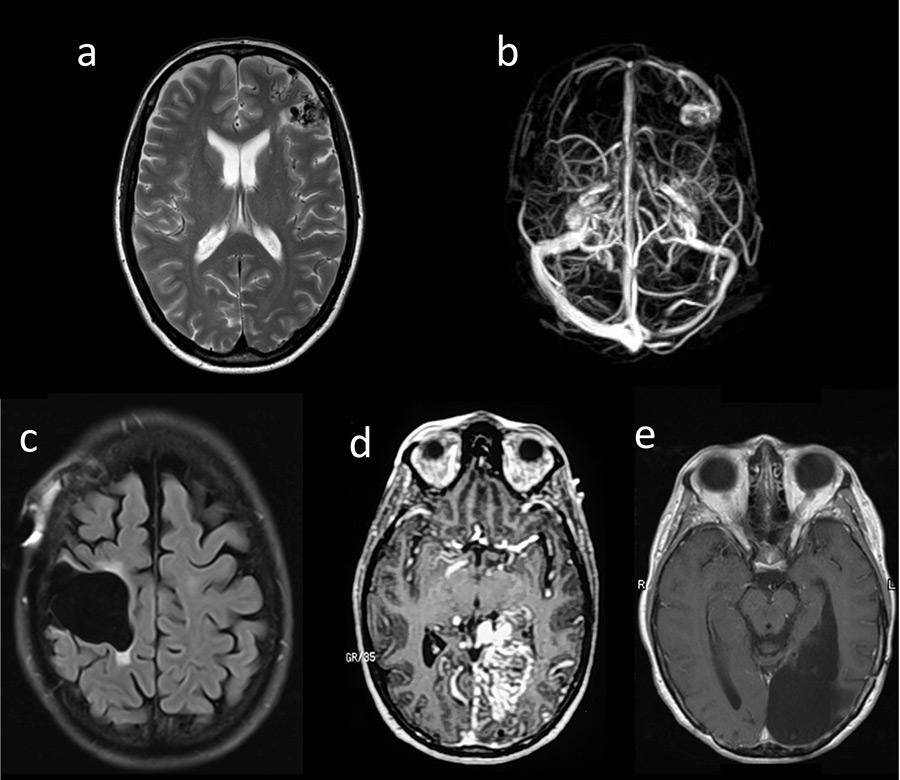
Figure 1
(a, b) Patient 1: T1-MRI (magnetic resonance imaging ) (a) and MRI-venogram (b); (c) Patient 2: MRI (FLAIR) after surgery; (d, e) Patient 3: MRI-angiography prior to surgery (d) and MRI (T1) after surgery (e).
DOI: https://doi.org/10.4414/smw.2016.14361
We reported previously that seven patients with cerebral arteriovenous malformations (AVM) for whom embolisation was utilised as the form of treatment developed amyotrophic lateral sclerosis (ALS) after a median latency of 14 years [1]. The patients were unusually young at ALS onset (median age 38 years) and had an AVM with perinidal angiogenesis and multiple embolisation sessions. We speculated that a reduction of the synthesis of vascular endothelial growth factor (VEGF) after AVM embolisation [2] may have been the underlying mechanism. Meanwhile, Katsavarou and colleagues reported two other young patients who developed ALS 11 and 14 years after their first cerebral AVM embolisation [1]. Here, we describe three novel cases of ALS which developed 13–34 years after treatment of cerebral AVM.
A 34-year-old woman developed a secondarily generalised epileptic seizure and was diagnosed with an AVM with perinidal angiogenesis in the left middle frontal gyrus. Two partial embolisations were performed: one shortly thereafter and another 19 years later (fig. 1a, b). At the age of 54, she developed bulbar onset ALS meeting the criteria for definitive ALS [3]. No ALS or AVM was reported in her family history. Sanger sequencing of the entire coding region and flanking intronic regions of the VEGFA gene (ENST00000372055, CCDS34457) in accordance with standard protocols revealed no variation.
Figure 1
(a, b) Patient 1: T1-MRI (magnetic resonance imaging ) (a) and MRI-venogram (b); (c) Patient 2: MRI (FLAIR) after surgery; (d, e) Patient 3: MRI-angiography prior to surgery (d) and MRI (T1) after surgery (e).
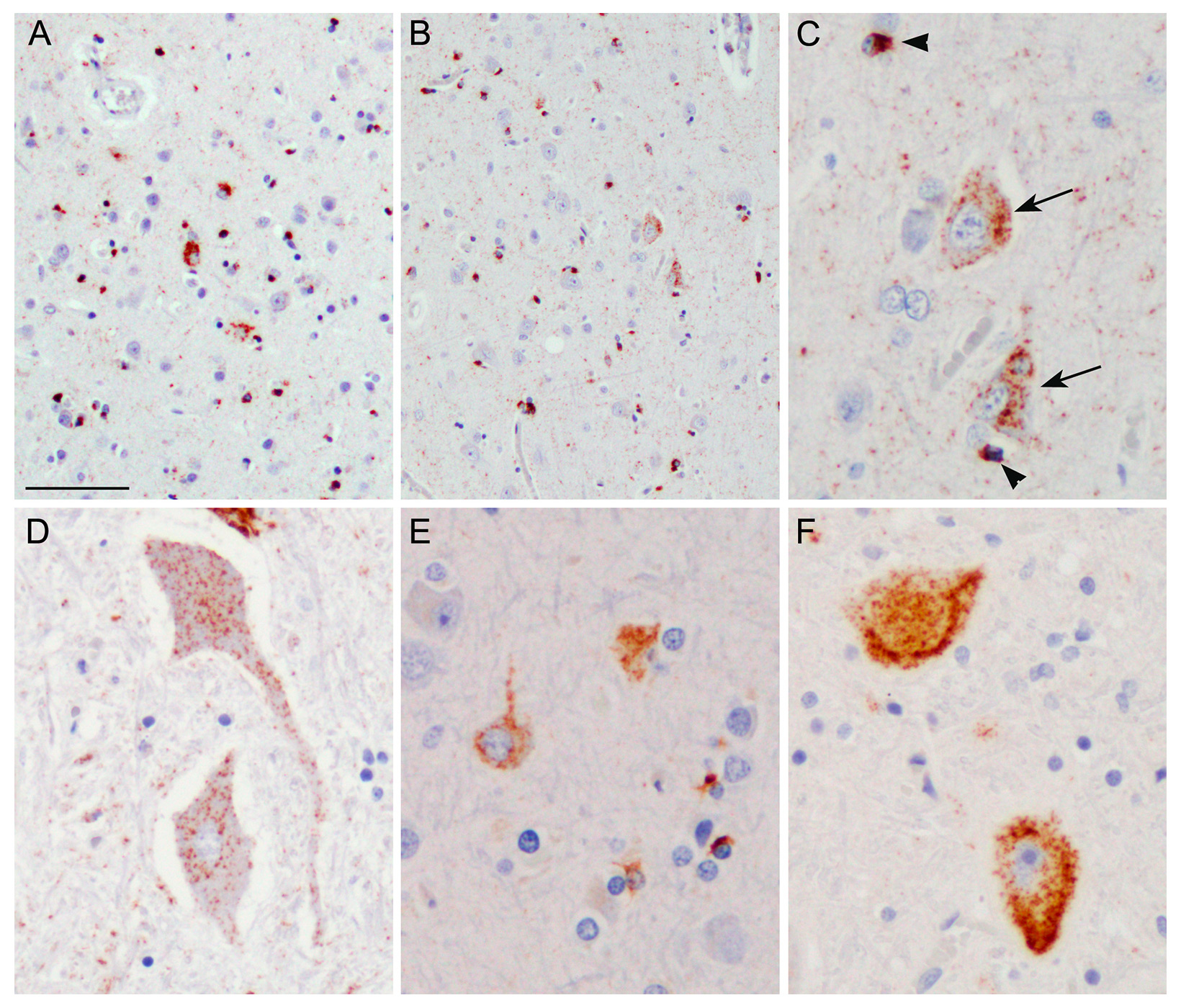
Figure 2
Immunohistochemistry with an antibody against phosphorylated TDP-43.
A–D: Case 2 showing classical amyotrophic lateral sclerosis (ALS) with TDP-43 pathology. A: Left precentral gyrus. B: Right precentral gyrus. C: Higher magnification of B demonstrating TDP-43 positive neuronal cytoplasmic inclusions (arrows) and glial cytoplasmic inclusions (arrowheads). D: Anterior horn of the spinal cord with neuronal cytoplasmic TDP-43 inclusions. E–F: For comparison, TDP-43 staining from the precentral gyrus (E) and spinal cord (F) from an ALS case without an arteriovenous malformation are shown. Scale bar: 80 µm (A, B); 30 µm (C, E), 40 µm (D, F).
A 37-year-old woman experienced headaches resulting in the diagnosis of an AVM of the precentral gyrus. The AVM was embolised in four sessions before being surgically removed (fig. 1c). Thirteen years later, at the age of 50, she developed ALS starting in her left arm, with fatal outcome after 3 years. Post-mortem examination of the central nervous system confirmed classical ALS pathology with neuronal and glial TAR-DNA binding protein (TDP)-43 inclusions predominantly in the upper and lower motor neurons (fig. 2). Notably, no obvious difference in the extent and severity of TDP-43 pathology was seen between the right and left precentral gyrus (fig. 2A, B). No immunoreactive inclusions were detected with antibodies against dipeptide repeat proteins (poly-GA), the highly characteristic feature of C9orf72 repeat expansion carriers or with antibodies against FUS (an RNA binding protein), thereby excluding relevant C9orf72 and FUS gene mutations. There was no mutation of the VEGFA gene.
A woman with headaches and a visual field deficit was found to have a left-sided medial occipital AVM, and at the ages 20 and 21 years, respectively, two embolisations with silastic spheres were done. She returned at the age of 36 with severe headaches and complete right homonymous hemianopia (fig. 1d). She then underwent three embolisation sessions over a 1-week period with a combination of cyanoacrylate, polyvinyl alcohol particles and platinum microcoils followed by complete surgical resection. At age 53, she began to develop right arm twitching, atrophy and weakness. A diagnosis of definite ALS was made. The patient refused genetic analyses.
We extend the existing descriptions of an association between cerebral AVM embolisation and deferred ALS with an onset ranging up to 34 years after the procedure and without correlation between the side of ALS limb onset and the location of the AVM in the brain. Therefore, the connection between AVMs and ALS cannot be explained solely by local, but rather by systemic factors. Interestingly, no ALS has so far been reported in AVM patients treated with surgery or radiotherapy alone or with complete embolisation. AVMs lead to increased local angiogenic activity associated with increased VEGF expression. This was apparent in the perinidal angiogenesis in all seven cases of AVM and ALS in the initial study [1]. Our results extend these numbers by two further cases (no information for patient 3). We conclude that embolisation of cerebral AVMs with perinidal angioneogenesis might induce mechanisms such as lowering of the VEGF level [2] and thereby might influence the risk of ALS development. In addition to AVM or embolisation procedures, cerebrovascular injury from a variety of causes has been suggested to be a risk factor for ALS “within the context of a more complex multiple-hit model of pathogenesis” by Turner et al. [5]. Thus, the mechanism underlying the association of AVM or AVM embolisation and ALS development remains speculative and might depend on specific influences on, for example, VEGF production or on less specific consequences from (vessel-associated?) brain injury.
1 Valavanis A, Schwarz U, Baumann CR, Weller M, Linnebank M. Amyotrophic lateral sclerosis after embolization of cerebral arterioveneous malformations. J Neurol. 2014;261:732–7.
2 Kim GH, Hahn DK, Kellner CP, Hickman ZL, Komotar RJ, et al. Plasma levels of vascular endothelial growth factor after treatment for cerebral arteriovenous malformations. Stroke. 2008;39:2274–9.
3 Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
4 Katsavarou O, Ntampos S, Sarmas I, Triantafyllou N, Giannopoulos S, Kyritsis AP. Embolization treatment of cerebral arteriovenous malformations and amyotrophic lateral sclerosis. Neurol Sci. 2015;36:483–4.
5 Turner MR, Goldacre R, Talbot K, Goldacre M. Cerebrovascular injury as a risk factor for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:244–6.
Authors’ contribution: M. Linnebank: description of patient 1, draft of manuscript. C. McDougall: description of patient 2. S. Krueger and S. Biskup: genetic analyses. M. Neumann: neuropathology. M. Weller and A. Valavanis: contributed to design of the study and finaliziton of the manuscript. J. Prudlo: description of patient 2, organization of analyses, contribution to finalize the manuscript.
Dislosure statement:M. Linnebank received funding, grants or honoraria from Almirall, Bayer, Biogen, Genzyme, Merck, Novartis, Teva. C. McDougall is consultant to Medtronic (eV3). S. Krueger reported no disclosures. S. Biskup reported no disclosures. M. Neumann reported no disclosures. M. Weller has received research grants from Acceleron, Actelion, Alpinia Institute, Bayer, Isarna, MSD, Merck & Co, Novocure, PIQUR and Roche and honoraria for lectures or advisory board participation or consulting from Celldex, Immunocellular Therapeutics, Isarna, Magforce, MSD, Merck & Co, Northwest Biotherapeutics, Novocure, Pfizer, Roche and Teva. A. Valavanis reported no disclosures. J. Prudlo reported no disclosures.