Connexins and pannexins: from biology towards clinical targets
DOI: https://doi.org/10.4414/smw.2016.14365
Paolo
Meda, Jacques-Antoine
Haefliger
Summary
Efficient cell communication is a prerequisite for the coordinated function of tissues and organs. In vertebrates, this communication is mediated by a variety of mechanisms, including the exchange of molecules between cells, and between cells and the extracellular medium, via membrane channels made of connexin and pannexin proteins. These channels are a necessary component of all human tissues. Here, we review the biological essentials of the connexin and pannexin families, and the roles of these proteins in the function of cells which are central to major human diseases. We then discuss how connexins and pannexins participate in human pathology, and the clinical perspectives that this knowledge opens.
Introduction
Proper adaptation of human tissues and organs to the changing conditions of the environment and of the milieu intérieur, of necessity depends on a communication network between the component cells, which cross-talk to coordinate and, at times, synchronise their activities [1, 2]. With evolution, this network has progressively diversified to form a complex array of cross-talking and to some extent overlapping cell communication mechanisms [3]. These mechanisms provide for the neural and hormonal signalling between distant cells, and for the more local signalling ensured by extracellular molecules that either interact with specific cell receptors, or enter cells equipped with ion-selective channels [4]. It is now appreciated that most cells are also equipped with connexins and pannexins, which form channels permeable to a large variety of molecules, and that either link two adjacent cytoplasms (connexins) or allow for the communication of cytoplasm with the extracellular fluid (pannexins and, occasionally, connexins) [5–7]. The striking evolutionary conservation of these proteins, their constant expression in all living animals, and the specific phenotypes resulting from their experimental modulation, indicate that connexins and pannexins are indispensable for the functioning of most vertebrate tissues. The increasing numbers of human diseases that have been associated with specific mutations and variants of these proteins, or with their quantitative and distribution changes, further stress their relevance for the clinic [8–14].
Here, we review the biological essentials of connexins and pannexins, and the roles these proteins play in the insulin-producing β-cells of the endocrine pancreas, the renin-producing cells of the kidney cortex and the vascular cells of tumours, three cell types that are central to the pathogenesis of major human diseases. We then discuss the evidence implicating connexins and pannexins in human pathology, and their potential clinical interest.
The connexins
Twenty connexins (Cx), encoded by as many genes, are expressed in humans [5, 6]. Most cell types of our body simultaneously co-express multiple connexin species, in patterns that differ in various cell types owing to the control of gene transcription by cell-specific promoters [5, 6]. The expression of the cognate proteins is largely regulated at the transcriptional level, even though some connexins may further undergo a variety of post-translational modifications [5, 6]. Along the trafficking from the endoplasmic reticulum to the cell membrane, all connexins acquire a similar membrane topography, featuring four membrane-spanning domains, linked by two extracellular and one intracellular loop, and both N- and C-terminal regions in the cytoplasm (fig. 1a). The amino acid sequence of the four α-helical trans-membrane domains, the two extracellular loops and the N-terminus are highly conserved in different mammalian species, whereas the sequence of the intracellular loop and the C-terminus are highly variable [5, 6]. During their transport within endoplasmic reticulum- and Golgi-derived vesicles, six connexin molecules, of either the same or a different type, oligomerise to form a doughnut-shaped structure referred to as a connexon, which provides the wall of a central hydrophilic space of 2–3 nm in diameter.
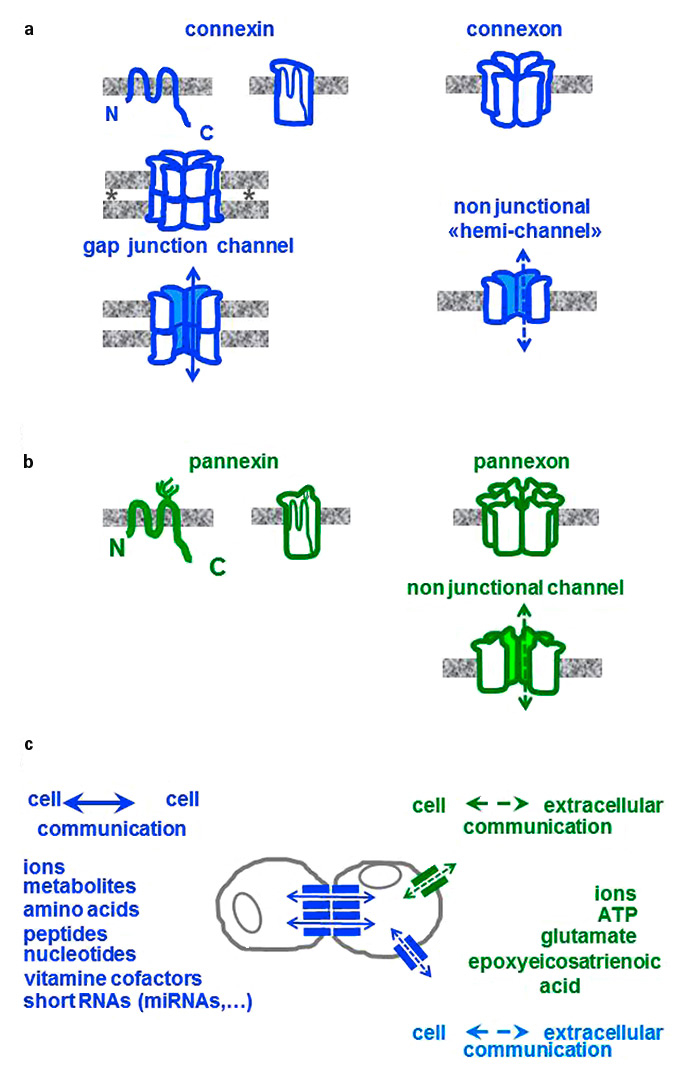
Figure 1
Connexins and pannexins. (a)All connexins (blue) feature 4 domains, which cross the cell membrane (grey) and are connected by one cytoplasmic loop and two extracellular loops. Six connexins oligomerise to form tubular structures, referred to as connexons. Connexons of one cell usually pair with the connexons of an adjacent cell to bridge the intercellular space (asterisks), thus establishing a cell-to-cell, or gap junctional channel. Infrequently, connexons may also be inserted in a non-junctional region of the cell membrane to form “hemi-channels”, which connect cytoplasms to the extracellular medium. (b) Pannexins (green) show a membrane topography similar to that of connexins, and also oligomerise to form pannexons, similar to connexons. However, pannexins feature glycosylated residues on the second extracellular loop that prevent the close apposition of two cells, which is required to form intercellular channels. Hence, pannexons only form non-junctional channels. (c)Connexins mediate cell-to-cell communications by allowing for a bidirectional, diffusional exchange of several cytosolic molecules, including those listed (blue). Pannexins mediate the efflux of cytosolic molecules (green), thus allowing for paracrine signalling, or the uptake by the cells of molecules present in the extracellular medium.
Connexons are usually inserted into lipid rafts of the cell membrane, wherefrom they diffuse to cluster in regions where calcium-dependent cell adhesion molecules permit the close apposition of the membranes of adjacent cells. At these sites, the intercellular space is reduced to a “gap” (hence these sites are referred to as gap junctions) of about 2 nm thickness, where the connexons of two adjacent cells join, extracellular loop-to-extracellular loop, to form an intercellular, gap junctional channel (fig. 1a) [5, 6]. Through these channels, adjacent cells rapidly and bi-directionally exchange a variety of cytosolic molecules, including ions, metabolites, nucleotides, morphogens, vitamin cofactors, small peptides and microRNAs (miRNAs), which move from cell-to-cell (fig. 1c) by diffusion, along electrochemical gradients, without escaping into the extracellular fluid [5, 6]. This event, which is referred to as ionic and metabolic coupling or cell-to-cell coupling, is controlled by the opening and closing of the connexin channels, which are gated by several factors, including cytosolic pH and free calcium concentration. The conductance and permeability of the individual channels is a function of their connexin composition, and of the size, shape and charge of the permeant molecules. Contrasting with many other membrane proteins, connexins have a rather short half-life. Their turnover usually involves the invagination of plaques of gap junction channels into one of the interacting cells, and their degradation by both the lysosomal and the proteasomal pathways [5, 6].
Connexins have been implicated in many prenatal, neonatal and adult cell functions, including cell division, apoptosis, migration, organ growth and morphogenesis during embryogenesis, cell differentiation, gene expression, electrical and metabolic synchronisation, cell contraction, secretion of exocrine and endocrine glands, growth and renewal of normal tissues, resistance of tissues to cytotoxic pharmacological, immune and infectious agents, immune reactions, cell transformation, tumour growth and metastasis [5, 6]. The in vivo relevance of these functions is supported by the specific phenotypes observed in a variety of transgenic models featuring a knock-in, a knock out or a mutation of selected connexin isoforms [5, 6]. The clinical relevance of at least some of these phenotypes is stressed by the increasing identification of human diseases which are associated to mutations and single nucleotide polymorphisms of specific connexins [5, 6, 8–12]. These studies have further documented that, if most connexins allow for cell-to-cell coupling, other functions are dependent on specific connexin isoforms [6, 7]. Most of these functions have been attributed to the connexin-dependent cell coupling. However, some effects may be intracellular, i.e. accounted for by the interaction between connexins and other proteins, and/or their effects on gene expression [5, 6].
Some connexons infrequently remain in non-junctional domains of the cell membrane, where they function as unicellular “hemi-channels” (fig. 1a), for the bi-directional exchange of ions and second messengers between cytosol and the extracellular space (fig. 1c). The biophysical properties of “hemi-channels” differ from those of the junctional channels made by the same connexin species, suggesting distinct roles. However, whether such roles are of physiological relevance remains to be assessed, inasmuch as function of connexin hemi-channels has so far been demonstrated only under pathological conditions [6]. The function of such hemi-channels remains under debate, since it is difficult to distinguish their contribution from that of gap junction channels and the non-junctional channels made of pannexins. Most cell types simultaneously express gap junction plaques made of connexins and second messenger-permeated non-junctional channels, made by pannexins and, occasionally, by connexins (fig. 2a,c,d and fig. 3a,b,c) [6].
The pannexins
Three pannexins (Panx), encoded by as many genes, are also expressed in humans [6, 7]. Pannexins also display four membrane-spanning segments, two extracellular and one cytoplasmic loop, and feature both N- and C-terminal domains within the cytoplasm (fig. 1b) [6, 7]. However, pannexins do not show sequence homology with connexins, and display consensus sequences for glycosylation on the second extracellular loop (fig. 1b). Most cell types express Panx1, in combination with various connexin patterns, whereas the distribution of Panx2 and Panx3 is restricted to selected cell types [6, 7]. During their transport in the cytoplasm, six pannexins oligomerise to form pannexons, which resemble connexons. However, pannexons do not usually form cell-to-cell channels, since the extracellular loop glycosylation impedes a sufficiently close approximation and pairing of the pannexons made by adjacent cells [6, 7]. The predominant role of pannexons is to form channels in non-junctional domains of the cell membrane, which show little selectivity for current-carrying ions, and provide for the leakage of cytosolic second messengers, including ATP, glutamate and epoxyeicosatrienoic acid into the extracellular medium (fig. 1c) [6, 7]. Pannexons also permit the reverse uptake into cells of extracellular and membrane-impermeant molecules (fig. 1c) [8, 9]. Opening of pannexin channels is induced by membrane depolarisation, mechanical stress, and activation of purinergic receptors, whereas their closure can be experimentally induced by a number of drugs that also turn off connexin channels [6, 7]. Under physiological conditions, pannexons open frequently but briefly, thereby preventing the dialysis of vital cell moieties [6, 7].
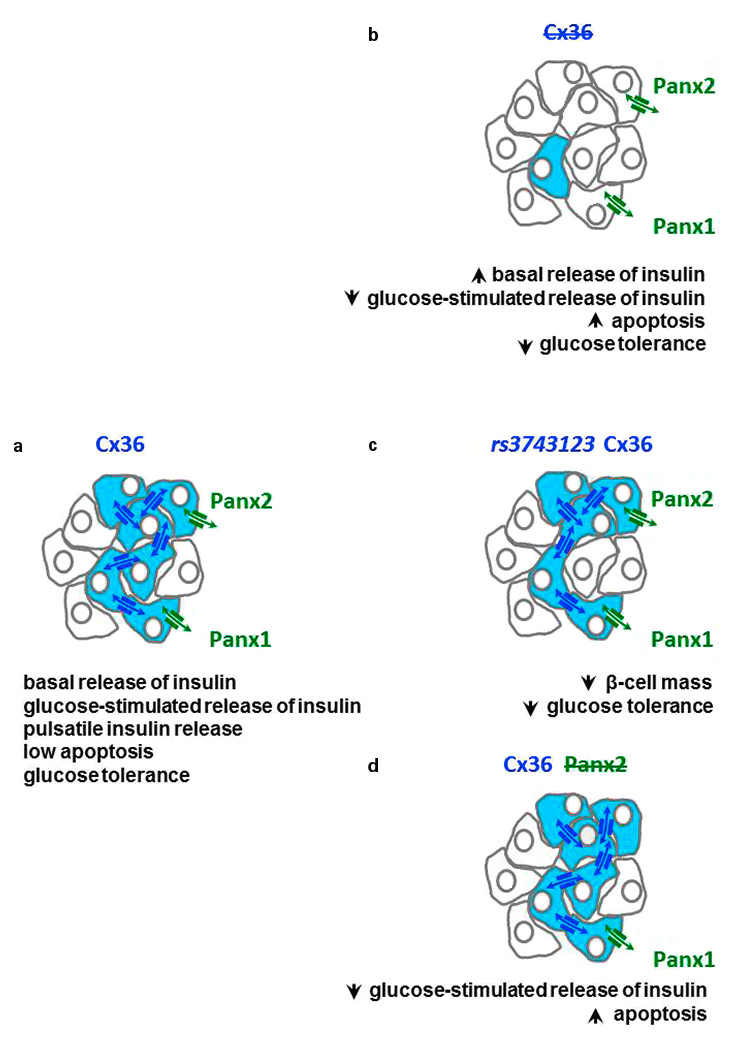
Figure 2
Cx36 and the insulin-producing β-cells. (a)The most abundant endocrine cell type of pancreatic islets, named β-cells, are only coupled by Cx36 channels, and also express Panx2 and Panx1. (b) Loss of Cx36 increases (upward arrow heads) or decreases (downward arrow heads) the major characteristics of β-cells (Cx36-coupled cells are shown in blue), notably insulin secretion and apoptosis. (c) These alterations are also observed in rodents and humans when β-cells express the rs3743123variant of Cx36.The β-cell changes observed under such conditions mimic the major defects of type 2 and type 1 diabetes. In vitro, cells expressing this Cx36 variant are less coupled than cells expressing comparable levels of the wild type Cx36 form. (d) Loss of Panx2 also results in a decrease of glucose-stimulated insulin secretion and in an increase of β-cell apoptosis. Reverse transcriptase-polymerase chain reaction analysis indicates that Panx2 and Panx1 may be co-expressed with Cx36 by individual β-cells, although a validation of this tentative conclusion awaits the development of fully specific antibodies against the two pannexin species.
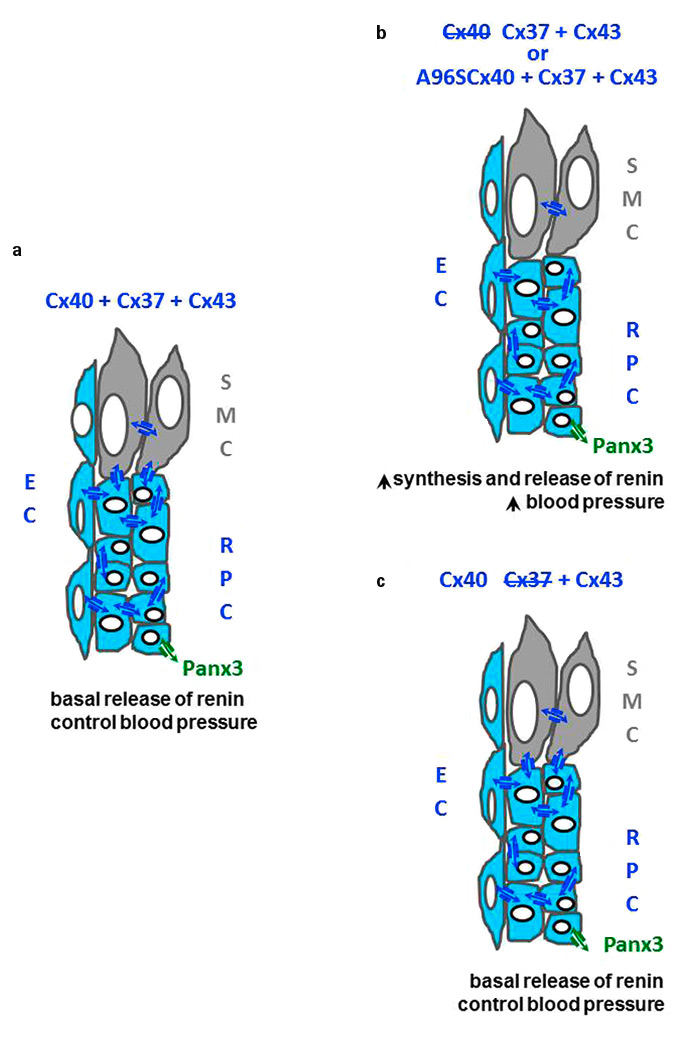
Figure 3
Cx40, Cx37, Cx43 and the renin-producing cells. (a)Within the renal cortex, renin-producing cells (RPC) are coupled by Cx37 and Cx40 channels, to companion cells and nearby endothelial cells (EC), and also express Panx3. The smooth muscle cells (SMC, grey) of the afferent arterioles are coupled by Cx43 channels to companion cells and to RPC. (b) Loss of Cx40 increases the major function of RPC, i.e. renin biosynthesis and secretion, resulting in persistent hypertension. These changes are also observed when cells express the A96Svariant of Cx40 (b), but not after deletion of Cx37 (c). The alterations in renin secretion observed under such conditions mimic those observed in most forms of hypertension.
Pannexin channels have been implicated in several cell functions [8, 9], including the intercellular propagation of calcium waves mediated by the release of cytosolic ATP, which activates metabotropic and ionotropic purinergic receptors both on the cells that release ATP (autocrine pathway) and in neighbouring cells (paracrine pathway) [6, 7, 13, 14]. Pannexins also contribute to the immune response, ischaemia-induced cell death, oxidative stress, cell growth and tumorigenesis, including in humans [6, 7, 13, 14]. Circumstantial evidence also indicates intracellular functions of pannexins, including the homeostasis of intracellular calcium, the formation of the inflammosome, and the release of bacterial catabolites [6, 7]. In many cell types, including those that we discuss in this review, individual cells simultaneously express various pannexins together with one (e.g. β-cells) or several different connexin species (e.g. renin-producing cells, endothelial cells, smooth muscle cells) (fig. 2a,c,d and 3a,b,c).
Cx36 and the insulin-producing β-cells of pancreatic islets
Cx36 gap junctions mediate the electrical coupling of most pancreatic β-cells within each islet of the rodent and human pancreas, as indicated by the rhythmic and synchronised bursts of electrical activity and cytosolic free calcium, and also provide for the exchange of nucleotides and metabolites between small numbers of β-cells (fig. 2a) [5, 15-17]. Full uncoupling and electrical desynchronisation of β-cells are observed after either the homologous recombination of the Gjd2 gene, which encodes Cx36 (fig. 2b), or its conditional deletion in β-cells [5, 15–17]. Physically and pharmacologically uncoupled β-cells show decreased expression of the insulin gene and proinsulin biosynthesis, increased basal release of insulin, and poor to nil glucose-induced insulin release, as a result of a sustained increase in free cytosolic calcium. These effects are largely reversible after reaggregation or wash-out of the blocking drugs, two conditions that also promote the recruitment of secretory and biosynthetically active β-cells. Cx36 expression increases with the neonatal acquisition by mouse β-cells of a normal sensitivity to glucose, and is reduced after a high-fat diet, which induces glucose intolerance. In contrast, transgenic mice whose β-cells overexpress the ectopic Cx32 become intolerant to glucose as a result of decreased insulin secretion [5, 15–17].
The β-cells of Cx36-null mice lack gap junctions and coupling, demonstrating that, in this system, Cx36 is the only connexin species forming functional cell-to-cell channels [15, 16, 18]. The pancreatic islets of these animals do not release insulin in the normal pulsatile fashion owing to loss of the normal intercellular synchronisation of the cytosolic calcium transients induced by glucose [15, 16]. These islets also show increased basal release of insulin [5, 16] resulting in glucose intolerance of the transgenic animals (fig. 2a) due to alterations of both first and second phases of insulin release [18]. The excessive basal secretion is consistent with the finding that uncoupled β-cells can no longer be inhibited by hyperpolarising currents generated in nearby cells [19], and with the prolonged decay in their off-response once glucose stimulation ceases [5, 16]. The loss of glucose responsiveness is consistent with the loss of the calcium oscillations that drive the regular oscillatory output of insulin during stimulation of insulin release [5, 15–18]. The intolerance to post-prandial glucose levels is likely due to the in vivo decrease in the oscillations of circulating insulin, which associates with lower amplitude and longer decay of the first phase of glucose-induced insulin secretion, as well as with reduced insulin oscillations during the second phase. The data indicate that Cx36 occupies a prominent hierarchical position in the multi-factorial regulation of insulin dynamics, which is central to glycaemic control [5, 15–18].
In vivo, loss of Cx36 also sensitises β-cells to pharmacological and immunological conditions resulting in β-cell death (fig. 2b), specifically in the presence of the cytokines that induce apoptosis at the onset of type 1 diabetes [5, 15, 19, 20]. Conversely, transgenic mice overexpressing Cx36 are fully protected against the same insults [5, 15]. Pro-apoptotic cytokines and oxidised low density lipoprotein particles reduce the expression of Cx36 via mechanisms dependent on inducible cAMP early repressor (ICER-1) and AMP-activated protein kinase (AMPK)- [20, 21], and Cx36 plays a central role in the oxidative and the endoplasmic reticulum stress induced by cytokines [19, 20], two effects that enhance the mitochondria-dependent apoptosis of β-cells. The mechanism underlying the protective role of Cx36 remains to be unravelled. The data show that physiological Cx36 signalling contributes to the survival of β-cells.
Cx36 may also form “hemi-channels” on non-junctional portions of the β-cell membrane [16] which, by permitting a leakage of cytosolic molecules into the extra-cellular islet fluid, may have an autocrine effect on β-cells and/or a paracrine action on their neighbours. However, there is as yet no evidence that these channels are opened under physiological conditions. Beta-cells further express Panx2 and Panx1 [16] (fig. 2a,b,c), which may provide channels for a glutamate-dependent paracrine pathway. Whether these proteins form functional channels in pancreatic islets remains to be shown. Still, pancreatic islets from Panx2-null mice show a decrease in basal and glucose-induced insulin secretion, and an increase in both basal and cytokine-induced β-cell apoptosis (fig. 2d). Thus, Panx2 may contribute to autocrine and/or paracrine purinergic mechanisms involving ADP and P2Y receptors, which could contribute to control the survival and function of insulin-producing cells under pathologic conditions, notably in diabetes [16].
Cx40, Cx37 and the renin-producing cells of kidneys
Adult renin-producing cells are coupled to companion cells, as well as to the nearby endothelial cells of the juxtaglomerular apparatus, by gap junction channels [5, 22] made of Cx40 and Cx37 [5, 22] (fig. 3a). This coupling is implicated in the transmission along the afferent arteriole of vasomotor signals generated in the macula densa cells, and also accounts for so-called “nephron coupling” [5, 22]. Invalidation of the Cx40 gene results in a marked and sustained hypertension of mice (fig. 3b), a phenotype largely explained by an increased synthesis and release of renin [5, 22]. This increase results from the loss of the normal feedback mechanisms whereby the circulating levels of angiotensin II, as well as an increased intrarenal blood pressure, normally suppress the expression of the renin gene and the release of the hormone in the afferent arteriole [5, 22]. The inhibitory effect of the macula densa on renin secretion is also attenuated in Cx40-deficient mice [5, 22]. The deletion of Cx40 only in renin-secreting cells induces a hypertension similar to that of the Cx40-null mice (fig. 3b) [5, 22], whereas the restoration of Cx40 only in the renin-secreting cells of the latter animals reduces their renin and hypertension levels [23]. In contrast, the endothelium-specific deletion of Cx40 does not alter blood pressure [5, 22], and the restoration of Cx40 expression only in the endothelial cells of Cx40-null mice fails to reduce their hypertension, in spite of a normal expression of Cx37 and endothelial nitric oxide synthase (eNOS) [23]. Cx37-null mice [24], which feature a normal renal expression of Cx40, Cx43 and Cx45, are normotensive, and show normal values of renin secretion (fig. 3c), even though this secretion is stimulated by a low salt diet more in these mice than in control animals [25]. In situ,renin-producing cells also interact with the smooth muscle cells of the afferent arterioles, which are coupled by Cx43 [26], and the renal levels of this protein are increased in both the hypertensive Cx40-null mice and in control mice made hypertensive by clipping one renal artery. Even though mice with a smooth muscle cell-specific deletion of Cx43 are normotensive, the replacement of Cx43 by Cx32 reduces renin production and protects mice from the hypertension that is normally induced by a decrease in the blood supply to one kidney or by a high-salt diet [26].
The data show the prominent role of Cx40 in the regulation of renin secretion and, thus, of blood pressure, and also stress the additional contribution of Cx43. Panx3 has been reported in the juxtaglomerular apparatus [27], but the possible involvement of non-junctional connexin and pannexin channels in renin-dependent hypertension has not yet been directly evaluated.
Cx40, Cx37, Cx43 and the endothelial and smooth muscle cells of vessels
The hypertension of Cx40-deficient mice is partly accounted for by peripheral vascular effects of the renin-dependent activation of the renin-angiotensin system [5]. Thus, Cx40-null mice feature an altered vasomotion of small arterioles, and blockade of either angiotensin converting enzyme or the angiotensin II receptor AT1 reduces their hypertension. The obligatory participation of peripheral vascular factors is further supported by the observation that the replacement of Cx40 by Cx45 induces modest hypertension, in spite of normal levels of plasma renin [22]. The vascular component of hypertension appears to be a result of an interruption of the cell-to-cell coupling mediated by Cx40, which normally controls the vasomotor tone, since Cx40-null mice feature an abnormal reactivity of the peripheral vasculature [28], variably attributed to the loss of Cx40, the concurrent decrease of Cx37 [5, 22, 27], or the up-regulation of Cx43 [29].
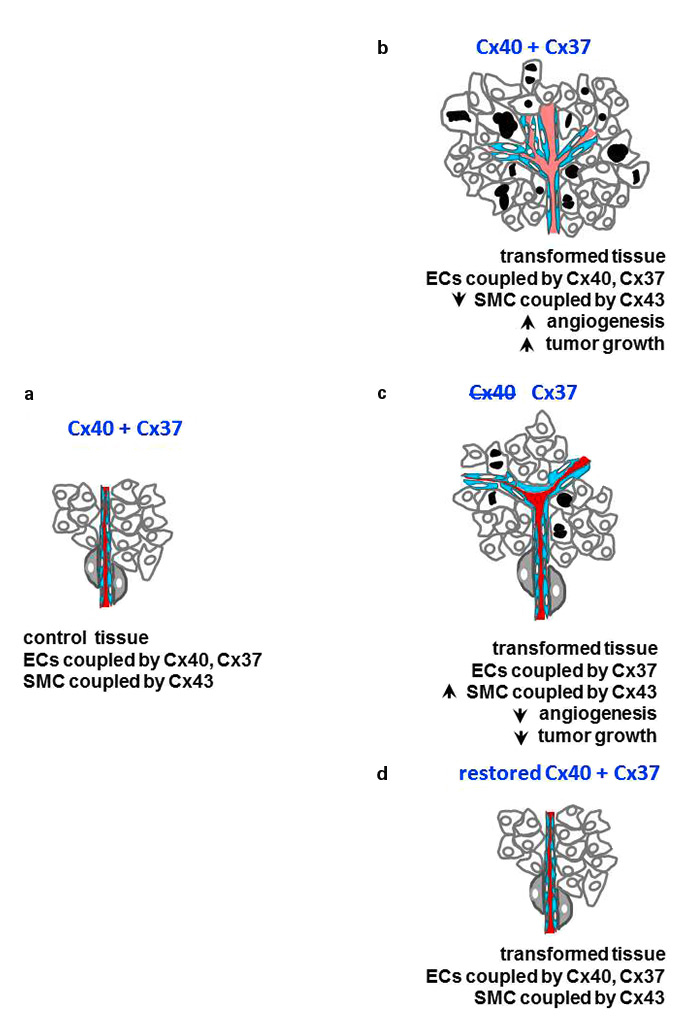
Figure 4
Cx40, cancer growth and angiogenesis. (a)In most tissues, the endothelial cells of micro vessels are coupled (blue) by Cx37 and Cx40 channels. (b) The tumoral growth of transformed tissues is usually associated with the proliferation of endothelial cells, resulting in the excessive development of new vessels, which feature abnormal walls lacking pericyte (grey cells) coverage. (c) These alterations, and the growth of transformed cells are decreased after loss of Cx40, resulting in a prolonged survival of the tumour-bearing animals. (d) Restoration of Cx40 expression in endothelial cells, prevents both excessive angiogenesis and tumour growth.
The endothelial cells of the afferent arterioles of kidneys, and of many other extra-renal arteries, are coupled by Cx40 and Cx37 (fig. 4a) [22, 29, 30]. The two connexins tightly interact with eNOS, increasing the activity of the enzyme and, thus, the production of NO. In turn, NO promotes the relaxation of smooth muscle cells, resulting in vasodilation. Accordingly, loss of either Cx40 or Cx37 impairs this mechanism [22, 30]. In contrast, the interactions of Cx40 and Cx37 with eNOS are increased in a murine model of volume-dependent hypertension [31]. The implementation of this model in Cx40-null mice, which represent a genetic model of renin-dependent hypertension, resulted in a further increase of their native hypertension and eNOS phosphorylation [31]. The data demonstrate that Cx37 and Cx40 modulate in vivo the production of NO and, thus, vasodilatation. The smooth muscle cells of many arterioles are coupled by Cx43 and Cx45 channels [5, 22], which ensure the rapid diffusion along the vascular tree of the calcium-dependent contraction waves that ultimately control vasomotor tone [32]. The Cx43 of smooth muscle cells increases with thickening of the media in different models of chronic renin-dependent hypertension [29], but not in models in which hypertension is caused by the pharmacological inhibition of eNOS. In contrast, all models of chronic hypertension induce Cx37 and increase Cx45 in the smooth muscle cells of the hypertrophied media layers [22, 29, 31].
The potential contribution of non-junctional connexin and pannexin channels has not yet been directly investigated in hypertension models. However, mice lacking Panx1 in vascular smooth muscle cells are hypotensive, suggesting that Panx1 expression contributes to the development of peripheral resistance [17].
Cx40 and cancer cells and angiogenesis
Angiogenesis is fundamental to the growth of most tumours [33]. During this process, quiescent endothelial cells migrate and proliferate to form new vessels, which are required to ensure the oxygenation and feeding of growing tumours. The newly formed vessels show a disorganised architecture, with a leaky intima layer and few, abnormal pericytes (fig. 4c), resulting in haemorrhages and increased interstitial fluid pressure, which limit blood perfusion [33, 34].
Endothelial cells are coupled by Cx40, Cx37 (fig. 4a) and Cx43 channels [29]. The latter connexin is also expressed by the smooth muscle cells and the pericytes of some vessels (fig. 4a,b,c) [35]. In the absence of Cx40, the growth of melanoma and urogenital model tumours is reduced in mice, a change associated with a decrease in angiogenesis (fig. 4b) and eNOS activity [36]. Under these conditions, newly-formed tumour vessels feature a normal structure (fig. 4b), providing for an improved perfusion of the growing tumour, which presumably accounted for the extended survival of the tumour-bearing animals observed after cyclophosphamide administration [36]. The data implicate Cx40 in the in vivo production of NO, and the ensuing regulation of angiogenesis which modulates tumour growth [36].
The combined invalidation of Cx40 and Cx37 causes lethal perinatal haemorrhages, indicating that both connexins are required for the normal development of the microvasculature. Given that both Cx37 and Cx40 directly interact with eNOS [30, 37], and that the levels of Cx37 expression are linked to those of Cx40 [30], a partial loss of Cx37 signalling could contribute to the alterations in tumour growth and angiogenesis. However, the angiogenesis and tumour alterations observed in mice lacking Cx40 are prevented in transgenic mice in which the connexin is solely restored in the endothelial cells (fig. 4c), and are mimicked in various angiogenesis models by a peptide that specifically interferes with Cx40 [36], indicating that this connexin isoforms plays a hierarchically predominant role in the control of both angiogenesis and tumour growth.
This tentative conclusion does not exclude participation of Cx43. Indeed, mice expressing about 50% of the control levels of Cx43 show an increased vascularisation of tumours resulting from implanted breast cancer cells, and still feature decreased mural cell coverage. Low levels of Cx43 also associate with increased expression of vascular endothelial growth factor (VEGF) and enhanced proliferation of endothelial cells in tumours generated by injection of mice with different types of transformed cells [38]. In contrast, when the same cells overexpressed Cx43, tumour growth and angiogenesis decrease [38]. Thus, Cx43 modulates the wall of neoangiogenetic vessels, whose alteration is one of the causes of the hypoxic environment of tumours, which usually increases their malignant phenotype and metastatic potential [35]. No direct experimental evidence has implicated the non-junctional channels made by connexins and pannexins in the control of angiogenesis. In contrast, increasing evidence implicates pannexins in tumour growth and metastasis [13, 14, 39].
The human situation
The observations summarised above provide compelling evidence that connexins and pannexins significantly contribute to the control of many cell functions in vivo. The finding that the cell-specific patterns of connexins, pannexins and coupling are remarkably conserved in different species, and that the phenotype of mice featuring cell-specific alterations of connexin/pannexin patterns mimics the signs of human diseases [5, 9, 10, 15–17, 35, 36], suggest that the signalling dependent on these proteins may be relevant in the pathogenesis of common clinical conditions. Specific polymorphisms and mutations of individual connexins and pannexins have now been linked to a variety of genetic diseases [8–14, 35, 36], and quantitative alterations in the two families of proteins have also been implicated in a variety of other diseases [5–7, 13]. Obviously, these implications cannot be validated in humans by the direct experimental testing that is feasible in rodents, and largely rely on genetic analysis of selected populations. In the autoimmune, type I form of diabetes, type 1 helper T cell (Th1) cytokines kill most pancreatic β-cells by apoptosis, leaving a residual mass insufficient to sustain the insulin demand of the organism. The loss of either Cx36 or Panx2 sensitises β-cells to the proapoptotic action of cytokines and, conversely, the overexpression of Cx36 protects against this lethal effect [5, 15, 19, 20].
Consistent with these findings, Th1 cytokines decrease Cx36 expression [5, 11, 15, 19, 20], and altered expression of the Cx36 transcript is detected in genome-wide scans of type I diabetes models [5, 11]. The phenotype of mice lacking Cx36 also mimics the β-cell alterations typical of prediabetes and type 2 diabetes, including loss of circulating insulin oscillations, glucose intolerance, increased basal secretion, decreased glucose-induced insulin secretion and increased β-cell apoptosis [5]. The GJD2 gene, which codes for human Cx36, is on a locus (15q14) that associates with type 2 diabetes [5, 11, 15, 16]. Hyperglycaemic individuals show lower levels of Cx36 messenger RNA than normoglycaemic individuals, particularly if they express the rs3743123 variant of Cx36 (fig. 2c) [11]. This variant differs from the wild type control Cx36 by a single nucleotide. In mice, this variant reduces the expression of Cx36 and the β-cell mass of adult animals, resulting in sustained hyperglycaemia in some, but not all, families [11]. A sulphonylurea, which stimulates insulin release from the glucose-unresponsive β-cells of type 2 diabetic patients, increases Cx36 coupling [5, 15, 20] and prevents the autoimmune apoptosis of β-cells in a model of type 1 diabetes [40]. Together with the findings that prolonged exposure of insulin-producing cells to high concentrations of glucose and fatty acids down regulates Cx36 expression, and that the levels of Cx36 transcript correlate with those of insulin [5, 15, 16], these observations indicate that reduced levels of Cx36 could be implicated in the pathogenesis of diabetes.
In humans, most forms of chronic hypertension result from altered functioning of the renin-angiotensin system, with or without alterations in resistance arteries. The distribution of connexins and pannexins is similar in the kidneys and vessels of humans and rodents, and the phenotype of animals submitted to the loss of either renal or vascular connexins, or to experimental models of renin-dependent and -independent hypertension, mimics the signs of the corresponding human diseases. On the renal side of the disease, two polymorphisms within the promoter region of the human Cx40 gene reduce the permeability of Cx40 channels and are associated with hypertension in mice [40] and selected human populations [5, 41, 42]. One of these polymorphisms also significantly increases the standing systolic blood pressure of normotensive individuals [42]. A mutation of Cx40 (A96SCx40), which reduces the permeability of the Cx40 channel, associates with patients suffering from cardiac arrhythmia and with hypertension in mice [41].
Circumstantial evidence indicates that connexin and pannexin channels are implicated in the establishment, growth and metastasis of a variety of cancers [13, 14, 17, 38]. Notably, a mutation of Panx1 is enriched in highly metastatic breast cancers, and Panx2 levels may correlate with the prognosis of glial tumours [17].
The translational perspectives
Screening of connexin mutations is now a standard in the diagnoses of congenital, prelingual deafness [8, 9] and skin disorders [9, 10].
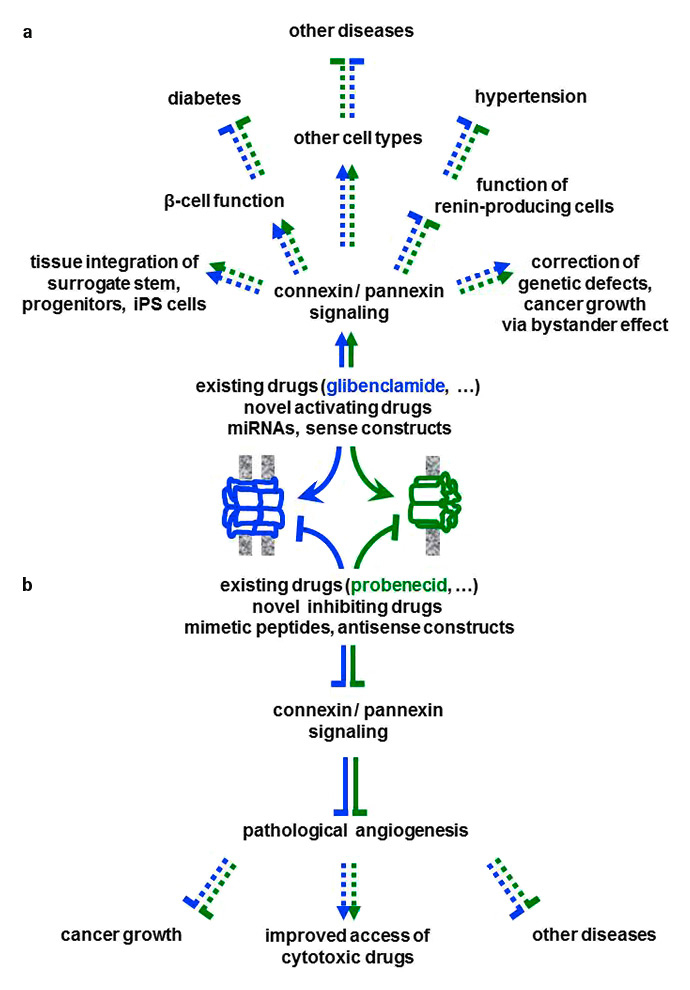
Figure 5
The translational perspectives. (a)The stimulation (arrows) of junctional and non-junctional channels (e.g. those formed by Cx36 and Panx2) is expected (dotted lines) to reproduce in humans the beneficial effects of connexin (blue) and pannexin (green) signalling observed in rodents. In turn, increasing the function of pancreatic β-cells is expected to improve the treatment of type 1 or type 2 diabetes. Increased connexin and pannexin signalling is also expected to foster the integration in the host tissues of surrogate cells designed for cell replacement therapies. (b) Conversely, the inhibition (flat-end lines) of selected connexin species (e.g. Cx40) is expected to prevent the over-stimulation of the renin-producing cells, thus contributing to mitigate several forms of hypertension. The specific down-regulation of Cx40 is also expected to prevent excessive angiogenesis, while preserving a normal vessel wall and function, thus improving the efficiency of current cytotoxic therapies. This combined approach could reduce the growth of some cancer forms and the development of other diseases due to excessive angiogenesis.
Clinical trials have begun in which retroviral vectors are used to express herpes simplex virus thymidine kinase in tumour cells [43]. The transduced cells can then be killed in vivo by ganciclovir, a guanosine analogue that, after phosphorylation by the viral enzyme, blocks cell proliferation by incorporating into nascent DNAs [43]. Strikingly, only a fraction of the transformed cells need to express the viral kinase to make the whole tumour sensitive to ganciclovir. Even if the mechanism of this "bystander effect" (fig. 5) [43] is still debated, the finding that the ganciclovir metabolite is transferred between infected and non-infected cells via gap junctional channels, indicates a likely role of connexin signalling [44]. An analogous, beneficial effect is “channel sharing”, whereby ionic fluxes into only a limited proportion of cells are sufficient to correct the defective functions of a much larger cell population. For example, less than 10% of the cells lacking the cystic fibrosis transmembrane conductance regulator protein (CFTR) need to be corrected, by CFTR transduction, to restore a normal fluid transport of intact epithelial sheets [45], given that connexin and pannexin channels are permeable to chloride anions [5]. Thus connexin and/or pannexin channels can amplify the effects of drugs and the correction of various genetic defects [46].
The studies reviewed here further demand that connexins and pannexins be considered in development of innovative approaches for the treatment of highly prevalent human diseases, by targeting junctional and non-junctional channels, using drugs altering the function of specific connexin and/or pannexins (fig. 5). For example, the antidiabetic sulphonylurea glibenclamide promotes the biosynthesis and function of Cx36 channels and is sufficiently safe for chronic administration in diabetic patients [5, 16]. The molecule is expected to promote connexin signalling, thus fostering insulin secretion and decreasing the apoptosis of pancreatic β-cells. On the other hand, the uricosuric probenecid, which blocks with some specificity Panx1 channels, may help to inhibit cancer growth [13, 14, 17]. It is likely that molecular tools will be instrumental to the development of mimetic peptides, miRNAs, and sense and antisense constructs (fig. 5), which have been shown to selectively interfere with the production and function of connexons and pannexons, including in human diseases [6, 47–49]. For example, a non-coding miRNA, which is down-regulated in the early stage of colorectal cancer [47] and acts as a tumour suppressor, is transferred via gap junctions between endothelial and transformed cells, limiting tumour angiogenesis and cancer growth [47]. As yet, however, few molecules match the obligatory specificity, reversibility and safety criteria required for use in the clinic, requiring the development of novel models for large-scale screening of better candidates [50]. This novel and most needed connexin/pannexin pharmacology is expected to foster the efficacy of existing treatments. For example, the association of drugs improving Cx36 signalling with drugs inhibiting the key metabolic enzyme AMPK is anticipated to improve the effects of the two individual therapies in decreasing β-cell apoptosis and in increasing insulin secretion [5, 15, 16]. Also, drugs inhibiting Cx40 signalling should potentiate the effects of a combined cytotoxic treatment, by favouring the delivery of the drug within tumours, via their beneficial effects on the structure of the newly formed vessels [48, 49].
The ongoing implementation of cell therapies, in which surrogate stem, progenitor or induced pluripotent stem cells are engineered for the in vivo replacement of damaged cells, implies that the transplanted cells become functionally integrated within the host tissue. In turn, this integration implies an adequate development of connexin- and pannexin-dependent cell interactions. Most of the embryonic stem cells and progenitor cells that are the basis of many such trials do not express the connexins found in adult tissues [5, 16], e.g. Cx36 for islet β-cells, possibly explaining the limited efficacy of the purported replacement. Proper expression of specific connexins/pannexins in the surrogate cells is expected to improve the efficacy of cell therapies (fig. 5), e.g. for the proper replenishment of β-cells in diabetic patients [51].
Conclusions
The signalling by connexins and pannexins has taken its place among the numerous mechanisms that collectively allow cells to ensure coherent functioning of tissues and organs. The available data point to a relevant role of both protein families in the physiological regulation of many cells, which are central to, if not causal of, prominent human diseases. These considerations now call for further research on the molecular organisation of connexin and pannexin signalling, on the cellular mechanisms that they activate, and on their hierarchical relationships. This information is a prerequisite for testing the pathogenetic role of connexins and pannexins, which, in turn, is needed to validate the potential usefulness of targeting these proteins for the development of innovative therapeutic strategies. In view of the variety of cell functions in which connexins and pannexins are involved, and of the complexity of the signalling arrays to which this signalling contributes in living tissues, these challenges are both considerable and exciting. Taking up these challenges is now timely, given the explosive worldwide prevalence of those human diseases to which connexins and pannexins may contribute, and for the treatment of which the existing therapies still have little efficiency and specificity and many secondary effects, and represent a major individual and economic burden.
Acknowledgments:We are grateful to the collaborators mentioned in the references. Our work largely took place at the Universities of Genève and Lausanne, and was generously supported by the Fonds National Suisse de la Recherche Scientifique, the Recherche Suisse Contre le Cancer, the European Union and the Juvenile Diabetes Foundation.
References
1 Bull L. On the evolution of multicelluarity and eusociality. Artif Life. 1999;5(1):1–15.
2 Michod RE, Roze D. Cooperation and conflict in the evolution of multicellularity. Heredity. 2001;86(Pt 1):1–7.
3 Edelman GM. Cell adhesion and the molecular processes of morphogenesis. Annu Rev Biochem. 1985;54:135–69.
4 LeRoith D, Delahunty G, Wilson GL, Roberts CT, Jr., Shemer J, Hart C, et al. Evolutionary aspects of the endocrine and nervous systems. Recent Prog HormRes. 1986;42:549–87.
5 Bosco D, Haefliger JA, Meda P. Connexins: key mediators of endocrine function. Physiol Rev. 2011;91(4):1393–445.
6 Esseltine JL, Laird DW. Next-generation connexin and pannexin cell biology. Trends Cell Biol. 2016.S0962-8924(16)30074-5. [Epub ahead of print]
7 Scemes E, Spray DC, Meda P. Connexins, pannexins, innexins: novel roles of “hemi-channels”. Pflugers Arch. 2009;457(6):1207–26.
8 Wingard JC, Zhao HB. Cellular and deafness mechanisms underlying connexin mutation-induced hearing loss – a common hereditary deafness. Front Cel Neurosci. 2015;9:202.
9 Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41(1):103–16.
10 Lilly E, Sellitto C, Milstone LM, White TW. Connexin channels in congenital skin disorders. Semin Cell Dev Biol. 2016;50:4–12.
11 Cigliola V, Populaire C, Pierri CL, Deutsch S, Haefliger JA, Fadista J, et al. A variant of GJD2, encoding for connexin 36, alters the function of insulin producing beta-cells. PLoS One. 2016;11(3):e0150880.
12 Mas C, Taske N, Deutsch S, Guipponi M, Thomas P, Covanis A, et al. Association of the connexin36 gene with juvenile myoclonic epilepsy. J Med Genet. 2004;41(7):e93.
13 Penuela S, Harland L, Simek J, Laird DW. Pannexin channels and their links to human disease. Biochem J. 2014;461(3):371–81.
14 Retamal MA, Reyes EP, Garcia IE, Pinto B, Martinez AD, Gonzalez C. Diseases associated with leaky hemichannels. Front Cell Neurosci. 2015;9:267.
15 Klee P, Allagnat F, Pontes H, Cederroth M, Charollais A, Caille D, et al. Connexins protect mouse pancreatic beta cells against apoptosis. J Clin Invest. 2011;121(12):4870–9.
16 Cigliola V, Allagnat F, Berchtold LA, Lamprianou S, Haefliger JA, Meda P. Role of connexins and pannexins in the pancreas. Pancreas. 2015;44(8):1234–44.
17 Furlow PW, Zhang S, Soong TD, Halberg N, Goodarzi H, Mangrum C, et al. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat Cell Biol. 2015;17(7):943–52.
18 Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RK. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes. 2012;61(7):1700–7.
19 Haefliger JA, Martin D, Favre D, Petremand Y, Mazzolai L, Abderrahmani A, et al. Reduction of connexin36 content by ICER-1 contributes to insulin-secreting cells apoptosis induced by oxidized LDL particles. PLoS One. 2013;8(1):e55198.
20 Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, et al. Lipotoxicity disrupts incretin-regulated human beta cell connectivity. J Clin Invest. 2013;123(10):4182–94.
21 Allagnat F, Klee P, Cardozo AK, Meda P, Haefliger JA. Connexin36 contributes to INS-1E cells survival through modulation of cytokine-induced oxidative stress, ER stress and AMPK activity. Cell Death Differ. 2013;20(12):1742–52.
22 Kurtz A. Connexins, renin cell displacement and hypertension. Curr Opin Pharmacol. 2015;21:1–6.
23 Le Gal L, Alonso F, Wagner C, Germain S, Nardelli Haefliger D, Meda P, et al. Restoration of connexin 40 (Cx40) in renin-producing cells reduces the hypertension of Cx40 null mice. Hypertension. 2014;63(6):1198–204.
24 Simon AM, Goodenough DA, Li E, Paul DL. Female infertility in mice lacking connexin 37. Nature. 1997;385(6616):525–9.
25 Wagner C, Kurtz L, Schweda F, Simon AM, Kurtz A. Connexin 37 is dispensable for the control of the renin system and for positioning of renin-producing cells in the kidney. Pflugers Arch. 2009;459(1):151–8.
26 Haefliger JA, Krattinger N, Martin D, Pedrazzini T, Capponi A, Doring B, et al. Connexin43-dependent mechanism modulates renin secretion and hypertension. J Clin Invest. 2006;116(2):405–13.
27 Abed AB, Kavvadas P, Chadjichristos CE. Functional roles of connexins and pannexins in the kidney. Cell Mol Life Sci. 2015;72(15):2869–77.
28 Jobs A, Schmidt K, Schmidt VJ, Lubkemeier I, van Veen TA, Kurtz A, et al. Defective Cx40 maintains Cx37 expression but intact Cx40 is crucial for conducted dilations irrespective of hypertension. Hypertension. 2012;60(6):1422–9.
29 Alonso F, Krattinger N, Mazzolai L, Simon A, Waeber G, Meda P, et al. An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc Res. 2010;87(1):166–76.
30 Alonso F, Boittin FX, Beny JL, Haefliger JA. Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am J Physiol Heart Circ Physiol. 2010;299(5):H1365–73.
31 Le Gal L, Alonso F, Mazzolai L, Meda P, Haefliger JA. Interplay between connexin40 and nitric oxide signaling during hypertension. Hypertension. 2015;65(4):910–5.
32 Koenigsberger M, Seppey D, Beny JL, Meister JJ. Mechanisms of propagation of intercellular calcium waves in arterial smooth muscle cells. Biophys J. 2010;99(2):333–43.
33 Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–121.
34 Fukumura D, Duda DG, Munn LL, Jain RK. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010;17(3):206–25.
35 Choudhary M, Naczki C, Chen W, Barlow KD, Case LD, Metheny-Barlow LJ. Tumor-induced loss of mural Connexin 43 gap junction activity promotes endothelial proliferation. BMC Cancer. 2015;15:427.
36 Alonso F, Domingos-Pereira S, Le Gal L, Derre L, Meda P, Jichlinski P, et al. Targeting endothelial connexin40 inhibits tumor growth by reducing angiogenesis and improving vessel perfusion. Oncotarget. 2016;7(12):14015–28.
37 Pfenniger A, Derouette JP, Verma V, Lin X, Foglia B, Coombs W, et al. Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30(4):827–34.
38 Wang WK, Chen MC, Leong HF, Kuo YL, Kuo CY, Lee CH. Connexin 43 suppresses tumor angogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1alpha. Int J Mol Sci. 2015;16(1):439–51.
39 Jiang JX, Penuela S. Connexin and pannexin channels in cancer. BMC cell biology. 2016;17(Suppl 1):12.
40 Lamprianou S, Gysemans C, Bou Saab J, Pontes H, Mathieu C, Meda P. Glibenclamide prevents diabetes in NOD mice, PLos One 2016, in press.
41 Lubkemeier I, Machura K, Kurtz L, Neubauer B, Dobrowolski R, Schweda F, et al. The connexin 40 A96S mutation causes renin-dependent hypertension. J Am Soc Nephrol. 2011;22(6):1031–40.
42 Firouzi M, Kok B, Spiering W, Busjahn A, Bezzina CR, Ruijter JM, et al. Polymorphisms in human connexin40 gene promoter are associated with increased risk of hypertension in men. J Hypertens. 2006;24(2):325–30.
43 van Dillen IJ, Mulder NH, Vaalburg W, de Vries EF, Hospers GA. Influence of the bystander effect on HSV-tk/GCV gene therapy. A review. Curr Gene Ther. 2002;2(3):307–22.
44 Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256(5063):1550–52.
45 Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2(1):21–5.
46 Hooper ML, Subak-Sharpe JH. Metabolic cooperation between cells. Int Rev Cytol. 1981;69:45–104.
47 Thuringer D, Jego G, Berthenet K, Hammann A, Solary E, Garrido C. Gap junction-mediated transfer of miR-145-5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget. 2016;7(19):28160-8.
48 Lemcke H, Steinhoff G, David R. Gap junctional shuttling of miRNA – A novel pathway of intercellular gene regulation and its prospects in clinical application. Cell Signal. 2015;27(12):2506–14.
49 Becker DL, Phillips AR, Duft BJ, Kim Y, Green CR. Translating connexin biology into therapeutics. Semin Cell Dev Biol. 2016;50:49–58.
50 Bavamian S, Pontes H, Cancela J, Charollais A, Startchik S, Van de Ville D, et al. The intercellular synchronization of Ca2+ oscillations evaluates Cx36-dependent coupling. PLoS One 2012;7(7):e41535.
51 Kahraman S, Okawa ER, Kulkarni RN. Is transforming stem cells to pancreatic beta cells still the holy grail for Type 2 diabetes? Curr Diab Rep. 2016;16(8):70.