Astrocyte power fuels neurons during stroke
DOI: https://doi.org/10.4414/smw.2016.14374
Stefano
Pluchino, Luca
Peruzzotti-Jametti, Christian
Frezza
This Viewpoint refers to “Transfer of mitochondria from astrocytes to neurons after stroke”, by Hayakawa et al. (Nature. 2016;535(7613):551–5. doi: 10.1038/nature18928).
In a paper published in Nature July 2016, a group of investigators coordinated by the Harvard neuroscientist Eng H. Lo set out to investigated the role of the intercellular transfer of mitochondria from astrocytes to neurons following ischemic stroke [1].
Mitochondria are intracellular organelles involved in several cellular processes, including oxidative phosphorylation, metabolism of amino acids, lipids, and steroids, and regulating multiple intracellular signalling cascades, such as cell-cycle, antiviral responses and cell death [2]. Mitochondria are also emerging players in intercellular communications. For instance, the generation of mitochondria-derived vesicles (MDVs) – a subset of small vesicular carriers that transport mitochondrial proteins and lipids to other intracellular organelles – drives the presentation of mitochondrial antigens on MHC class I molecules in immune cells, thus eliciting a local inflammatory response [3]. Similarly, the release of mitochondrial DNA or proteins in the extracellular space is highly immunogenic and represents a danger signal that activates leucocytes [4].
Intact mitochondria can also be exchanged between cells, with important biological and functional implications. Donor cells transfer mitochondria to target cells via gap junctions [5], or via a Rho GTPase-dependent transport through nanometer wide tubular extensions called tunnelling nanotubes that connect adjacent cells [6]. Another described mechanism of intercellular mitochondrial transfer is via the release of free organelles or their inclusion in extracellular membrane vesicles (EVs) [7]. The horizontal transfer of mitochondria is able to rescue impaired aerobic respiration, inhibit apoptosis, increase cell survival and proliferation, induce chemoresistance, and enhance phagocytic activity. Intercellular mitochondria exchange is multidirectional and associated with target cell-specific outcomes. For example, mitochondria derived from mesenchymal stem cells (MSC) promote partial de-differentiation and protection against ischaemia/reperfusion injury in target cardiomyocytes, and cardiomyocyte-derived mitochondria induce the expression of cardio-specific proteins in target MSCs.
Despite all this previously published evidence, the mechanisms regulating the intercellular transfer of mitochondria, its environmental triggers and its relevance in re-establishing tissue homeostasis after injury are yet to be fully understood.
Using a combination of electron microscopy, fluorescence-activated cell sorting (FACS) and tunable resistive pulse sensing (TRPS) analysis, Hayakawa and colleagues showed that the conditioned medium from astrocytes contains structurally intact mitochondrial particles capable of producing ATP and consuming O2, while concurrently conserving a functional membrane potential [1].
To investigate whether astrocyte-derived mitochondria play a role in re-establishing tissue homeostasis after an ischaemic injury, the authors used the in vitro oxygen/glucose deprivation (OGD) model of hypoxia, which impairs mitochondrial function in neurons. Under these experimental conditions, astrocyte-derived mitochondria were taken up by neurons subjected to OGD, increasing their viability and aerobic respiration, and dendrite length (fig. 1). This rescue effect was abolished when the mitochondrial particles were filtered from the conditioned medium, made dysfunctional via inhibition of the mitochondrial form of aconitase (which plays a key role in carbohydrate metabolism), or when mitochondria-free ATP-liposomes were delivered to OGD neurons.
To assess the mechanisms of mitochondria secretion by astrocytes the authors focused on the expression of CD38. CD38 catalyses the synthesis of cyclic ADP-ribose (cADPR) in mitochondrial membranes, increases in astrocytes in response to glutamate release from neurons [1] and has been implicated in neuroglial crosstalk. Of note, over-expression of CD38 in astrocytes led to a significant increase in cyclase activity, extracellular ATP and O2 consumption in secreted mitochondrial particles [1]. Importantly, the rescue effect of astrocyte-derived mitochondria on OGD neurons was also impaired when astrocytes were pre-treated with a small interfering RNA (siRNA) to silence Cd38 [1].
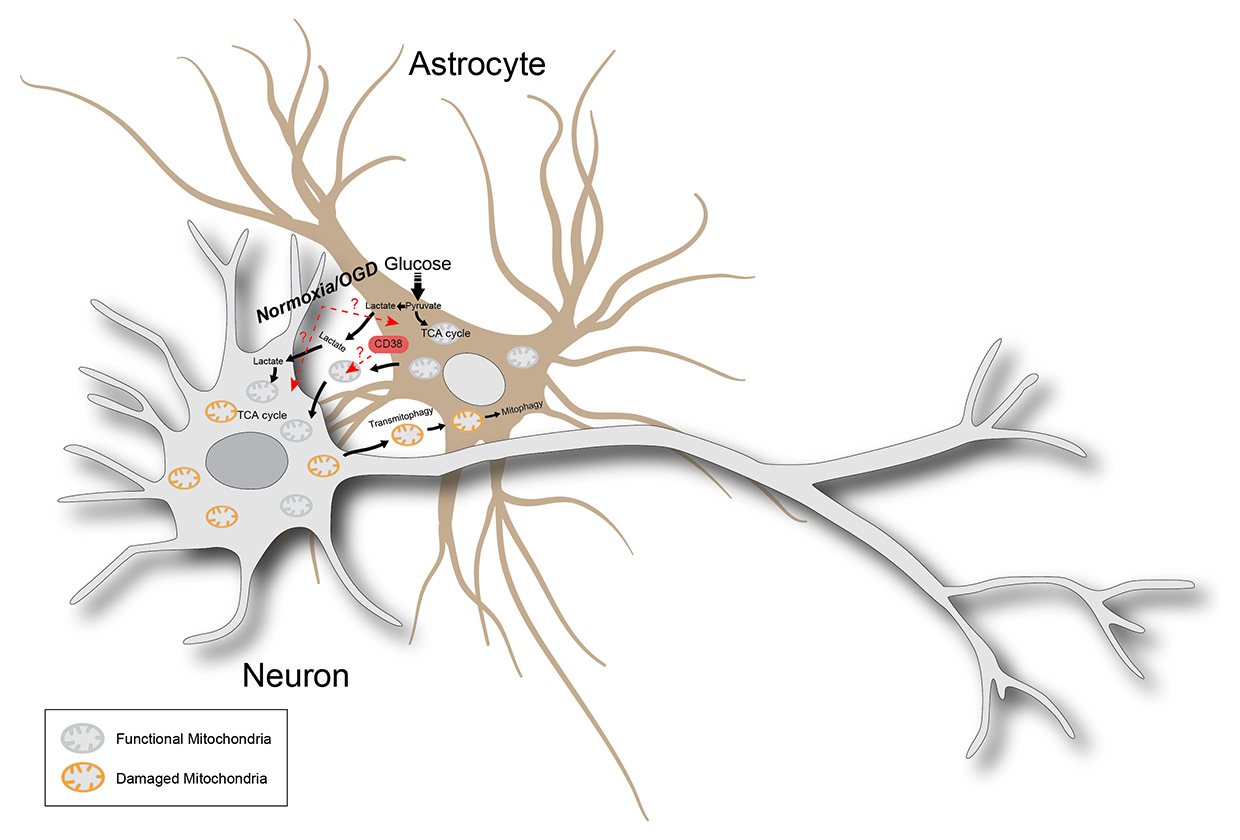
Figure 1
Metabolic-coupling between astrocytes and neurons following hypoxia. Astrocytes, which are highly glycolytic cells, are metabolically coupled with neurons in several ways. First, they can generate L-lactate from glucose and exchange it with neurons as a substrate for energy production (lactate shuttle). Second, they can transfer functional mitochondria (both via direct cell-to-cell contact and/or extracellular vesicles) to hypoxic neurons, thereby increasing aerobic respiration. Finally, damaged/dysfunctional mitochondria can be transferred from neurons to astrocytes to support their degradation via transmitophagy. Red dashed arrows with question marks indicate the main mechanistic questions left open following the breakthrough work by Hayakawa and colleagues: the role of hypoxia (vs normoxia) in regulating mitochondria release and transfer; the ability of hypoxic astrocytes to release functional mitochondria, and the role of CD38 in orchestrating the release of mitochondria.
Some of the most provocative data from this paper arise from the in vivo analysis of mitochondrial transfer. Injecting FACS-sorted astrocyte-derived mitochondrial particles into the cortex of mice subjected to middle cerebral artery occlusion (MCAO), as an animal model for brain stroke, led to mitochondria uptake by neighbouring neurons within 24 hours after injection [1]. Furthermore, by subjecting mutant mice in which astrocytes were fluorescently labelled (FVB/N-Tg (GFAPGFP)14Mes/J) to MCAO, they found that neurons from the ischaemic peri-infarct cortex showed an uptake of fluorescent (astroglial) particles and increased cell survival, which suggested an effective transfer of mitochondria between these two cell types in vivo. Finally, to further investigate the CD38-cADPR-calcium axis in astrocytes and their release of mitochondria in vivo, the authors injected control siRNA or Cd38 siRNA into the lateral ventricles of MCAO mice 5 days after stroke. As early as 48 hours after Cd38 siRNA delivery, significant attenuation of peri-infarct immunostaining of the surrogate marker of neuroplasticity GAP43, as well as worsened neurological outcomes, were observed [1]. Furthermore, Cd38 siRNA delivery reduced astroglial and neuronal mitochondria release, as well as O2 consumption in mitochondrial particles isolated from cerebrospinal fluid [1]. These finding suggest for the first time a real functional relevance of mitochondria transfer between astrocytes and neurons in vivo.
While providing an extremely novel and exciting perspective of mitochondria as extracellular signalling organelles, this work lacks some key controls and additional conditions that could have further supported the authors’ conclusions.
First, co-culture of astrocytes and neurons under normoxia was not performed, thus raising the key questions of whether mitochondria transfer could also occur under physiological conditions and to what extent hypoxia plays an added role. Second, exposure to OGD in vitro was limited to neurons only. Therefore, it is not known if hypoxic astrocytes – which are commonly found in the peri-ischaemic area – would still release functional mitochondria.
Also, the role played by CD38 in orchestrating the release of mitochondria is unclear. Indeed, CD38 being a protein involved in many cellular processes, its silencing could compromise astrocyte function, and the impairment of mitochondrial release could be a secondary (off-target) effect. Furthermore, the intracerebroventricular injection of naked Cd38 siRNA would affect various target cells, raising questions about the specificity and efficacy of the treatment. Finally, the behavioural studies on MCAO mice were limited to 7 days after Cd38 siRNA injection. This is an extremely short time-window for fully appreciating the functional relevance of the inhibition of mitochondrial release by astrocytes in the stroke brain in vivo.
In conclusion, whilst this work provides very compelling evidence that astrocytes transfer healthy mitochondria to damaged neurons during stroke, more work is required to understand the molecular mechanisms behind this transfer, and to fully assess its functional relevance in vivo.
Acknowledgements
The authors were supported by the European Research Council (ERC) under the ERC-2010-StG Grant agreement n° 260511-SEM_SEM (S.P.), by a research training fellowship from the Wellcome Trust [RRZA/057 RG79423 to L.P.J] and core support grant from the Wellcome Trust and MRC to the Wellcome Trust – Medical Research Council Cambridge Stem Cell Institute.
References
1 Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535(7613):551–5. doi:http://dx.doi.org/10.1038/nature18928 http://dx.doi.org/10.1038/nature18928 . http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16860735&dopt=Abstract
2 McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16(14):R551–60. doi:http://dx.doi.org/10.1016/j.cub.2006.06.054 http://dx.doi.org/10.1016/j.cub.2006.06.054 . http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16860735&dopt=Abstract
3 Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 2016;166(2):314–27. doi:http://dx.doi.org/10.1016/j.cell.2016.05.039 http://dx.doi.org/10.1016/j.cell.2016.05.039 . http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=27345367&dopt=Abstract
4 Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124(14):2173–83. doi:http://dx.doi.org/10.1182/blood-2014-05-573543 http://dx.doi.org/10.1182/blood-2014-05-573543 . PubMed
5 Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–65. doi:http://dx.doi.org/10.1038/nm.2736 http://dx.doi.org/10.1038/nm.2736 . http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22504485&dopt=Abstract
6 Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303(5660):1007–10. doi:http://dx.doi.org/10.1126/science.1093133 http://dx.doi.org/10.1126/science.1093133 . PubMed
7 Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun. 2015;6:8472. doi:http://dx.doi.org/10.1038/ncomms9472. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=26442449&dopt=Abstract