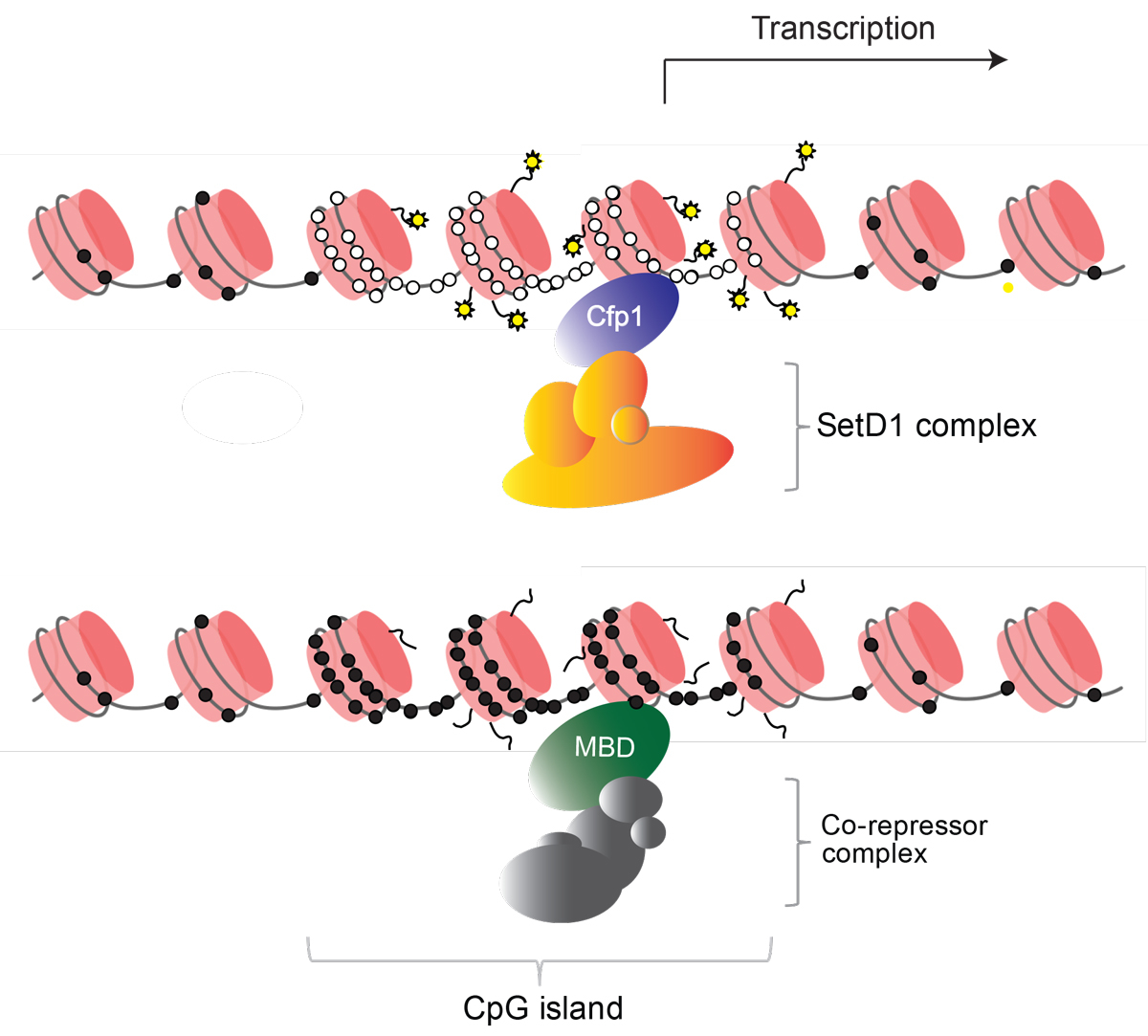
Figure 3 Non-methylated CpG islands recruit “active” histone marks, whereas methylated CpG islands lead to a repressive chromatin structure. CpGs are represented by circles that are either methylated (black) or non-methylated (white) on DNA that is wrapped around nucleosomes (pink cylinders). The CpG reader Cfp1 targets the SetD1 complex which methylates lysine 4 of histone H3. Other CpG readers also recruit “active” chromatin modifiers. MBD proteins bind methylated CpG, either in methylated CpG islands or elsewhere in the genome and recruit histone deacetylase complexes that inhibit transcription.
Interestingly, the high frequency of CpGs within CGIs does not appear to be the result of evolutionary selection on individual CpG sites [33]. It seems instead that a GC-rich base composition and lack of DNA methylation in these regions over millions of years has been sufficient to create by default a CpG-rich platform that is adaptive for gene regulation. Thus although CpGs may not be directly selected, there is compelling evidence that they are key to CGI function, as the ability to recruit both H3K4me3 and the polycomb group proteins depends on CpG density [17, 18, 20, 21, 34–36]. It follows that regions of GC-rich DNA with correspondingly high densities of CpG exert an effect on chromatin modification (including DNA methylation) via CpG binding proteins. This aspect of the epigenome is therefore directly influenced by DNA sequence context.
With the exception of CpG binders, little attention has been paid to factors recognising other low complexity sequence motifs. The following question arises: do short sequence motifs other than CpG behave as genomic signalling modules and, if so, how are they read and what are their biological effects? A pre-requisite for addressing this question is the existence of candidate DNA binding proteins that recognise sequence motifs that are sufficiently short or redundant. For example, diverse proteins that contain AT-hooks require only permutations of four As and Ts to bind [37]. To capture these and other such proteins, it is possible to screen nuclear protein extracts from embryonic stem cells for proteins that are reproducibly captured by AT-rich DNA sequences. Our preliminary evidence indicates that such a screen can recover proteins that bind AT-rich motifs and whose function is linked to differentiation and disease, as well as unstudied proteins about which little is known. The molecular mechanisms underlying the biological functions of these proteins are the focus of on-going research.
Is DNA base composition a signal that programmes the epigenome?
Apart from de novo methylation of DNA, there are several features of chromosome organisation that correlate with zones of differing base composition. Bernardi first recognised that the genome is a mosaic of domains or “isochores” with different sequence characteristics [38, 39]. Within an isochore, base composition is relatively homogeneous, ranging from GC-poor (~35%) to GC-rich (~55%) and the boundaries between isochores are relatively sharp. For several decades it has been recognised that isochores map onto a variety of interesting chromosomal features. These include regions of higher gene density and early replicating regions (both GC-rich) [40] as well as lamin-associated domains, regions of high LINE transposon density and G bands (all AT-rich) [41, 42]. Is the association causal or consequential? One possibility is that base composition is a passive by-product of features of chromosome structure and activity. Late replicating DNA, for example, may be subject to biased mutation due to the composition of nucleotide pools at this stage of the cell cycle [43]. An alternative view that has not so far been investigated is that DNA sequence composition drives aspects of chromosome organisation. In this case base composition would constitute a signal that could be read by DNA binding proteins. Since these global features of the epigenome are subject to modulation between cell types, a key component of this hypothesis is that sequence-specific factors recognising short motifs will be expressed in a tissue-restricted manner.
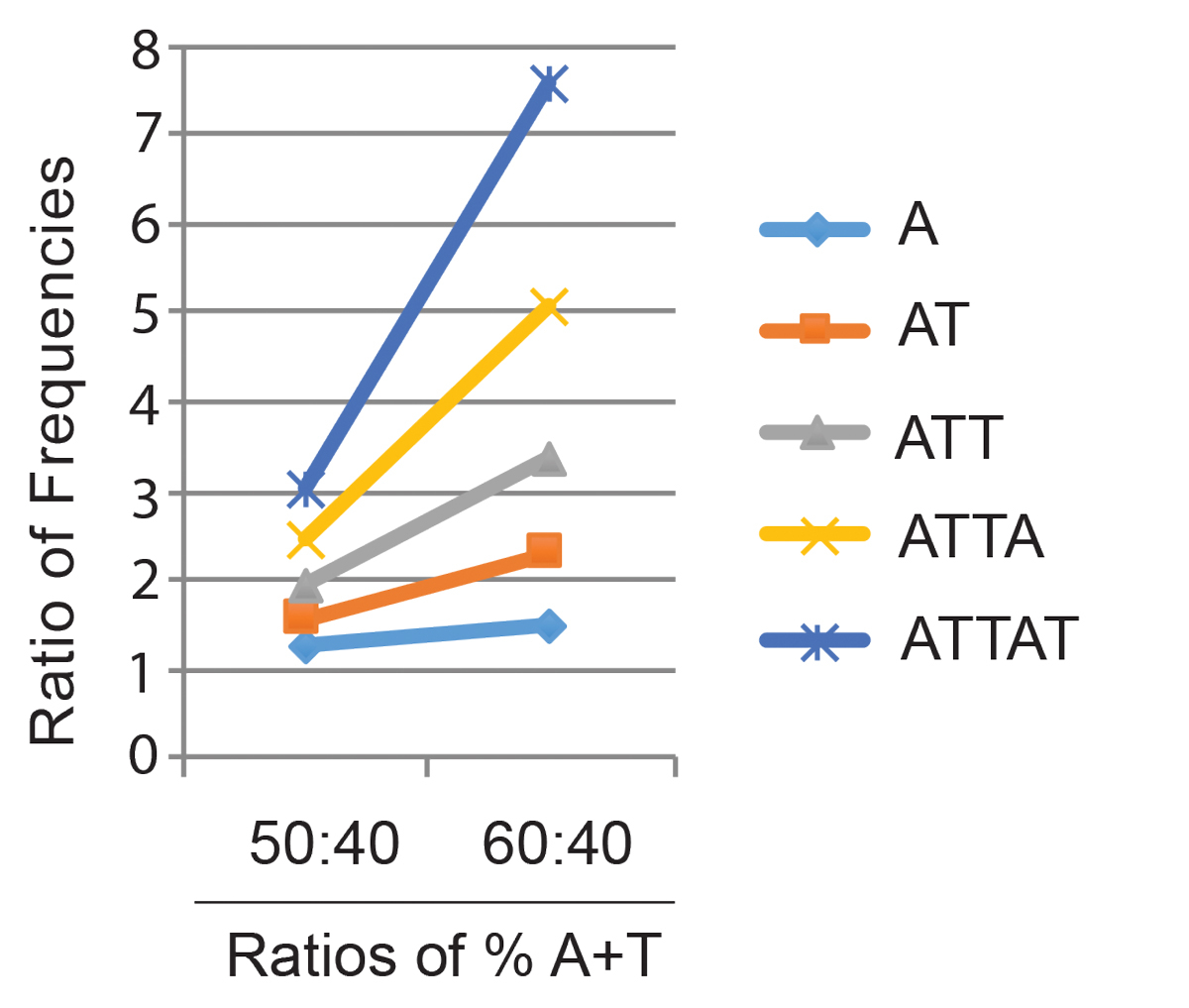
How might shared features of DNA base composition be read as a signal even though the detailed underlying base sequences are highly diverse? By analogy with CpG signalling in CGIs, we entertain the hypothesis that proteins recognising short DNA sequence motifs carry out this function. The rationale is that while the frequency of single bases is linearly dependent on the base composition, the frequency of runs of A/T tracts increases non-linearly as AT-richness rises. For example, the single base A is 1.5 times more frequent in random DNA of 60% AT DNA than in DNA of 40% AT, whereas the 5 base pair sequence AAAAA (or TTAAT etc.) is >7 times more frequent (fig. 4). This means that the frequency of A/T runs of four to six nucleotides is highly sensitive to base composition. There are conceptual parallels with CpG, which is a known signalling sequence that occurs on average every 100 base pairs in the bulk genome, but is locally an order of magnitude more frequent within CGIs. Candidate proteins recovered in a preliminary screen for generalised AT-rich DNA binding proteins are being tested in this laboratory to see if they fulfil these requirements. Some of these are AT-hook proteins, whereas others recognise AT-runs via unrelated DNA binding domains. Many, but not all, of the proteins are known and the biological effects of deficiency have been assessed either from studies of human disease or by gene disruption in mice. Molecular mechanisms underlying these effects are in nearly all cases not understood. Our current task is to select candidates whose role may be to mediate the effects of base composition on chromosome biology. In this way we hope to illuminate the role of genetic information in determining the epigenome, and hence in optimising gene expression.
The Charles Rodolphe Brupbacher Prize for Cancer Research
Biennially, the Charles Rodolphe Brupbacher Prize for Cancer Research is awarded to scientists who have made extraordinary contributions to basic oncological research. The Charles Rodolphe Brupbacher Prize for Cancer Research 2017 has been awarded to Sir Adrian Peter Bird, PhD for his contributions to our understanding of the role of DNA methylation in development and disease. This article is based on his award lecture held in Zurich, Switzerland, during the CRB Symposium 2017.
Acknowledgements
I thank laboratory members Timo Quante, Kashyap Chhatbar and Konstantina Skourti-Stathaki for advice and discussions, and the European Research Council and the Wellcome Trust for funding our research.
References
1
Ebrahimi
B
. Reprogramming barriers and enhancers: strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen (Lond). 2015;4(1):10 .https://doi.org/10.1186/s13619-015-0024-9
2
Berman
BP
,
Weisenberger
DJ
,
Aman
JF
,
Hinoue
T
,
Ramjan
Z
,
Liu
Y
, et al.
Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2011;44(1):40–6 .https://doi.org/10.1038/ng.969
3
Bird
AP
. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–13 .https://doi.org/10.1038/321209a0
4
Macleod
D
,
Charlton
J
,
Mullins
J
,
Bird
AP
. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8(19):2282–92 .https://doi.org/10.1101/gad.8.19.2282
5
Brandeis
M
,
Frank
D
,
Keshet
I
,
Siegfried
Z
,
Mendelsohn
M
,
Nemes
A
, et al.
Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371(6496):435–8 .https://doi.org/10.1038/371435a0
6
Lienert
F
,
Wirbelauer
C
,
Som
I
,
Dean
A
,
Mohn
F
,
Schübeler
D
. Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 2011;43(11):1091–7 .https://doi.org/10.1038/ng.946
7
Stadler
MB
,
Murr
R
,
Burger
L
,
Ivanek
R
,
Lienert
F
,
Schöler
A
, et al.
DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–5.
8
Ooi
SK
,
Qiu
C
,
Bernstein
E
,
Li
K
,
Jia
D
,
Yang
Z
, et al.
DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–7 .https://doi.org/10.1038/nature05987
9
Wachter
E
,
Quante
T
,
Merusi
C
,
Arczewska
A
,
Stewart
F
,
Webb
S
, et al.
Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife. 2014;3:e03397 .https://doi.org/10.7554/eLife.03397
10
Illingworth
RS
,
Gruenewald-Schneider
U
,
De Sousa
D
,
Webb
S
,
Merusi
C
,
Kerr
AR
, et al.
Inter-individual variability contrasts with regional homogeneity in the human brain DNA methylome. Nucleic Acids Res. 2015;43(2):732–44 .https://doi.org/10.1093/nar/gku1305
11
Hitchins
MP
. Constitutional epimutation as a mechanism for cancer causality and heritability?
Nat Rev Cancer. 2015;15(10):625–34 .https://doi.org/10.1038/nrc4001
12
Jones
PA
,
Issa
JP
,
Baylin
S
. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41 .https://doi.org/10.1038/nrg.2016.93
13
Hill
VK
,
Ricketts
C
,
Bieche
I
,
Vacher
S
,
Gentle
D
,
Lewis
C
, et al.
Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71(8):2988–99 .https://doi.org/10.1158/0008-5472.CAN-10-4026
14
Quante
T
,
Bird
A
. Do short, frequent DNA sequence motifs mould the epigenome?
Nat Rev Mol Cell Biol. 2016;17(4):257–62 .https://doi.org/10.1038/nrm.2015.31
15
Bird
A
. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21 .https://doi.org/10.1101/gad.947102
16
Bird
A
. The dinucleotide CG as a genomic signalling module. J Mol Biol. 2011;409(1):47–53 .https://doi.org/10.1016/j.jmb.2011.01.056
17
Thomson
JP
,
Skene
PJ
,
Selfridge
J
,
Clouaire
T
,
Guy
J
,
Webb
S
, et al.
CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464(7291):1082–6 .https://doi.org/10.1038/nature08924
18
Blackledge
NP
,
Farcas
AM
,
Kondo
T
,
King
HW
,
McGouran
JF
,
Hanssen
LL
, et al.
Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157(6):1445–59 .https://doi.org/10.1016/j.cell.2014.05.004
19
Blackledge
NP
,
Zhou
JC
,
Tolstorukov
MY
,
Farcas
AM
,
Park
PJ
,
Klose
RJ
. CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell. 2010;38(2):179–90 .https://doi.org/10.1016/j.molcel.2010.04.009
20
Farcas
AM
,
Blackledge
NP
,
Sudbery
I
,
Long
HK
,
McGouran
JF
,
Rose
NR
, et al.
KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife. 2012;1:e00205 .https://doi.org/10.7554/eLife.00205
21
Wu
X
,
Johansen
JV
,
Helin
K
. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol Cell. 2013;49(6):1134–46 .https://doi.org/10.1016/j.molcel.2013.01.016
22
Lee
JH
,
Voo
KS
,
Skalnik
DG
. Identification and characterization of the DNA binding domain of CpG-binding protein. J Biol Chem. 2001;276(48):44669–76 .https://doi.org/10.1074/jbc.M107179200
23
Bostick
M
,
Kim
JK
,
Estève
PO
,
Clark
A
,
Pradhan
S
,
Jacobsen
SE
. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317(5845):1760–4 .https://doi.org/10.1126/science.1147939
24
Hendrich
B
,
Bird
A
. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18(11):6538–47 .https://doi.org/10.1128/MCB.18.11.6538
25
Baubec
T
,
Ivánek
R
,
Lienert
F
,
Schübeler
D
. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell. 2013;153(2):480–92 .https://doi.org/10.1016/j.cell.2013.03.011
26
Nan
X
,
Campoy
FJ
,
Bird
A
. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–81 .https://doi.org/10.1016/S0092-8674(00)81887-5
27
Lee
JH
,
Skalnik
DG
. CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem. 2005;280(50):41725–31 .https://doi.org/10.1074/jbc.M508312200
28
Clouaire
T
,
Webb
S
,
Skene
P
,
Illingworth
R
,
Kerr
A
,
Andrews
R
, et al.
Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26(15):1714–28 .https://doi.org/10.1101/gad.194209.112
29
Lyst
MJ
,
Ekiert
R
,
Ebert
DH
,
Merusi
C
,
Nowak
J
,
Selfridge
J
, et al.
Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898–902 .https://doi.org/10.1038/nn.3434
30
Nan
X
,
Ng
HH
,
Johnson
CA
,
Laherty
CD
,
Turner
BM
,
Eisenman
RN
, et al.
Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–9 .https://doi.org/10.1038/30764
31
Wade
PA
,
Gegonne
A
,
Jones
PL
,
Ballestar
E
,
Aubry
F
,
Wolffe
AP
. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23(1):62–6 .https://doi.org/10.1038/12664
32
Zhang
Y
,
Ng
H-H
,
Erdjument-Bromage
H
,
Tempst
P
,
Bird
A
,
Reinberg
D
. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13(15):1924–35 .https://doi.org/10.1101/gad.13.15.1924
33
Cohen
NM
,
Kenigsberg
E
,
Tanay
A
. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145(5):773–86 .https://doi.org/10.1016/j.cell.2011.04.024
34
Straussman
R
,
Nejman
D
,
Roberts
D
,
Steinfeld
I
,
Blum
B
,
Benvenisty
N
, et al.
Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16(5):564–71 .https://doi.org/10.1038/nsmb.1594
35
Lynch
MD
,
Smith
AJ
,
De Gobbi
M
,
Flenley
M
,
Hughes
JR
,
Vernimmen
D
, et al.
An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J. 2012;31(2):317–29 .https://doi.org/10.1038/emboj.2011.399
36
Mendenhall
EM
,
Koche
RP
,
Truong
T
,
Zhou
VW
,
Issac
B
,
Chi
AS
, et al.
GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010;6(12):e1001244 .https://doi.org/10.1371/journal.pgen.1001244
37
Aravind
L
,
Landsman
D
. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;26(19):4413–21 .https://doi.org/10.1093/nar/26.19.4413
38
Bernardi
G
,
Olofsson
B
,
Filipski
J
,
Zerial
M
,
Salinas
J
,
Cuny
G
, et al.
The mosaic genome of warm-blooded vertebrates. Science. 1985;228(4702):953–8 .https://doi.org/10.1126/science.4001930
39
Bernardi
G
. The isochore organization of the human genome. Annu Rev Genet. 1989;23(1):637–61 .https://doi.org/10.1146/annurev.ge.23.120189.003225
40
Holmquist
GP
. Chromosome bands, their chromatin flavors, and their functional features. Am J Hum Genet. 1992;51(1):17–37.
41
Nora
EP
,
Dekker
J
,
Heard
E
. Segmental folding of chromosomes: a basis for structural and regulatory chromosomal neighborhoods?
BioEssays. 2013;35(9):818–28 .https://doi.org/10.1002/bies.201300040
42
Bickmore
WA
,
van Steensel
B
. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152(6):1270–84 .https://doi.org/10.1016/j.cell.2013.02.001
43
Wolfe
KH
,
Sharp
PM
,
Li
W-H
. Mutation rates differ among regions of the mammalian genome. Nature. 1989;337(6204):283–5 .https://doi.org/10.1038/337283a0