Virally vectored vaccine delivery: medical needs, mechanisms, advantages and challenges
DOI: https://doi.org/10.4414/smw.2017.14465
Division of Experimental Virology, Department of Biomedicine,
Virally vectored vaccine delivery: medical needs, mechanisms, advantages and challenges
w14465
Summary
Vaccines represent one of the most successful chapters in the history of medicine. Over the past decades, the advent of recombinant cDNA technology has enabled the biomedical community to genetically engineer viruses for vaccine delivery purposes. As a starting point, this review evaluates the unmet medical needs, which drive scientists and industry to exploit such fundamentally new technology for human vaccination.
The author discusses the molecular functioning, production and safety profile of replication-competent and -deficient viral vector systems, representing two fundamentally distinct classes of “genetic vaccines”. Building upon this knowledge, he dissects the immunological mechanisms rendering immune responses to viral vectors qualitatively and quantitatively distinct from those elicited by non-live vaccination approaches. These mechanisms comprise (1) the vectors’ innate immune recognition by the host cell, (2) potent priming of CD8+ cytotoxic T cells as a result of dendritic cell targeting and endogenous protein synthesis, (3) conformational antigen display for protective antibody induction as well as (4) prolonged availability of substantial quantities of antigen. Deduced from these features, preferential indications for virally vectored vaccines are discussed, taking into consideration specific medical needs as well as risk-benefit assessments of replicating vector systems. The limitations and challenges in virally vectored vaccination must also be given careful consideration. Pre-existing and vaccination-induced anti-vector immunity can interfere with vaccine immunogenicity and prime-boost vaccination, respectively. Additionally, the requirement for eukaryotic production systems imposes technological as well as regulatory hurdles. Existing strategies to overcome these challenges are outlined.
With the recent licensure of the first virally vectored vaccine this review seems timely to herald the introduction of virally vectored vaccines into daily medical practice.
The need for new vaccine delivery technology
Inactivated vaccines such as the Salk polio virus vaccine and subunit vaccines as represented by the hepatitis B virus (HBV) vaccine have a long history of clinical success in preventing infectious diseases [1, 2]. These “non-live” approaches often combine a favourable safety profile with limited reactogenicity, warranting broad acceptance in population-wide vaccination campaigns. Acceptable costs of goods and product stability provide additional practical arguments in favour of these approaches. Conversely, virally vectored vaccines, neither replicating nor replication-deficient, cannot reasonably claim to combine all of these advantages. One may therefore be wondering why academic researchers and the vaccine industry keep developing and refining virally vectored vaccine delivery strategies, despite the complexity, which is inherent to “live”, “genetic” products? Here, I intend to summarise the conceptual grounds and medical needs, which together form a strong rationale to pursue virally vectored vaccine approaches.
It is a commonly held notion that for disease indications that can efficiently be prevented by non-live vaccines, the corresponding products have already been brought to the market in the course of the 20th century. This “rule of thumb” in the perception of the “vaccine opportunity landscape” results predominantly from vaccine failures in highly challenging disease indications such as human immunodeficiency virus / acquired immunodeficiency syndrome (HIV/AIDS) [3, 4]. Missed opportunities for more non-live vaccines may, however, readily be identified amongst emerging diseases such as Chikungunya or Zika virus [5–7], for which medical needs and/or commercial opportunities have long provided insufficient incentive to drive serious vaccine development efforts. Moreover, the clinical efficacy of the human papilloma virus vaccine (for the prevention of cervical cancer), which was introduced to the clinic in the 21st century only, illustrates that some new vaccines of global impact and based on non-live vaccine technology remain to be developed [8]. Very encouraging recent phase III data on a subunit herpes zoster vaccine support this notion [9].
Still, the above rule of thumb analysis of vaccine opportunities is not overly pessimistic. Contemporary vaccine makers are indeed confronted with numerous challenging indications. Besides HIV/AIDS, hepatitis C, malaria and tuberculosis, which are most commonly cited in this context, other pressing “leftovers” of the 20th century vaccine makers comprise respiratory syncytial virus (RSV), cytomegalovirus (CMV) and the causative agents of haemorrhagic fever such as Ebola, Nipah and Lassa virus. It is in the context of these diseases that virally vectored vaccines find their primary application and represent strong development candidates.
Two main classes of virally vectored vaccines
Virally vectored vaccines can be categorised according to several criteria, such as the type or family of virus used for vectorisation, the medical target indication and population, prophylactic or therapeutic use, and so forth. For the purpose of this review, I propose to categorise virally vectored vaccines into two main classes, based on their ability (or the lack thereof) to replicate. “Replicating” will refer to the capacity of a viral vector to form infectious progeny and thereby establish a propagating infection in the vaccinee’s cells (see below). Conversely, the sole ability to express viral genes and amplify the viral genome intracellularly would not qualify as “replicating”. The term “vectored” will be used in a fairly restricted manner here. It will refer to the delivery of a piece of genetic information (viral, bacterial, parasitic or other), originating from a different species from that of the viral backbone used for delivery. Accordingly, live-attenuated vaccines, including the cold-adapted influenza virus vaccine, will not be considered as “vectored”.
Replication-deficient viral vectors
Molecular functioning
Viral vectors, both replication-competent and -deficient, are viral particles that can infect or transduce target cells in a vaccinee to deliver and express their genetic information. Whereas the term “infection” requires that viral replication takes place inside a target cell, the mere introduction of genetic information is referred to as “transduction”. Accordingly, viral vectors infecting or transducing target cells both represent “genetic vaccines”. In marked contrast to wild type or live-attenuated viruses, however, replication-deficient viral vectors fail to produce new infectious progeny particles when infecting or transducing the cell of a vaccinee. Still, and despite the fact that viruses do not represent living organisms, replication-deficient viral vectors are commonly classified as “live” vaccines, since the immunogenic principle of “genetic vaccines” consists in de novo expression of vaccine antigen(s) in the vaccinee’s cells. The antigen, which should induce a protective immune response, is not necessarily contained in the formulated particle. Antigen expression is a “live” process occurring in vivo. Thus, strictly speaking, “replication-deficient” viral vector systems are able to replicate their genetic information in the vaccinee’s cells but are unable to propagate infectivity in the vaccinee, and thus can also be referred to as “propagation-deficient”. For the purpose of this review, virus-like particles and other forms of non-live viral derivatives, which merely constitute virally adjuvanted protein, will not be counted amongst replication-deficient viral vectors.
Production
Replication-deficient viral vectors commonly require a specific cellular substrate for production (production cells), which, unlike the cells of a vaccinee, permit the formation of infectious progeny. Most commonly, this cell substrate is made to express one or multiple viral protein(s), which are necessary for the assembly of infectious viral particles (fig. 1). Such expression can be obtained by transient transfection or by stable integration of genetic elements into the production cell line. Thereby, the cell line complements in trans one or multiple viral gene products, which have been deleted from the viral genome, converting the virus into a replication-deficient vector. Accordingly, the vector is replication-deficient because formation of infectious particles depends on trans-complementation of one (or multiple) gene product(s), and these are missing in the vaccinee’s cells. This mechanistic principle applies to most types of replication-deficient vectors such as those of the alphavirus [10], adenovirus [11] and arenavirus-based systems [12]. Recombinant versions of modified vaccinia virus Ankara (MVA) should also be counted amongst the replication-deficient viral vectors. Unlike the aforementioned vector types, however, MVA only is replication-deficient in mammalian cells such as those of the vaccinee. MVA replicates in chicken embryonic fibroblasts as a production cell substrate, and trans-complementation of a viral gene product is not needed [13].
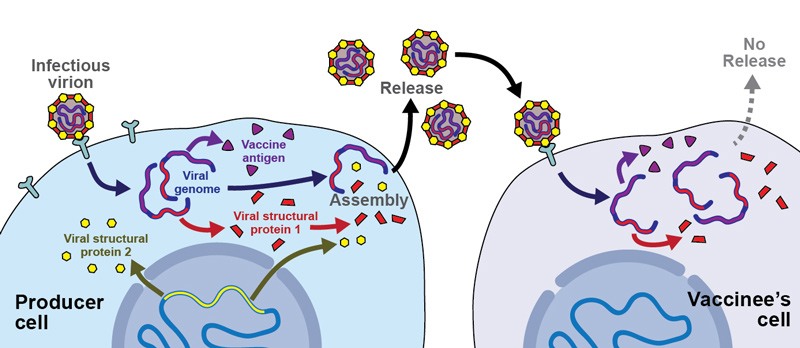
Figure 1 Molecular functioning of replication-deficient viral vectors.
Replication-deficient viral vectors are composed of all structural viral proteins (here schematically: structural viral proteins 1 and 2), and contain the viral genome. Batch production is performed in so-called “producer cells”, which express one or multiple viral proteins for trans-complementation of the vector’s defective genome. In this example, stably transfected producer cells express from their genomic expression cassette the viral “structural protein 2”. The vector genome lacks “structural protein 2”, and encodes only for “structural protein 1” and the “vaccine antigen”. During batch production (left and centre), infectious vector particles infect producer cells, amplify their genome and produce “structural protein 1” as well as the “vaccine antigen”. New infectious particles are formed, containing the genome and structural protein 1 (expressed from the vector genome) as well as structural protein 2 (expressed from the producer cell genome). Upon administration to a vaccinee, these replication-deficient viral particles will infect the vaccinee’s cells, resulting in the expression of structural protein 1 as well as of the vaccine antigen. Owing to the lack of structural protein 2 in the vaccinee’s cells, however, further infectious particles cannot be formed, and the infection does not propagate. Expression of the vaccine antigen in the vaccinee’s cells leads to the desired immune response.
Safety profile
The lack of in vivo replication after administration to a vaccinee has important implications for clinical exploitation. If viral reversion is excluded [14, 15], the lack of replication itself warrants for a basic level of patient safety. Although adverse events in vectored gene delivery can be unpredictable [16], overwhelming infection and resulting disease represent a primary concern surrounding all virus-based products and are excluded if a vector is replication-deficient. This facilitates clinical trials and eventually also licensure for human use. After all, safety is first priority, particularly for vaccines to be used in a healthy population that is not at imminent risk of vaccine-preventable severe disease. For the development of vaccines in disease indications with the aforementioned characteristics, replication-deficient viral vector systems are thus usually preferred over replicating ones.
Replication-competent viral vectors
Molecular functioning
Analogously to replication-deficient viral vectors, replication-competent ones are genetic vaccines and require eukaryotic cell substrates for production. By definition, however, they do not depend on complementation, rendering any genetic modification of production cells unnecessary.
Production
Essentially, replication-competent viral vectors are produced on Good Manufacturing Practice (GMP)-grade eukaryotic cell lines, analogously to the widely used live-attenuated measles, mumps and rubella vaccines. These processes have been industrial routine for long and will not be the subject of this review. Accordingly, live-attenuated human vaccines can represent attractive opportunities for vectorisation, one reason being that production processes are already established, warranting reasonably low costs of goods.
Safety profile
Safety considerations are in the spotlight of every clinical development programme relying on replicating viral vector technology. Potential concerns and regulatory limitations can emerge from two independent angles: patient safety and environmental safety. It depends on each specific vector and its context of use whether spread to other individuals or into the environment represents a possibility and/or a risk. The natural host range and transmission routes of the vectorised virus will help to establish an environmental risk assessment [17]. The fact that these vectors are genetically modified organisms must also be given consideration, and implies additional legislation. Patient safety is dictated by the hazard profile of the parental virus in combination with any potentially attenuating modifications introduced. At times, the process of genetic modification for vectorisation, i.e., the introduction of a vaccine antigen, can by itself result in some level of viral attenuation [18]. In other instances, specific attenuating mutations or deletions are made, with the goal of improving patient safety [19, 20]. The use of animal viruses, which are known or expected not to cause disease in humans, can represent an additional attractive option [21]. One of the most commonly pursued strategies consists, however, in the exploitation of clinically validated live-attenuated vaccines as vectors. Besides established production processes (see above) and well-documented immunogenicity, the clinical safety profile of live-attenuated vaccines such as those against yellow fever, measles or polio represent a strong incentive to their use as vectors [22–24]. Depending on the clinical context of use, however, fairly human-pathogenic viruses such as replicating cytomegalovirus are also being considered for vaccine delivery purposes ([25] see below).
Mechanisms underlying the exquisite immunogenicity of virally vectored vaccines
Virally vectored vaccines are self-adjuvanted
The mammalian immune system has evolved to efficiently recognise viral intruders, and to mount potent innate and adaptive responses to infection. Accordingly, multiple innate sensors and pathways that coordinate immunity to viruses have been identified (fig. 2). Depending on their nature and genome replication strategy, diverse pattern recognition receptors are involved. Nucleic acid sensors include Toll-like receptor (TLR) 3, TLR7, TLR8 and TLR9 in the endosome, as well as cytosolic recognition molecules such as RIG-I-like receptors RIG-I (retinoic acid inducible gene I), MDA5 (melanoma differentiation-associated gene 5) [26–30] and DNA sensors such as cGAS (cyclic GMP-AMP synthase) ([31–34], reviewed in [35]). Upon binding of viral genomes, these receptors signal through the NFκB (nuclear factor “kappa-light-chain-enhancer” of activated B-cells) and mitogen-activated protein (MAP) kinase pathways, resulting in the induction of pro-inflammatory cytokines and chemokines. Additionally, interferon regulatory factor (IRF) 3 and IRF7 signalling leads to type I interferon (IFN-I) induction (reviewed in [36]). The primary evolutionary purpose of these pathways consists in antimicrobial defence, with the IFN-I-induced “antiviral state” as a paradigm [37]. Additionally, these virus-induced transcriptional programmes activate infected antigen-presenting cells, which instruct the adaptive immune system to mount potent antibody and helper T cell type 1 (Th1)-biased cellular responses including CD8+ T cells. Accordingly, virus-induced inflammation is optimally suited to induce potent vaccine responses, and virally vectored vaccines are generally “self-adjuvanted”. The meaning of this term refers to the finding that adjuvant activity is “inbuilt”, with no need for supplementation. Attenuation and replication deficiency can profoundly impact or virtually abrogate systemic inflammation, thus preventing, or at least substantially reducing, the subjective feeling of disease that would result from natural infection. At the cellular level and in the tissue microcompartments, to which live vaccines are delivered and drained, the cellular pathways of innate activation can, however, operate fairly normally. In select cases, attenuation can even unleash immunostimulatory properties not evident with the wild type virus. Vaccinia virus is a classic example: the virus expresses an IFN-I decoy receptor [38] and antagonists of intracellular IFN-I induction. In contrast, modified vaccinia virus Ankara (MVA), a replication-deficient vaccinia virus variant with a large genome deletion, has lost these virulence factors and thus cannot suppress the host IFN-I response [39]. This and other examples demonstrate that the replication capacity and level of attenuation cannot readily predict the self-adjuvanting abilities of live virus vaccines.
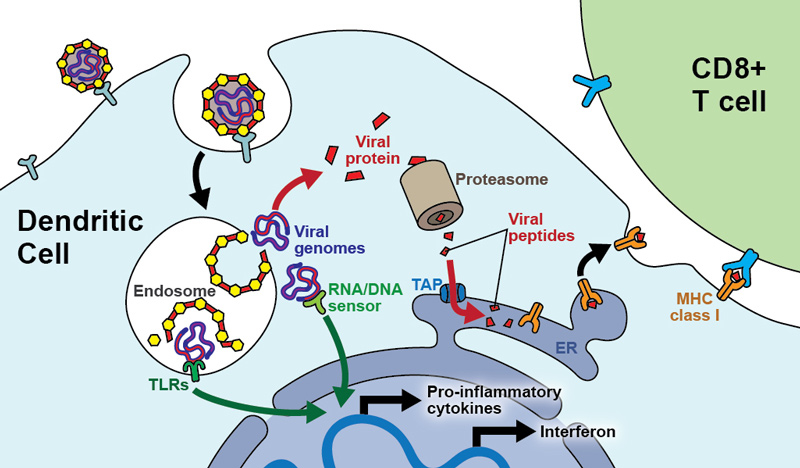
Figure 2 Mechanisms of immune activation by virally vectored vaccines.
Vaccine vector particles infect dendritic cells (DCs) of a vaccinee. In this schematic, the vector particles are taken up by receptor-mediated endocytosis and inside the endosome release their genome into the cytoplasm of the DC. If the viral genome gets exposed inside endosome rather than being released into the cytoplasm, Toll-like receptors (TLRs) sense it. Once inside the cytoplasm, the viral genome is amplified and sensed by cytoplasmic sensors of viral nucleic acids (“RNA/DNA sensor”). Both pathways signal through common pathways such as the NFκB and MAPK pathways, resulting in the transcriptional activation of pro-inflammatory cytokines but also in type I interferon production. Altogether, these events lead to functional activation of the dendritic cell. Simultaneously, the viral genomic information will be expressed, leading to synthesis of viral proteins. A proportion thereof is degraded by the proteasome and the resulting peptidic fragments are channelled through the transporter associated with antigen presentation (TAP) into the endoplasmic reticulum (ER). Inside the ER, these viral peptides are loaded onto MHC class I molecules, which are then exported to the cell surface for presentation to virus-specific CD8+ T cells. Simultaneous functional activation of the DC by the aforementioned innate sensors warrants efficient CD8+ T cell priming.
Viral particles: nature’s delivery system for cytotoxic T cell priming
Cytotoxic CD8+ T cells (CTLs) represent an important correlate of protection against many types of intracellular pathogens, as well as against tumours [40]. Accordingly, robust CTL immunity is a key element that should be supplied for potent protection against a range of pathogens (see below), and CTL immunity is most potently induced by viruses. As “nature’s own gene delivery system”, viruses exploit the host cell’s translational machinery for gene expression (fig. 2). Gene products are synthesised in the cytosol of infected cells and peptidic fragments thereof, through the proteasome and the transporter associated with antigen (TAP) complex, have direct access to the host major histocompatibility complex (MHC) class I peptide loading machinery. This process is referred to as “direct presentation” [41]. It contrasts with non-live vaccines, which rely exclusively upon crosspresentation, a process whereby phagocytic uptake delivers peptide fragments onto MHC class I complexes of professional antigen presenting cells (APCs) [42]. Accordingly, potent activation, expansion and differentiation of CTL responses (CTL induction) by live viral vaccines is, at least in part, due to expression of vectored antigens in professional APCs such as dendritic cells. Additionally, if direct presentation occurs, the very APCs, which present antigenic peptide, are functionally activated by the viral infection [12]. Simultaneous functional activation will augment, if not critically decide, the ability of APCs to prime potent CTL responses [43]. Interestingly, dendritic cells can apparently be infected or at least transduced by a very broad range of viruses, even by some, such as hepatitis B, that otherwise exhibit a fairly restricted cell type- and tissue-tropism ([44], reviewed in [45]). Additionally, viral vectors can, of course, also exploit the crosspresentation machinery [46]. In conjunction with the self-adjuvanted inflammatory milieu, all of these mechanisms synergise to potentiate CTL induction by virally vectored vaccines.
Conformational antigen display on vector-infected host cell membranes
For challenging antibody targets such as viral envelope proteins, correct three-dimensional folding and mammalian glycosylation patterns are often essential to display conformational epitopes for protective antibody induction. Full-length protein expression and display on host cell membranes can also be of importance to warrant intact epitope display [47]. Membranous antigens, not only on virions but also on APCs, represent an efficient source for antigen recognition and uptake by B cells [48, 49]. Live viral vaccines direct conformational antigen expression on the cell surface of infected APCs and thus are optimally suited to meet these requirements. Conversely, the incorporation of vaccine antigens into virions does not seem a prerequisite for antibody induction. Even non-enveloped vaccine vectors can induce antibody responses against conformational antigens that are membrane-anchored [50]. Accordingly, the “genetic vaccines” concept applies also to antibody induction.
Sustained supply of significant amounts of antigen
Virally vectored vaccines have the potential to express vaccine antigen in significant quantities and over prolonged periods of time, which represents an important basis for optimal immune stimulation [51]. Whereas this applies first and foremost to replicating live vaccines [52], also the potency of immune responses to replication-deficient vector systems can be governed by the amount of antigen expressed in the vaccinee [53]. The principle of genetic vaccines warrants that the persistence of vaccine antigen is not a simple function to the antigen’s in vivo half-life. Protracted production of vaccine antigen enables its supply to the ensuing immune response for critical periods of time.
Areas of preferential application for virally vectored vaccines
From these considerations one can deduce several circumstances and conditions, in which virally vectored vaccines find preferential use. Three shall be discussed here.
When classical vaccine technology falls short in efficacy
HIV represents a classic example where subunit vaccines have failed to provide clinical efficacy [3, 4]. In addition, pilot studies in nonhuman primates, which aimed at testing live-attenuated vaccines based on viral genome mutations or deletions, evidenced a rapid reversion to virulence [54]. For tuberculosis, a live-attenuated vaccine (bacille Calmette Guérin, BCG) exists, but does not afford consistent protection in adult populations [55]. Several candidate vaccines against CMV have been tested but did not provide the desired level of protection [56]. A partially protective malaria subunit vaccine is about to enter clinical use in highly endemic areas [57], but formulations of higher efficacy should be sought. For these and similar indications, virally vectored delivery technologies represent attractive strategies owing to the mechanistic considerations outlined above in the section “Mechanisms underlying the exquisite immunogenicity of virally vectored vaccines”.
When CD8+ T cell immunity is key.
Control of primary infection with HIV and hepatitis C virus critically depends on CD8 T cell responses [58–61]. Also for Mycobacterium tuberculosis, an intracellular parasite, a role for CD8+ T cells has been firmly established in recent years [62], and the control of certain protozoa such as liver stage plasmodium can be T cell-dependent [63].
When inactivated vaccines augment disease severity upon infection
Respiratory syncytial virus (RSV) represents the classic example where an inactivated vaccine has led to disease enhancement rather than protection [64]. Underlying mechanisms consist in insufficient potency and/or durability of immune responses, in combination with the type of immune response induced. Dengue virus bears the potential for disease enhancement if immunity does not cover all four serotypes and/or if antibody levels drop below critical thresholds [65, 66]. An inactivated measles virus vaccine has led to disease enhancement analogously to the RSV vaccine [67]. The currently used live-attenuated vaccine against measles virus may eventually have to be replaced by an alternative product in order to eradicate the disease.
Indications for replicating vaccine vectors with imperfect safety or reactogenicity profiles
Both patient safety and environmental safety considerations argue rather in favour of replication-deficient viral vectors and against replicating ones. Depending on virus type, degree of attenuation and administered dose, the spread of replication-competent viral vectors can cause significant reactogenicity, together with the potential for severe adverse events even at sites that are distant from vaccine administration [68]. In certain circumstances, however, these drawbacks may be well in balance with advantages such as augmented immunogenicity. The latter can result from higher levels and more prolonged vaccine antigen expression or from desirable effects of inflammation (see below). The following situations come to mind:
Prophylactic vaccination in epidemic outbreaks with high lethality: In an Ebola outbreak, the risk-benefit evaluation of vaccination speaks clearly in favour of using a reactogenic vaccine, provided no equally effective product with a better safety/reactogenicity profile is available [21, 50, 68, 69].
When a vaccine must induce rapid protection: Postexposure prophylaxis after exposure to a deadly pathogen requires that immunity be installed before the pathogen has replicated to critical thresholds [70, 71]. Under these circumstances, vaccine safety and reactogenicity are of lesser importance than efficacy, unless the expected risks are comparable to the one imposed by the pathogen itself.
When a single vaccine dose must induce durable protection: Vaccination and notably pathogen eradication campaigns in resource-limited circumstances and/or in remote locations can render repeated booster vaccinations logistically challenging, if not impossible. Lassa virus vaccination in rural areas of West Africa may represent such a case. Lassa virus seroprevalence rates approach 50% [72], such that the risk – benefit assessment and logistical constraints may justify the use of replicating virally vectored vaccines with an imperfect safety profile [73].
Endemic diseases with substantial public health impact: The first Dengue vaccine (CYD-TDV, see also section below “Challenges and limitations in virally vectored vaccination”), a vectorised form of the live-attenuated Yellow fever vaccine virus, has been licensed in a handful of highly Dengue-endemic countries [74, 75]. This is noteworthy for two reasons: First, the Yellow fever vaccine can, albeit very rarely, lead to severe disease [76, 77]. Secondly, the World Health Organization recommends vaccination while acknowledging the possibility that vaccination may increase the risk for severe Dengue illness in select groups of vaccinees [78]. This concern is, however, outweighed by the strong evidence that the vaccine will reduce Dengue illness at the population level. Similar considerations might apply to a hypothetical HIV vaccine to be used in highly endemic areas. The cumulated lifetime infection risk of an individual might outweigh significant side effects of vaccination. Accordingly, even replicating recombinant cytomegalovirus, a life-long persistent infection with considerable disease potential in humans, has been proposed as a vector for an HIV vaccine [25].
Therapy of persistently infected or terminally diseased individuals: The aforementioned CMV-based vaccine candidate is given particularly serious consideration for therapeutic application in persistently HIV infected individuals [79]. Analogously, a replication-competent herpes simplex virus-based oncolytic product has been licensed for the treatment of cancer [80]. With the clinical introduction of immune checkpoint inhibitors in cancers, active immunisation also approaches [81–83]. Replicating virally vectored vaccines clearly have their place in this rapidly evolving field [84], and virus-induced inflammation – in prophylactic vaccination an unwanted side-effect – may even benefit tumour control [85].
Challenges and limitations in virally vectored vaccination
Recombinant DNA technology, which allows for the engineering of viral genomes as live vaccine delivery systems, has been available for more than three decades [86, 87]. Given the numerous advantages of these technologies, as outlined, the paucity of human products on the market may seem surprising. The currently first and only one, the tetravalent dengue vaccine CYD-TDV [74, 75], is a recombinant, replicating live vaccine for the prevention of severe Dengue fever and has been licensed in a handful of highly Dengue-endemic countries areas over the past two years. The broad pipeline of products, both replicating and replication-deficient (reviewed in [88]), that are fairly advanced in clinical testing, raises hope that many more products will enter the market in the coming years. Clinical development costs in the range of several hundred million dollars are common to most prophylactic vaccines, and GMP production, release testing and regulatory approval are particularly onerous for virally vectored products. Moreover, certain limitations and challenges are inherently linked to virally vectored vaccine approaches. Considering that some of these have considerably slowed down the clinical translation of virally vectored vaccine technology, two of them will be summarised below.
Anti-vector immunity
From the standpoint of immunogenicity and efficacy, one limitation of viral vaccine delivery systems consists in the interference resulting from so-called “anti-vector immunity”. The same phenomenon can manifest in essentially two ways:
Pre-existing anti-vector immunity
Individuals who are immune to the wild type parent virus, from which a viral vaccine vector is derived, tend to respond poorly to vaccination. This phenomenon was prominently noted in HIV vaccine trials relying on adenovirus 5- (Ad5-) based vectors. When compared to Ad5-seronegative individuals, study participants with antibody immunity to Ad5 responded poorly to vaccination [89, 90]. The underlying mechanism consists in immunological interference by so-called “anti-vector immunity”, which can comprise humoral as well as cellular mechanisms [12, 91, 92]. Adoptive serum transfer experiments in animals demonstrated that infection with Ad5, which in certain areas of the globe is fairly common, elicits “interfering antibodies” that virtually abrogate the immunogenicity of Ad5-based vectors [12]. Supposedly they operated by neutralising the incoming viral vector particles, i.e., rendering them non-infectious and triggering their degradation, the primary purpose antibodies serve in nature to prevent viral re-infection [93].
Vaccination-induced anti-vector immunity
Viral vectors induce not only immune responses against the vectored antigen (i.e., the antigen of the pathogen, against which immunity should be induced) but result also in immunity to the vector backbone itself – anti-vector immunity. Most vaccines require, however, booster administrations in order to elicit optimal immune protection. Moreover, regular refresher vaccinations are often needed to maintain immunity over the years. The induction of anti-vector immunity upon primary vaccination, most prominently represented by interfering antibodies, can, however, dramatically curtail the effectiveness of booster immunizations.
Vaccine makers can draw a number of conclusions from these observations. First, it seems advantageous to exploit viruses with low seroprevalence in the human population, thereby avoiding pre-existing anti-vector immunity [50, 94]. To minimise detrimental effects of vaccination-induced anti-vector immunity on prime-boost vaccination, one should preferentially vectorise viruses that fail to elicit significant amounts of interfering antibodies [12, 95]. Alternatively, strategies can be developed to render vaccine vector particles less susceptible to antibody neutralisation [92, 96]. A combination of these approaches should, in principle, overcome the limitations imposed by pre-existing or vaccination-induced anti-vector immunity, allowing us to leverage the full potential of virally vectored delivery systems in homologous prime-boost vaccination. An additional strategy to overcome vaccination-induced anti-vector immunity, which has become fairly popular in recent years, consists in heterologous prime-boost combinations. Responses to adenoviral vector primary immunisation are, for example, commonly boosted by alternative viral delivery vehicles such as poxvirus-based or rhabdovirus-based ones in so-called “heterologous prime-boost” vaccination [50, 97, 98]. The combination of two active principles in one vaccine product introduces, however, an additional layer of complexity notably with respect to vaccine production, licensure and administration in the field.
Complexity of production
By nature, mammalian viruses generally require eukaryotic and often mammalian cells for production. Although large-scale GMP bioreactor work has become an industrial routine over the past decades, it can still represent a major cost driver. The balance between the vaccine’s effective dose and production yields per culture volume (including purification processes, see below) is usually indicative of the commercial viability of a vaccine vector in a given disease programme. Replication-deficient vector systems tend to show more marked dose dependency than replication-deficient ones and, accordingly, production yields are often a more prominent bottleneck. Purification processes pose additional challenges since viral infectivity, the mechanistic principle of live vaccine activity, must be preserved. This excludes virus inactivation steps, which are a routine for most mammalian cell culture products such as monoclonal antibodies. To exclude the presence of adventitious infectious agents in vaccine batches [99], extensive testing of cell lines and products is required, narrowing the range of eligible cell lines to a select few commonly available ones. New immortalised cell lines could be generated, in principle, but the effort and time required for their generation from primary human tissue, their selection based on favourable biological characteristics, their characterisation and eventual regulatory acceptance are substantial, and success is uncertain. Accordingly, the process of generating a new cell line is dissuasive for most vaccine programmes, and a small number of rather old but well-characterised cell lines dominate the field. As a consequence, cell line development has been somewhat neglected in the recent past. Ultimately, the ongoing revolution in mammalian genome engineering [100] may thus represent a game changer also to virally vectored vaccination.
Concluding remarks
Viruses represent nature’s gene delivery system, and thus are optimally suited to deliver genetic information also for the purpose of vaccination. Additionally, our immune system was trained in evolution to fight viral intruders, having led to a range of mechanisms that specifically potentiate immune responses against antigens encountered in a viral infection context. These conceptual considerations underscore the utility of viruses as delivery vehicles of genetic vaccines. Preclinical and clinical experience supports these mechanistic considerations, positioning virally vectored delivery systems amongst the most promising technologies to fight infectious diseases and cancers. Given the considerable unmet medical needs in both aforementioned domains of medicine, the likelihood is that virally vectored vaccines will become integrated into daily medical routine in the not too far future.
Acknowledgements
I wish to thank Paul-Henri Lambert and Claire-Anne Siegrist for insightful discussions and mentoring in vaccine-related topics.
Prof. Daniel D. Pinschewer, MD, Division of Experimental Virology, Department of Biomedicine, Haus Petersplatz, University of Basel, Petersplatz 10, CH-4009 Basel, Switzerland., Daniel.Pinschewer[at]unibas.ch
References
1WHO. Hepatitis B fact sheet. http://wwwwhoint/mediacentre/factsheets/fs204/en/. 2015;(Updated July 2016).
2WHO. Poliomyelitis fact sheet. http://wwwwhoint/mediacentre/factsheets/fs114/en/. 2015;(Updated July 2016).
3
Flynn
NM
,
Forthal
DN
,
Harro
CD
,
Judson
FN
,
Mayer
KH
,
Para
MF
; rgp120 HIV Vaccine Study Group. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J Infect Dis. 2005;191(5):654–65. https://doi.org/10.1086/428404
4
Pitisuttithum
P
,
Gilbert
P
,
Gurwith
M
,
Heyward
W
,
Martin
M
,
van Griensven
F
, et al.; Bangkok Vaccine Evaluation Group. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194(12):1661–71. https://doi.org/10.1086/508748
5
Erasmus
JH
,
Auguste
AJ
,
Kaelber
JT
,
Luo
H
,
Rossi
SL
,
Fenton
K
, et al.
A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat Med. 2017;23(2):192–9. https://doi.org/10.1038/nm.4253
6
Akahata
W
,
Yang
ZY
,
Andersen
H
,
Sun
S
,
Holdaway
HA
,
Kong
WP
, et al.
A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat Med. 2010;16(3):334–8. https://doi.org/10.1038/nm.2105
7
Marston
HD
,
Lurie
N
,
Borio
LL
,
Fauci
AS
. Considerations for Developing a Zika Virus Vaccine. N Engl J Med. 2016;375(13):1209–12. https://doi.org/10.1056/NEJMp1607762
8
Harper
DM
,
Franco
EL
,
Wheeler
CM
,
Moscicki
AB
,
Romanowski
B
,
Roteli-Martins
CM
, et al.; HPV Vaccine Study group. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. Lancet. 2006;367(9518):1247–55. https://doi.org/10.1016/S0140-6736(06)68439-0
9
Lal
H
,
Cunningham
AL
,
Godeaux
O
,
Chlibek
R
,
Diez-Domingo
J
,
Hwang
SJ
, et al.; ZOE-50 Study Group. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med. 2015;372(22):2087–96. https://doi.org/10.1056/NEJMoa1501184
10
Lundstrom
K
. Alphavirus-based vaccines. Viruses. 2014;6(6):2392–415. https://doi.org/10.3390/v6062392
11
Imperiale
MJ
,
Kochanek
S
. Adenovirus vectors: biology, design, and production. Curr Top Microbiol Immunol. 2004;273:335–57. https://doi.org/10.1007/978-3-662-05599-1_10
12
Flatz
L
,
Hegazy
AN
,
Bergthaler
A
,
Verschoor
A
,
Claus
C
,
Fernandez
M
, et al.
Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat Med. 2010;16(3):339–45. https://doi.org/10.1038/nm.2104
13
Volz
A
,
Sutter
G
. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. Adv Virus Res. 2017;97:187–243. https://doi.org/10.1016/bs.aivir.2016.07.001
14
Hyvärinen
A
,
Yongabi
F
,
Mäkinen
K
,
Wahlfors
J
,
Pellinen
R
. Recombination of replicon and helper RNAs and the emergence of propagation-competent vectors upon Sindbis virus vector production. Int J Mol Med. 2013;32(2):410–22. doi:.https://doi.org/10.3892/ijmm.2013.1395
15
Bett
AJ
,
Prevec
L
,
Graham
FL
. Packaging capacity and stability of human adenovirus type 5 vectors. J Virol. 1993;67(10):5911–21.
16
Wilson
JM
. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96(4):151–7. https://doi.org/10.1016/j.ymgme.2008.12.016
17
Baldo
A
,
Galanis
E
,
Tangy
F
,
Herman
P
. Biosafety considerations for attenuated measles virus vectors used in virotherapy and vaccination. Hum Vaccin Immunother. 2016;12(5):1102–16. https://doi.org/10.1080/21645515.2015.1122146
18
Buller
RM
,
Smith
GL
,
Cremer
K
,
Notkins
AL
,
Moss
B
. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317(6040):813–5. Published online October 06, 1985. https://doi.org/10.1038/317813a0
19
Brun
J
,
McManus
D
,
Lefebvre
C
,
Hu
K
,
Falls
T
,
Atkins
H
, et al.
Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18(8):1440–9. https://doi.org/10.1038/mt.2010.103
20
Mineta
T
,
Rabkin
SD
,
Yazaki
T
,
Hunter
WD
,
Martuza
RL
. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–43. https://doi.org/10.1038/nm0995-938
21
Agnandji
ST
,
Huttner
A
,
Zinser
ME
,
Njuguna
P
,
Dahlke
C
,
Fernandes
JF
, et al.
Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N Engl J Med. 2016;374(17):1647–60. https://doi.org/10.1056/NEJMoa1502924
22
Mandl
S
,
Sigal
LJ
,
Rock
KL
,
Andino
R
. Poliovirus vaccine vectors elicit antigen-specific cytotoxic T cells and protect mice against lethal challenge with malignant melanoma cells expressing a model antigen. Proc Natl Acad Sci USA. 1998;95(14):8216–21. https://doi.org/10.1073/pnas.95.14.8216
23
Monath
TP
,
Seligman
SJ
,
Robertson
JS
,
Guy
B
,
Hayes
EB
,
Condit
RC
, et al.; Brighton Collaboration Viral Vector Vaccines Safety Working Group (V3SWG). Live virus vaccines based on a yellow fever vaccine backbone: standardized template with key considerations for a risk/benefit assessment. Vaccine. 2015;33(1):62–72. https://doi.org/10.1016/j.vaccine.2014.10.004
24
Tangy
F
,
Naim
HY
. Live attenuated measles vaccine as a potential multivalent pediatric vaccination vector. Viral Immunol. 2005;18(2):317–26. https://doi.org/10.1089/vim.2005.18.317
25
Hansen
SG
,
Vieville
C
,
Whizin
N
,
Coyne-Johnson
L
,
Siess
DC
,
Drummond
DD
, et al.
Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15(3):293–9. https://doi.org/10.1038/nm.1935
26
Querec
T
,
Bennouna
S
,
Alkan
S
,
Laouar
Y
,
Gorden
K
,
Flavell
R
, et al.
Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203(2):413–24. https://doi.org/10.1084/jem.20051720
27
Gitlin
L
,
Barchet
W
,
Gilfillan
S
,
Cella
M
,
Beutler
B
,
Flavell
RA
, et al.
Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006;103(22):8459–64. https://doi.org/10.1073/pnas.0603082103
28
Kato
H
,
Takeuchi
O
,
Sato
S
,
Yoneyama
M
,
Yamamoto
M
,
Matsui
K
, et al.
Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–5. https://doi.org/10.1038/nature04734
29
Hornung
V
,
Ellegast
J
,
Kim
S
,
Brzózka
K
,
Jung
A
,
Kato
H
, et al.
5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–7. https://doi.org/10.1126/science.1132505
30
Pichlmair
A
,
Schulz
O
,
Tan
CP
,
Näslund
TI
,
Liljeström
P
,
Weber
F
, et al.
RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. https://doi.org/10.1126/science.1132998
31
Ablasser
A
,
Goldeck
M
,
Cavlar
T
,
Deimling
T
,
Witte
G
,
Röhl
I
, et al.
cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380–4. https://doi.org/10.1038/nature12306
32
Li
XD
,
Wu
J
,
Gao
D
,
Wang
H
,
Sun
L
,
Chen
ZJ
. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341(6152):1390–4. https://doi.org/10.1126/science.1244040
33
Gao
D
,
Wu
J
,
Wu
YT
,
Du
F
,
Aroh
C
,
Yan
N
, et al.
Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341(6148):903–6. https://doi.org/10.1126/science.1240933
34
Gao
P
,
Ascano
M
,
Wu
Y
,
Barchet
W
,
Gaffney
BL
,
Zillinger
T
, et al.
Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153(5):1094–107. https://doi.org/10.1016/j.cell.2013.04.046
35
Xia
P
,
Wang
S
,
Gao
P
,
Gao
G
,
Fan
Z
. DNA sensor cGAS-mediated immune recognition. Protein Cell. 2016;7(11):777–91. https://doi.org/10.1007/s13238-016-0320-3
36
Pichlmair
A
,
Reis e Sousa
C
. Innate recognition of viruses. Immunity. 2007;27(3):370–83. https://doi.org/10.1016/j.immuni.2007.08.012
37
Isaacs
A
,
Lindenmann
J
. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258–67. https://doi.org/10.1098/rspb.1957.0048
38
Symons
JA
,
Alcamí
A
,
Smith
GL
. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81(4):551–60. https://doi.org/10.1016/0092-8674(95)90076-4
39
Waibler
Z
,
Anzaghe
M
,
Frenz
T
,
Schwantes
A
,
Pöhlmann
C
,
Ludwig
H
, et al.
Vaccinia virus-mediated inhibition of type I interferon responses is a multifactorial process involving the soluble type I interferon receptor B18 and intracellular components. J Virol. 2009;83(4):1563–71. https://doi.org/10.1128/JVI.01617-08
40
Schreiber
RD
,
Old
LJ
,
Smyth
MJ
. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. https://doi.org/10.1126/science.1203486
41
Neefjes
J
,
Jongsma
ML
,
Paul
P
,
Bakke
O
. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–36. doi:.https://doi.org/10.1038/nri3084
42
Rock
KL
,
Shen
L
. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005;207(1):166–83. https://doi.org/10.1111/j.0105-2896.2005.00301.x
43
Probst
HC
,
Lagnel
J
,
Kollias
G
,
van den Broek
M
. Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8+ T cell tolerance. Immunity. 2003;18(5):713–20. https://doi.org/10.1016/S1074-7613(03)00120-1
44
Arima
S
,
Akbar
SM
,
Michitaka
K
,
Horiike
N
,
Nuriya
H
,
Kohara
M
, et al.
Impaired function of antigen-presenting dendritic cells in patients with chronic hepatitis B: localization of HBV DNA and HBV RNA in blood DC by in situ hybridization. Int J Mol Med. 2003;11(2):169–74.
45
Freigang
S
,
Probst
HC
,
van den Broek
M
. DC infection promotes antiviral CTL priming: the ‘Winkelried’ strategy. Trends Immunol. 2005;26(1):13–8. https://doi.org/10.1016/j.it.2004.11.013
46
Bernhard
CA
,
Ried
C
,
Kochanek
S
,
Brocker
T
. CD169+ macrophages are sufficient for priming of CTLs with specificities left out by cross-priming dendritic cells. Proc Natl Acad Sci USA. 2015;112(17):5461–6. https://doi.org/10.1073/pnas.1423356112
47
Rujas
E
,
Caaveiro
JM
,
Partida-Hanon
A
,
Gulzar
N
,
Morante
K
,
Apellániz
B
, et al.
Structural basis for broad neutralization of HIV-1 through the molecular recognition of 10E8 helical epitope at the membrane interface. Sci Rep. 2016;6(1):38177. https://doi.org/10.1038/srep38177
48
Batista
FD
,
Iber
D
,
Neuberger
MS
. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411(6836):489–94. https://doi.org/10.1038/35078099
49
Sanderson
NS
,
Zimmermann
M
,
Eilinger
L
,
Gubser
C
,
Schaeren-Wiemers
N
,
Lindberg
RL
, et al.
Cocapture of cognate and bystander antigens can activate autoreactive B cells. Proc Natl Acad Sci USA. 2017;114(4):734–9. https://doi.org/10.1073/pnas.1614472114
50
Ewer
K
,
Rampling
T
,
Venkatraman
N
,
Bowyer
G
,
Wright
D
,
Lambe
T
, et al.
A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N Engl J Med. 2016;374(17):1635–46. https://doi.org/10.1056/NEJMoa1411627
51
Zinkernagel
RM
. Localization dose and time of antigens determine immune reactivity. Semin Immunol. 2000;12(3):163–71, discussion 257–344. https://doi.org/10.1006/smim.2000.0253
52
Akondy
RS
,
Johnson
PL
,
Nakaya
HI
,
Edupuganti
S
,
Mulligan
MJ
,
Lawson
B
, et al.
Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc Natl Acad Sci USA. 2015;112(10):3050–5. https://doi.org/10.1073/pnas.1500475112
53
Quinn
KM
,
Zak
DE
,
Costa
A
,
Yamamoto
A
,
Kastenmuller
K
,
Hill
BJ
, et al.
Antigen expression determines adenoviral vaccine potency independent of IFN and STING signaling. J Clin Invest. 2015;125(3):1129–46. https://doi.org/10.1172/JCI78280
54
Sawai
ET
,
Hamza
MS
,
Ye
M
,
Shaw
KE
,
Luciw
PA
. Pathogenic conversion of live attenuated simian immunodeficiency virus vaccines is associated with expression of truncated Nef. J Virol. 2000;74(4):2038–45. https://doi.org/10.1128/JVI.74.4.2038-2045.2000
55
Moliva
JI
,
Turner
J
,
Torrelles
JB
. Prospects in Mycobacterium bovis Bacille Calmette et Guérin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis?
Vaccine. 2015;33(39):5035–41. https://doi.org/10.1016/j.vaccine.2015.08.033
56
Plotkin
S
. The history of vaccination against cytomegalovirus. Med Microbiol Immunol (Berl). 2015;204(3):247–54. https://doi.org/10.1007/s00430-015-0388-z
57
Agnandji
ST
,
Lell
B
,
Soulanoudjingar
SS
,
Fernandes
JF
,
Abossolo
BP
,
Conzelmann
C
, et al., RTS,S Clinical Trials Partnership. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365(20):1863–75. https://doi.org/10.1056/NEJMoa1102287
58
Carrington
M
,
Nelson
GW
,
Martin
MP
,
Kissner
T
,
Vlahov
D
,
Goedert
JJ
, et al.
HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283(5408):1748–52. https://doi.org/10.1126/science.283.5408.1748
59
Migueles
SA
,
Sabbaghian
MS
,
Shupert
WL
,
Bettinotti
MP
,
Marincola
FM
,
Martino
L
, et al.
HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci USA. 2000;97(6):2709–14. https://doi.org/10.1073/pnas.050567397
60
Neumann-Haefelin
C
,
McKiernan
S
,
Ward
S
,
Viazov
S
,
Spangenberg
HC
,
Killinger
T
, et al.
Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology. 2006;43(3):563–72. Published online February 24, 2006. https://doi.org/10.1002/hep.21049
61
Neumann-Haefelin
C
,
Timm
J
,
Schmidt
J
,
Kersting
N
,
Fitzmaurice
K
,
Oniangue-Ndza
C
, et al.
Protective effect of human leukocyte antigen B27 in hepatitis C virus infection requires the presence of a genotype-specific immunodominant CD8+ T-cell epitope. Hepatology. 2010;51(1):54–62. Published online December 25, 2009. https://doi.org/10.1002/hep.23275
62
Chen
CY
,
Huang
D
,
Wang
RC
,
Shen
L
,
Zeng
G
,
Yao
S
, et al.
A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 2009;5(4):e1000392. https://doi.org/10.1371/journal.ppat.1000392
63
Fernandez-Ruiz
D
,
Ng
WY
,
Holz
LE
,
Ma
JZ
,
Zaid
A
,
Wong
YC
, et al.
Liver-Resident Memory CD8(+) T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity. 2016;45(4):889–902. https://doi.org/10.1016/j.immuni.2016.08.011
64
Kim
HW
,
Canchola
JG
,
Brandt
CD
,
Pyles
G
,
Chanock
RM
,
Jensen
K
, et al.
Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89(4):422–34. https://doi.org/10.1093/oxfordjournals.aje.a120955
65
Halstead
SB
. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J Infect Dis. 1979;140(4):527–33. https://doi.org/10.1093/infdis/140.4.527
66
Murphy
BR
,
Whitehead
SS
. Immune response to dengue virus and prospects for a vaccine. Annu Rev Immunol. 2011;29(1):587–619. https://doi.org/10.1146/annurev-immunol-031210-101315
67
Fulginiti
VA
,
Eller
JJ
,
Downie
AW
,
Kempe
CH
. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA. 1967;202(12):1075–80. https://doi.org/10.1001/jama.1967.03130250057008
68
Huttner
A
,
Dayer
JA
,
Yerly
S
,
Combescure
C
,
Auderset
F
,
Desmeules
J
, et al.; VSV-Ebola Consortium. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect Dis. 2015;15(10):1156–66. https://doi.org/10.1016/S1473-3099(15)00154-1
69
Henao-Restrepo
AM
,
Longini
IM
,
Egger
M
,
Dean
NE
,
Edmunds
WJ
,
Camacho
A
, et al.
Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 2015;386(9996):857–66. https://doi.org/10.1016/S0140-6736(15)61117-5
70
Günther
S
,
Feldmann
H
,
Geisbert
TW
,
Hensley
LE
,
Rollin
PE
,
Nichol
ST
, et al.
Management of accidental exposure to Ebola virus in the biosafety level 4 laboratory, Hamburg, Germany. J Infect Dis. 2011;204(Suppl 3):S785–90. https://doi.org/10.1093/infdis/jir298
71
Feldmann
H
,
Jones
SM
,
Daddario-DiCaprio
KM
,
Geisbert
JB
,
Ströher
U
,
Grolla
A
, et al.
Effective post-exposure treatment of Ebola infection. PLoS Pathog. 2007;3(1):e2. https://doi.org/10.1371/journal.ppat.0030002
72
McCormick
JB
,
Webb
PA
,
Krebs
JW
,
Johnson
KM
,
Smith
ES
. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis. 1987;155(3):437–44. https://doi.org/10.1093/infdis/155.3.437
73
Lukashevich
IS
. Advanced vaccine candidates for Lassa fever. Viruses. 2012;4(11):2514–57. https://doi.org/10.3390/v4112514
74
Capeding
MR
,
Tran
NH
,
Hadinegoro
SR
,
Ismail
HI
,
Chotpitayasunondh
T
,
Chua
MN
, et al.; CYD14 Study Group. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet. 2014;384(9951):1358–65. https://doi.org/10.1016/S0140-6736(14)61060-6
75
Villar
L
,
Dayan
GH
,
Arredondo-García
JL
,
Rivera
DM
,
Cunha
R
,
Deseda
C
, et al.; CYD15 Study Group. Efficacy of a tetravalent dengue vaccine in children in Latin America. N Engl J Med. 2015;372(2):113–23. https://doi.org/10.1056/NEJMoa1411037
76
Martin
M
,
Tsai
TF
,
Cropp
B
,
Chang
GJ
,
Holmes
DA
,
Tseng
J
, et al.
Fever and multisystem organ failure associated with 17D-204 yellow fever vaccination: a report of four cases. Lancet. 2001;358(9276):98–104. https://doi.org/10.1016/S0140-6736(01)05327-2
77
Vasconcelos
PF
,
Luna
EJ
,
Galler
R
,
Silva
LJ
,
Coimbra
TL
,
Barros
VL
, et al.; Brazilian Yellow Fever Vaccine Evaluation Group. Serious adverse events associated with yellow fever 17DD vaccine in Brazil: a report of two cases. Lancet. 2001;358(9276):91–7. https://doi.org/10.1016/S0140-6736(01)05326-0
78WHO. Dengue vaccine: WHO position paper – July 2016. http://wwwwhoint/wer/2016/wer9130pdf?ua=1. 2016.
79
Hansen
SG
,
Piatak
M, Jr
,
Ventura
AB
,
Hughes
CM
,
Gilbride
RM
,
Ford
JC
, et al.
Immune clearance of highly pathogenic SIV infection. Nature. 2013;502(7469):100–4. https://doi.org/10.1038/nature12519
80
Andtbacka
RH
,
Kaufman
HL
,
Collichio
F
,
Amatruda
T
,
Senzer
N
,
Chesney
J
, et al.
Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–8. https://doi.org/10.1200/JCO.2014.58.3377
81
Madan
RA
,
Mohebtash
M
,
Arlen
PM
,
Vergati
M
,
Rauckhorst
M
,
Steinberg
SM
, et al.
Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):501–8. https://doi.org/10.1016/S1470-2045(12)70006-2
82
van den Eertwegh
AJ
,
Versluis
J
,
van den Berg
HP
,
Santegoets
SJ
,
van Moorselaar
RJ
,
van der Sluis
TM
, et al.
Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(5):509–17. https://doi.org/10.1016/S1470-2045(12)70007-4
83
Hodi
FS
,
O’Day
SJ
,
McDermott
DF
,
Weber
RW
,
Sosman
JA
,
Haanen
JB
, et al.
Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. https://doi.org/10.1056/NEJMoa1003466
84
Kantoff
PW
,
Schuetz
TJ
,
Blumenstein
BA
,
Glode
LM
,
Bilhartz
DL
,
Wyand
M
, et al.
Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–105. https://doi.org/10.1200/JCO.2009.25.0597
85
Zamarin
D
,
Holmgaard
RB
,
Subudhi
SK
,
Park
JS
,
Mansour
M
,
Palese
P
, et al.
Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32. https://doi.org/10.1126/scitranslmed.3008095
86
Smith
GL
,
Mackett
M
,
Moss
B
. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature. 1983;302(5908):490–5. https://doi.org/10.1038/302490a0
87
Mackett
M
,
Smith
GL
,
Moss
B
. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc Natl Acad Sci USA. 1982;79(23):7415–9. https://doi.org/10.1073/pnas.79.23.7415
88
Draper
SJ
,
Heeney
JL
. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8(1):62–73. https://doi.org/10.1038/nrmicro2240
89
Quirk
EK
,
Mogg
R
,
Brown
DD
,
Lally
MA
,
Mehrotra
DV
,
DiNubile
MJ
, et al.
HIV seroconversion without infection after receipt of adenovirus-vectored HIV type 1 vaccine. Clin Infect Dis. 2008;47(12):1593–9. https://doi.org/10.1086/593313
90
McElrath
MJ
,
De Rosa
SC
,
Moodie
Z
,
Dubey
S
,
Kierstead
L
,
Janes
H
, et al.; Step Study Protocol Team. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008;372(9653):1894–905. https://doi.org/10.1016/S0140-6736(08)61592-5
91
Schirmbeck
R
,
Reimann
J
,
Kochanek
S
,
Kreppel
F
. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol Ther. 2008;16(9):1609–16. https://doi.org/10.1038/mt.2008.141
92
Roberts
DM
,
Nanda
A
,
Havenga
MJ
,
Abbink
P
,
Lynch
DM
,
Ewald
BA
, et al.
Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441(7090):239–43. https://doi.org/10.1038/nature04721
93
Sumida
SM
,
Truitt
DM
,
Lemckert
AA
,
Vogels
R
,
Custers
JH
,
Addo
MM
, et al.
Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174(11):7179–85. https://doi.org/10.4049/jimmunol.174.11.7179
94
Barouch
DH
,
Pau
MG
,
Custers
JH
,
Koudstaal
W
,
Kostense
S
,
Havenga
MJ
, et al.
Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172(10):6290–7. https://doi.org/10.4049/jimmunol.172.10.6290
95
Penaloza MacMaster
P
,
Shields
JL
,
Alayo
QA
,
Cabral
C
,
Jimenez
J
,
Mondesir
J
, et al.
Development of novel replication-defective lymphocytic choriomeningitis virus vectors expressing SIV antigens. Vaccine. 2017;35(1):1–9. https://doi.org/10.1016/j.vaccine.2016.11.063
96
Kreppel
F
,
Kochanek
S
. Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol Ther. 2008;16(1):16–29. https://doi.org/10.1038/sj.mt.6300321
97
Pol
JG
,
Zhang
L
,
Bridle
BW
,
Stephenson
KB
,
Rességuier
J
,
Hanson
S
, et al.
Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22(2):420–9. https://doi.org/10.1038/mt.2013.249
98
Flatz
L
,
Cheng
C
,
Wang
L
,
Foulds
KE
,
Ko
SY
,
Kong
WP
, et al.
Gene-based vaccination with a mismatched envelope protects against simian immunodeficiency virus infection in nonhuman primates. J Virol. 2012;86(15):7760–70. https://doi.org/10.1128/JVI.00599-12
99
Victoria
JG
,
Wang
C
,
Jones
MS
,
Jaing
C
,
McLoughlin
K
,
Gardner
S
, et al.
Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J Virol. 2010;84(12):6033–40. https://doi.org/10.1128/JVI.02690-09
100
Komor
AC
,
Badran
AH
,
Liu
DR
. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell. 2017;168(1-2):20–36. https://doi.org/10.1016/j.cell.2016.10.044