Patent foramen ovale: a novel cardiovascular risk factor in patients with sleep disordered breathing and high altitude dwellers?
DOI: https://doi.org/10.4414/smw.2016.14371
Emrush
Rexhai, Urs
Scherrer, Stefano F
Rimoldi
Summary
Diseases associated with chronic hypoxaemia are a leading cause of morbidity and mortality in Western countries. Epidemiological data indicate that cardiovascular diseases contribute substantially to this problem, but the underlying mechanisms are incompletely understood. Sleep disordered breathing and high altitude exposure are frequent conditions associated with hypoxaemia. Recent evidence suggests that in these conditions the concomitant presence of a patent foramen ovale plays an important pathogenic role. For example, in patients with obstructive sleep apnoea the presence of a patent foramen ovale is associated with more severe sleep disordered breathing, nocturnal oxygen desaturation, generalised endothelial dysfunction and arterial hypertension. After patent foramen ovale closure, both sleep disordered breathing and cardiovascular phenotype improve, suggesting the existence of a possible causal link.
During short-term high altitude exposure, the presence of a patent foramen ovale, by aggravating altitude-induced hypoxaemia, facilitates exaggerated pulmonary hypertension. Interestingly, there is increasing evidence showing that in high-altitude dwellers a patent foramen ovale also alters the cardiovascular phenotype.
In this article we will summarise recent evidence demonstrating how a patent foramen ovale alters the cardiovascular phenotype and increases cardiovascular risk in patients with sleep disordered breathing and high-altitude dwellers.
Introduction
Diseases associated with chronic hypoxaemia are a leading cause of morbidity and mortality in Western countries and epidemiological data suggest that cardiovascular diseases substantially contribute to this problem, but the underlying mechanisms are poorly understood. Sleep disordered breathing (SDB, with a reported prevalence of 5–49% in the general population) [1, 2] is an important example illustrating this problem, since there is abundant evidence that it is associated with an increased prevalence of arterial hypertension, stroke, myocardial ischaemia, heart failure, arrhythmia and pulmonary hypertension [3, 4]. Recent data suggest that in patients with SDB, the concomitant presence of a patent foramen ovale (PFO) may contribute to increased cardiovascular risk.
High-altitude-induced hypoxaemia is another important example of this problem, with >120 million persons worldwide who live at high altitude and are chronically exposed to hypoxia. Studies of high-altitude populations, and in particular of maladapted subgroups, provide important insight into underlying mechanisms involved in the pathogenesis of hypoxaemia-related disease in general. Recent experimental evidence suggests that at high altitude, the presence of patent PFO, by aggravating altitude-induced hypoxemia, may contribute to cardiovascular morbidity in these populations. Here, we will review recent evidence demonstrating how PFO alters the cardiovascular phenotype in patients with SDB and high-altitude dwellers and then show how its closure may represent a possibility to reduce cardiovascular risk in these patients.
Sleep disordered breathing, definition and epidemiology
The term “sleep disordered breathing” (SDB) is used to describe four distinct pathological patterns of breathing: obstructive sleep apnoea (OSA), central sleep apnoea (CSA) sleep-related hypoventilation disorders and sleep-related hypoxaemia disorder. In this review, we focus on the first two disorders. OSA is a very frequent condition characterised by intermittent upper airway obstruction causing a reduction (hypopnoea) or cessation (apnoea) of inspiratory airflow leading to intermittent hypoxaemia. During apnoea/hypopnoea respiratory efforts are present in OSA, and the episode generally ends in arousal. CSA is much less frequent than OSA, since it accounts for <1% of all cases of SDB in the general population [5]. In CSA, by definition, there is no respiratory effort during hypopnoea or apnoea, and CSA is generally associated with chronic heart failure or found during high-altitude exposure. The prevalence of SDB has generally been estimated to be between 5 and 15% [2]. However, recent data suggest that its prevalence in the general population may be much higher (23.4% in women and 49.7% in men), a fact related to the increased sensitivity of current recording techniques and scoring criteria [1].
The main symptoms of OSA are snoring, excessive daytime sleepiness, morning headaches, daytime fatigue and concentration difficulties. Clinical findings often associated with OSA include obesity, enlarged neck circumference, retrognathia and nasal obstruction [4]. Diagnosis of OSA requires sleep recording demonstrating >5 episodes of apnoea or hypopnoea per hour of sleep, and its severity is assessed with the apnoea/hypopnoea index (AHI: mild 3–15; moderate 15–30; severe >30 events/h) [6, 7].
Sleep disordered breathing as a cardiovascular risk factor
Systemic circulation
SDB-induced intermittent oxygen desaturation, decreased intrathoracic pressure [8] and arousals have been shown to trigger sympathetic activation [9], increase oxidative stress [10, 11], decrease plasma nitric oxide bioavailability [12] and induce inflammation [13, 14], alterations that all may contribute to endothelial dysfunction [15, 16]. In line with this concept, OSA patients without any traditional cardiovascular risk factor have been shown to display systemic endothelial dysfunction [17]. Endothelial dysfunction is the first step in the development of arteriosclerosis. In line with this concept, OSA patients show signs of arteriosclerosis, including increased coronary artery calcium, arterial stiffness and carotid intima-media thickness [18–20], that are thought to contribute to increased cardiovascular mortality and morbidity in these patients.

Figure 1
Pathophysiological relationship between obstructive sleep apnoea (OSA) and cardiovascular diseases. OSA is characterised by intermittent hypoxaemia, arousals and intrathoracic pressure swings inducing sympathetic activation, exaggerated oxidative stress and chronic inflammation leading to systemic and pulmonary endothelial dysfunction, the first step in the development of generalised arteriosclerosis and pulmonary hypertension.
In OSA desaturation/reoxygenation sequences follow a typical pattern and are thought to trigger sympathetic activation, oxidative stress and inflammation, which, in turn, cause cardiovascular disease and increase cardiovascular risk (fig. 1). Several cardiovascular diseases are associated with OSA (fig. 1). Among these diseases, arterial hypertensionis the most frequent. Notably, OSA is the most common cause of secondary hypertension and a frequent cause of drug-resistant hypertension [21, 22], as suggested by a prevalence of OSA >30% in drug-resistant hypertension compared with a prevalence of 5 to 15% in the general population of patients with hypertension [23, 24]. Accordingly, patients with hypertension, in particular those with drug-resistant hypertension, should be routinely evaluated for OSA [25]. Arterial hypertension is the major risk factor for stroke and contributes to coronary artery disease. In line with this concept, OSA has been reported to be associated with an increased prevalence of stroke and myocardial ischaemia [26–28]. Finally, SDB has been reported to be associated with systolic heart failure [29, 30] and arrhythmias, such as nocturnal bradycardia, atrioventricular block, atrial fibrillation, ventricular ectopy and ventricular tachycardia [31, 32].
Pulmonary circulation
Hypoxaemia induced by sleep disordered breathing causes haemodynamic and structural alterations of the pulmonary circulation. Hypoxaemia-induced pulmonary vasoconstriction increases precapillary pulmonary artery pressure [33]. Negative intrathoracic pressure during apnoea increases right ventricular preload and pulmonary blood flow by increasing venous return, [34], and increases pulmonary venous pressure by reducing left ventricular compliance (via an increase in left ventricular transmural pressure) [35]. Moreover, as detailed above, intermittent hypoxaemia promotes inflammation, sympathetic activation and oxidative stress, which, in turn, cause pulmonary endothelial dysfunction [36, 37] and vascular remodelling [38], which ultimately may lead to persistent pulmonary hypertension [3].
Patent foramen ovale, definition and epidemiology
Patent foramen ovale (PFO) is an embryological remnant characterised by insufficient postnatal adhesion of the cardiac atrial septum primum and secundum. The gold standard diagnostic test is transoesophageal echocardiography. In the general population, the prevalence of PFO is estimated to be approximately 25% [39]. PFO has been suggested to play a pathogenic role in various conditions, such as decompression illness, migraine, transient global amnesia, platypnoea orthodeoxia, high-altitude pulmonary oedema, cerebrovascular ischaemia, paradoxical emboli during air travel [40] and OSA [41–45]. The prevalence of PFO has been found to be increased in patients with OSA; Shanoudy et al. reported a prevalence of PFO of 67% (vs 17% in controls) [46] whereas Beelke et al. found a prevalence of 27% (vs 15% in controls) [47].
PFO and cardiovascular function in sleep disordered breathing
SDB induces vascular dysfunction, but the underlying mechanisms are incompletely understood [48]. Recent evidence suggests that the presence of a PFO may contribute to this problem [16, 49, 50]. In a prospective open-label interventional clinical trial we searched for PFO with transoesophageal echocardiography in 40 consecutive patients with newly diagnosed OSA [16, 50]. PFO was found in 14 OSA patients (prevalence 33%), whereas it was not detectable in the remaining 26 patients. OSA patients with PFO underwent closure, whereas patients without this problem served as controls. Conventional therapy (i.e. continuous positive airway pressure therapy) was postponed in both groups, and cardiovascular assessment was performed at baseline and at 3-month follow-up; endothelial function was assessed by use of flow mediated dilation of the brachial artery, arterial blood pressure by 24-hour ambulatory blood pressure measurement and carotid stiffness by measuring parameters of local elasticity; cardiac function and pulmonary artery pressure were evaluated with transthoracic echocardiography. At baseline, endothelial function, carotid stiffness, arterial blood pressure and cardiac function were similar in patients with and without PFO, whereas the right ventricular to right atrial pressure gradient (RV-RA gradient) was roughly 20% higher in OSA patients with PFO than in those without this problem (fig. 2, left panel) [16]. At follow-up, PFO closure significantly improved the apnoea-hypopnoea index (ΔAHI –7.9 ± 10.4 vs +4.7 ± 13.1 events/h, p = 0.0009, PFO closure versus control) and the oxygen desaturation index (ΔODI –7.6 ± 16.6 vs +7.6 ± 17.0 events/h, p = 0.01). The number of patients with severe OSA decreased significantly after PFO closure (from 79% to 21%, p = 0.007). Figure 2 shows that these favourable effects of PFO closure on nocturnal breathing and oxygen saturation translated into improved pulmonary and systemic cardiovascular function, as evidenced by a 4-mm Hg decrease of the RV-RA pressure gradient (fig. 2, left panel) [47], a significant decrease of nocturnal systolic (–7 mm Hg) and diastolic (–3 mm Hg) blood pressure (fig. 2, right panel) together with the restoration of normal circadian blood pressure variability and improved left ventricular diastolic function [16].
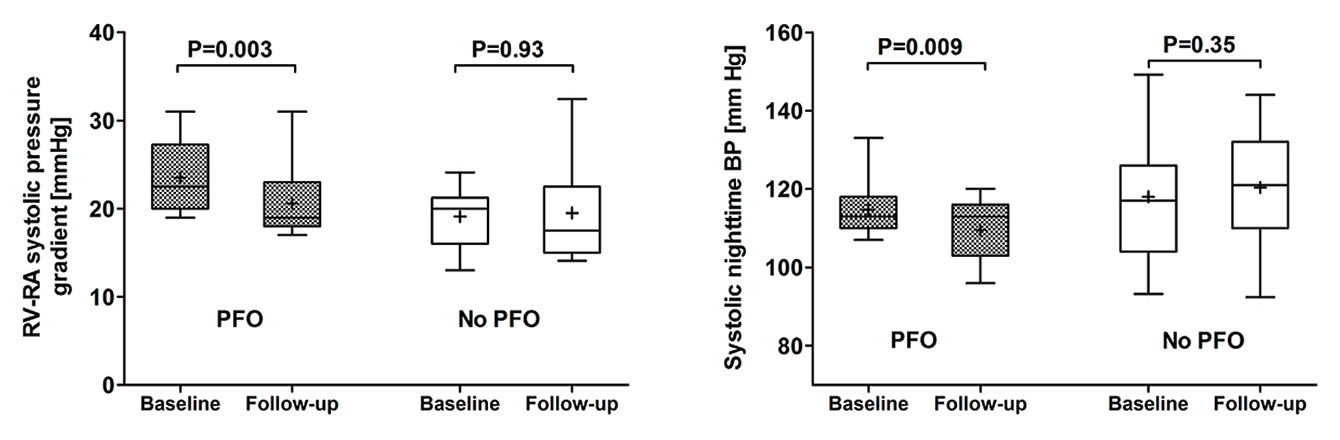
Figure 2
Pulmonary artery pressure and systolic night-time blood pressure in obstructive sleep apnoea (OSA) patients with or without patent foramen ovale (PFO). Effects of PFO closure. Pulmonary artery pressure (right-ventricular to right-atrial [RV-RA] systolic pressure gradient) is significantly higher in OSA patients with PFO than in OSA patients without PFO (no PFO) at baseline and significantly decreases after PFO closure at follow-up (left panel). Systolic night-time blood pressure (BP) significantly decreases after PFO closure at follow-up (right panel).
In summary, these data provide the first direct experimental evidence that in patients with OSA, a PFO contributes to altered systemic and pulmonary blood pressure regulation, and altered left ventricular diastolic function. These detrimental effects appear to be related, at least in part, to a PFO-induced aggravation of SDB and nocturnal oxygen desaturation. PFO closure interrupts this vicious cycle. The significant decrease of average night-time heart rate could suggest that the favourable effects of PFO closure were related, at least in part, to a reduction of sympathetic nerve activity [16].
PFO and cardiovascular function during acute, short-term and chronic high-altitude exposure
There is evidence that exaggerated hypoxaemia contributes to vascular dysfunction during high-altitude exposure [51, 52], but the effects of the presence of a PFO on cardiovascular regulation has been little investigated. In mountaineers susceptible to high-altitude pulmonary oedema (HAPE), patent foramen ovale was reported to be roughly four times more frequent than in participants resistant to this condition. During acute exposure to high altitude (4559 m), HAPE-susceptible participants with a large PFO had more severe hypoxaemia and higher pulmonary artery pressure than those with a small or no PFO. These findings suggest that at high altitude, a large PFO may contribute to exaggerated arterial hypoxaemia and pulmonary hypertension and, in turn, facilitate HAPE [41]. Moreover, there is direct experimental evidence in humans showing that, in addition to aggravating hypoxaemia by right-to-left shunting, PFO also aggravates hypoxaemia by impairing pulmonary gas exchange efficiency [53]. Taken together, these studies suggest that the presence of PFO, by aggravating altitude-induced hypoxaemia, facilitates exaggerated pulmonary hypertension during short-term high-altitude exposure.
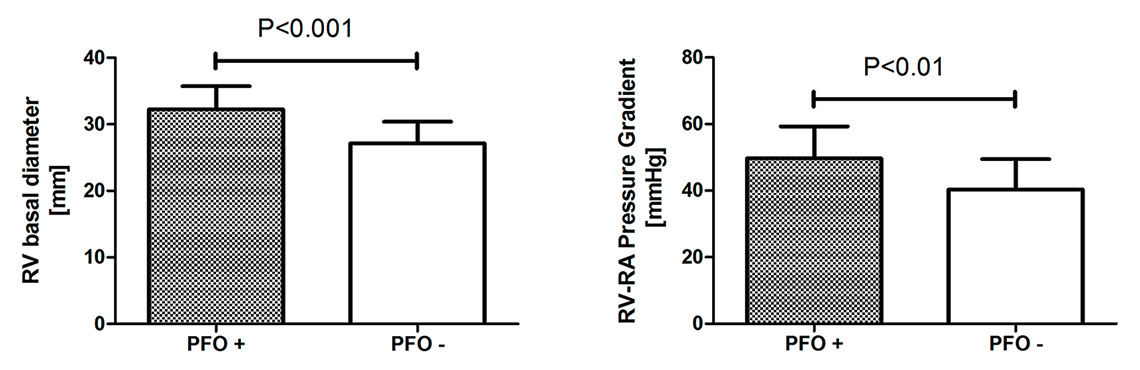
Figure 3
Right ventricular (RV) basal diameter and right-ventricular to right-atrial (RV-RA) systolic pressure gradient in high-altitude dwellers with (+) or without (-) patent foramen ovale (PFO). High-altitude dwellers with PFO display RV enlargement (left panel) and higher pulmonary artery pressure (right panel) compared with high-altitude dwellers without PFO.
Interestingly, there is increasing evidence to suggest that a PFO also alters the cardiovascular phenotype during long-term high-altitude exposure. For example, in a recent study, we searched for PFO with transoesophageal echocardiography in healthy high-altitude dwellers and patients with chronic mountain sickness who were born and had been permanently living between 3600 and 4000 m [54]. The prevalence of PFO in these high-altitude dwellers was 32% (18/57 participants) and thus did not differ from the prevalence reported at low altitude. Importantly, however, the presence of PFO in high altitude dwellers was associated with right ventricular enlargement at rest (p <0.01, fig. 3, left panel), and exaggerated pulmonary hypertension (fig. 3, right panel) and right ventricular dysfunction during mild exercise [54]. In line with these findings, in a subsequent study we found that in high-altitude dwellers suffering from SDB, pulmonary artery pressure was significantly higher in patients with PFO than in those without it [49]. Finally, preliminary data (Rexhaj et al., not published) suggest that in high-altitude dwellers, the presence of a PFO is associated with systemic vascular dysfunction.
In summary, these data suggest that in high-altitude dwellers the presence of a PFO is associated with systemic and pulmonary vascular dysfunction. In the pulmonary circulation, this vascular dysfunction leads to increased pulmonary artery pressure and right heart dysfunction. Further studies need to examine whether in high-altitude dwellers systemic vascular dysfunction associated with a PFO increases the prevalence of cardiovascular endpoints in the systemic circulation [16, 49].
Effects of a PFO on arterial oxygenation in patients with sleep disordered breathing at low and high altitude
In patients with OSA at low altitude, the presence of PFO is associated with a marked increase of both the frequency and severity of nocturnal oxygen desaturation [16, 53, 56]. In high-altitude dwellers (3600 m), the presence of PFO is associated with more severe SDB and nocturnal hypoxaemia [49]. Taken together, these data suggest that in SDB, the presence of a PFO puts these patients at risk for more severe nocturnal hypoxaemia. This could be related, at least in part, to right-to-left shunting across a PFO, as evidenced by invasive haemodynamic measurements in healthy humans, demonstrating that during the onset of simulated OSA, in response to the steep decline in intrathoracic pressure, right atrial pressure exceeds left atrial pressure (fig. 4) [57]. Accordingly, Schäfer et al. found that in OSA patients, during apnoea the pulmonary artery pressure increased by about 10 mm Hg; the main contributors to this response were hypoxaemia, mechanical factors and an increase in negative intrathoracic pressure [58].
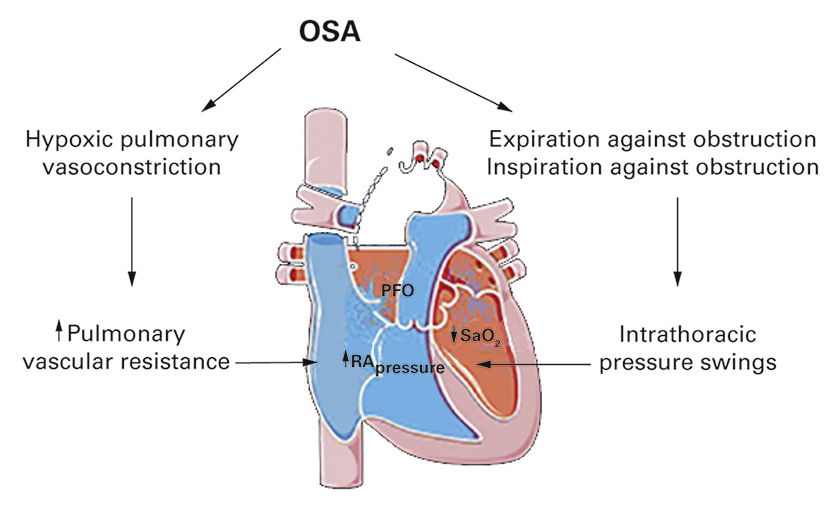
Figure 4
Pathophysiological link between obstructive sleep apnoea (OSA) and patent foramen ovale (PFO). OSA is characterised by intermittent hypoxaemia that causes hypoxic pulmonary vasoconstriction and increases pulmonary vascular resistance. During apnoea/hypopnoea the patient expires or inspires against the collapsed upper airways producing intrathoracic pressure swings. Augmented pulmonary vascular resistance and intrathoracic pressure swings increase right atrial (RA) pressure over left atrial pressure. The presence of a PFO allows right-to-left shunting which further exacerbates hypoxaemia, thereby completing a vicious circle.
Is there a direct causal link between sleep disordered breathing and PFO?
One possibility could be that SDB induces/reopens a PFO, i.e. SDB-induced exaggerated nocturnal hypoxaemia during apnoea/hypopnoea triggers pulmonary vasoconstriction, elevates pulmonary artery pressure and increases right atrial pressure above left atrial pressure, which, in turn, promotes PFO. The increased prevalence of PFO in patients with chronic obstructive pulmonary disease could be consistent with this hypothesis [59]. Alternatively, and more probably, a PFO appears to have an exacerbating effect on phasic breathing in OSA. The crucial element in this context is short episodes of atrial right-to-left shunt with ensuing episodes of hypoxaemia, which aggravate the already disturbed central breathing regulation in OSA. The initiating incident in the cascade of phasic breathing in OSA is the first apnoea in the context of muscular relaxation during rapid eye movement sleep. Rising arterial pressure of carbon dioxide (PCO2) in the context of this apnoea induces breathing efforts against the closed glottis, which briefly elevate right atrial pressure above left atrial pressure and leads to shunting of deoxygenated blood to the systemic side in the presence of a PFO. Hypoxaemia impairs endothelial function in the pulmonary and in the systemic circulation, and it acts as a central respiratory stimulant that influences PCO2-steered respiratory regulation differently from other stimulants. Hypoxaemia lowers the eupnoeic PCO2 level in the context of hyperventilation without concomitant reduction in the apnoeic PCO2 threshold, thus destabilising the system and rendering it more prone to ensuing apnoeic phases [50, 60]. In line with this hypothesis, circumstantial evidence suggested that PFO closure may improve symptoms and SDB in patients with OSA [61–63]. This concept is markedly strengthened by a recent prospective study showing that PFO closure in OSA patients improved SDB and nocturnal oxygenation, and significantly decreased the number of patients suffering from severe OSA from 79 to 21% [16, 50].
These data show a pathophysiological interaction between OSA and PFO, where right-to-left shunting across a PFO aggravates SDB and nocturnal oxygen desaturation, leading to more severe cardiovascular alterations, as evidenced by the improvement of both SDB and cardiovascular phenotype after PFO closure in patients with OSA. These data strongly suggest that PFO represents a novel cardiovascular risk factor in patients with sleep disordered breathing (and high-altitude dwellers). If confirmed in larger prospective randomised studies, PFO closure in SBD may emerge as a valid alternative to conventional therapy for decreasing the cardiovascular risk (and, by attenuating nocturnal oxygen desaturation, possibly also for attenuating clinical symptoms), particularly in patients who are intolerant of nocturnal ventilatory and/or dental device treatment.
References
1 Heinzer R, Vat S, Marques-Vidal P, Marti-Soler H, Andries D, Tobback N, et al. Prevalence of sleep-disordered breathing in the general population: the HypnoLaus study. Lancet Respir Med. 2015;3(4):310–8. doi:http://dx.doi.org/10.1016/S2213-2600(15)00043-0.
2 Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328(17):1230–5. doi:http://dx.doi.org/10.1056/NEJM199304293281704.
3 Kessler R, Chaouat A, Weitzenblum E, Oswald M, Ehrhart M, Apprill M, et al. Pulmonary hypertension in the obstructive sleep apnoea syndrome: prevalence, causes and therapeutic consequences. Eur Respir J. 1996;9(4):787–94. doi:http://dx.doi.org/10.1183/09031936.96.09040787.
4 Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thorac Dis. 2015;7(9):E298–310.
5 Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am J Respir Crit Care Med. 1998;157(1):144–8. doi:http://dx.doi.org/10.1164/ajrccm.157.1.9706079.
6 Berry RB, Budhiraja R, Gottlieb DJ, Gozal D, Iber C, Kapur VK, et al.; American Academy of Sleep Medicine; Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. J Clin Sleep Med. 2012;8(5):597–619.
7 Iber C, Ancoli-Israel S, Chesson ALJ, et al. The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. 1st ed Westchester, IL: American Academy of Sleep Medicine 2007.
8 Tkacova R, Rankin F, Fitzgerald FS, Floras JS, Bradley TD. Effects of continuous positive airway pressure on obstructive sleep apnea and left ventricular afterload in patients with heart failure. Circulation. 1998;98(21):2269–75. doi:http://dx.doi.org/10.1161/01.CIR.98.21.2269.
9 Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96(4):1897–904. doi:http://dx.doi.org/10.1172/JCI118235.
10 Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest. 2003;124(4):1386–92. doi:http://dx.doi.org/10.1378/chest.124.4.1386.
11 Yamauchi M, Nakano H, Maekawa J, Okamoto Y, Ohnishi Y, Suzuki T, et al. Oxidative stress in obstructive sleep apnea. Chest. 2005;127(5):1674–9. doi:http://dx.doi.org/10.1378/chest.127.5.1674.
12 Alonso-Fernández A, García-Río F, Arias MA, Hernanz A, de la Peña M, Piérola J, et al. Effects of CPAP on oxidative stress and nitrate efficiency in sleep apnoea: a randomised trial. Thorax. 2009;64(7):581–6. doi:http://dx.doi.org/10.1136/thx.2008.100537.
13 Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005;112(17):2660–7. doi:http://dx.doi.org/10.1161/CIRCULATIONAHA.105.556746.
14 Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, et al. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation. 2003;107(8):1129–34. doi:http://dx.doi.org/10.1161/01.CIR.0000052627.99976.18.
15 Rexhaj E, Rimoldi SF, Pratali L, Brenner R, Andries D, Soria R, et al. Sleep-Disordered Breathing and Vascular Function in Patients With Chronic Mountain Sickness and Healthy High-Altitude Dwellers. Chest. 2016;149(4):991–8. doi:http://dx.doi.org/10.1378/chest.15-1450.
16 Rimoldi SF, Ott S, Rexhaj E, de Marchi SF, Allemann Y, Gugger M, et al. Patent Foramen Ovale Closure in Obstructive Sleep Apnea Improves Blood Pressure and Cardiovascular Function. Hypertension. 2015;66(5):1050–7. doi:http://dx.doi.org/10.1161/HYPERTENSIONAHA.115.06303.
17 Bruno RM, Rossi L, Fabbrini M, Duranti E, Di Coscio E, Maestri M, et al. Renal vasodilating capacity and endothelial function are impaired in patients with obstructive sleep apnea syndrome and no traditional cardiovascular risk factors. J Hypertens. 2013;31(7):1456–64, discussion 1464. doi:http://dx.doi.org/10.1097/HJH.0b013e328360f773.
18 Nadeem R, Harvey M, Singh M, Khan AA, Albustani M, Baessler A, et al. Patients with obstructive sleep apnea display increased carotid intima media: a meta-analysis. Int J Vasc Med. 2013;2013:839582.
19 Nagahama H, Soejima M, Uenomachi H, Higashi Y, Yotsumoto K, Samukawa T, et al. Pulse wave velocity as an indicator of atherosclerosis in obstructive sleep apnea syndrome patients. Intern Med. 2004;43(3):184–8. doi:http://dx.doi.org/10.2169/internalmedicine.43.184.
20 Weinreich G, Wessendorf TE, Erdmann T, Moebus S, Dragano N, Lehmann N, et al.; Heinz Nixdorf Recall (HNR) study group. Association of obstructive sleep apnoea with subclinical coronary atherosclerosis. Atherosclerosis. 2013;231(2):191–7. doi:http://dx.doi.org/10.1016/j.atherosclerosis.2013.09.011.
21 Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2000;342(19):1378–84. doi:http://dx.doi.org/10.1056/NEJM200005113421901.
22 Rimoldi SF, Messerli FH, Bangalore S, Scherrer U. Resistant hypertension: what the cardiologist needs to know. Eur Heart J. 2015;36(40):2686–95. doi:http://dx.doi.org/10.1093/eurheartj/ehv392.
23 Logan AG, Perlikowski SM, Mente A, Tisler A, Tkacova R, Niroumand M, et al. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J Hypertens. 2001;19(12):2271–7. doi:http://dx.doi.org/10.1097/00004872-200112000-00022.
24 Pedrosa RP, Drager LF, Gonzaga CC, Sousa MG, de Paula LK, Amaro AC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension. 2011;58(5):811–7. doi:http://dx.doi.org/10.1161/HYPERTENSIONAHA.111.179788.
25 Rimoldi SF, Scherrer U, Messerli FH. Secondary arterial hypertension: when, who, and how to screen? Eur Heart J. 2014;35(19):1245–54. doi:http://dx.doi.org/10.1093/eurheartj/eht534.
26 Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Nieto FJ, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163(1):19–25. doi:http://dx.doi.org/10.1164/ajrccm.163.1.2001008.
27 Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353(19):2034–41. doi:http://dx.doi.org/10.1056/NEJMoa043104.
28 Yumino D, Tsurumi Y, Takagi A, Suzuki K, Kasanuki H. Impact of obstructive sleep apnea on clinical and angiographic outcomes following percutaneous coronary intervention in patients with acute coronary syndrome. Am J Cardiol. 2007;99(1):26–30. doi:http://dx.doi.org/10.1016/j.amjcard.2006.07.055.
29 Arikawa T, Toyoda S, Haruyama A, Amano H, Inami S, Otani N, et al. Impact of Obstructive Sleep Apnoea on Heart Failure with Preserved Ejection Fraction. Heart Lung Circ. 2016;25(5):435–41. doi:http://dx.doi.org/10.1016/j.hlc.2015.09.011.
30 Javaheri S, Blackwell T, Ancoli-Israel S, Ensrud KE, Stone KL, Redline S; Osteoporotic Fractures in Men Study Research Group. Sleep-disordered Breathing and Incident Heart Failure in Older Men. Am J Respir Crit Care Med. 2016;193(5):561–8. doi:http://dx.doi.org/10.1164/rccm.201503-0536OC.
31 Becker H, Brandenburg U, Peter JH, Von Wichert P. Reversal of sinus arrest and atrioventricular conduction block in patients with sleep apnea during nasal continuous positive airway pressure. Am J Respir Crit Care Med. 1995;151(1):215–8. doi:http://dx.doi.org/10.1164/ajrccm.151.1.7812557.
32 Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, et al.; Sleep Heart Health Study. Association of nocturnal arrhythmias with sleep-disordered breathing: The Sleep Heart Health Study. Am J Respir Crit Care Med. 2006;173(8):910–6. doi:http://dx.doi.org/10.1164/rccm.200509-1442OC.
33 Ismail K, Roberts K, Manning P, Manley C, Hill NS. OSA and pulmonary hypertension: time for a new look. Chest. 2015;147(3):847–61. doi:http://dx.doi.org/10.1378/chest.14-0614.
34 Guyton AC, Lindsey AW, Abernathy B, Richardson T. Venous return at various right atrial pressures and the normal venous return curve. Am J Physiol. 1957;189(3):609–15.
35 Buda AJ, Pinsky MR, Ingels NB, Jr, Daughters GT, 2nd, Stinson EB, Alderman EL. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301(9):453–9. doi:http://dx.doi.org/10.1056/NEJM197908303010901.
36 Lattimore JD, Wilcox I, Adams MR, Kilian JG, Celermajer DS. Treatment of obstructive sleep apnoea leads to enhanced pulmonary vascular nitric oxide release. Int J Cardiol. 2008;126(2):229–33. doi:http://dx.doi.org/10.1016/j.ijcard.2007.04.001.
37 Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, et al. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol. 2011;301(1):H247–52. doi:http://dx.doi.org/10.1152/ajpheart.01309.2010.
38 Sajkov D, Wang T, Saunders NA, Bune AJ, Neill AM, Douglas Mcevoy R. Daytime pulmonary hemodynamics in patients with obstructive sleep apnea without lung disease. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1518–26. doi:http://dx.doi.org/10.1164/ajrccm.159.5.9805086.
39 Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59(1):17–20. doi:http://dx.doi.org/10.1016/S0025-6196(12)60336-X.
40 Messerli FH, Rimoldi SF, Scherrer U, Meier B. Economy Class Syndrome, patent foramen ovale and stroke. Am J Cardiol. 2016;S0002-9149(16)31256-5.
41 Allemann Y, Hutter D, Lipp E, Sartori C, Duplain H, Egli M, et al. Patent foramen ovale and high-altitude pulmonary edema. JAMA. 2006;296(24):2954–8. doi:http://dx.doi.org/10.1001/jama.296.24.2954.
42 Lau EM, Jaijee SK, Melehan KL, Wong KK, Yee BJ, Grunstein RR, et al. Prevalence of patent foramen ovale and its impact on oxygen desaturation in obstructive sleep apnea. Int J Cardiol. 2013;165(1):35–40. doi:http://dx.doi.org/10.1016/j.ijcard.2011.07.050.
43 Schwerzmann M, Seiler C, Lipp E, Guzman R, Lövblad KO, Kraus M, et al. Relation between directly detected patent foramen ovale and ischemic brain lesions in sport divers. Ann Intern Med. 2001;134(1):21–4. doi:http://dx.doi.org/10.7326/0003-4819-134-1-200101020-00009.
44 Seiler C. Patent foramen ovale (PFO): is there life before death in the presence of PFO? Eur J Clin Invest. 2015;45(8):875–82. doi:http://dx.doi.org/10.1111/eci.12469.
45 Soliman OI, Geleijnse ML, Meijboom FJ, Nemes A, Kamp O, Nihoyannopoulos P, et al. The use of contrast echocardiography for the detection of cardiac shunts. Eur J Echocardiogr. 2007;8(3):S2–12. doi:http://dx.doi.org/10.1016/j.euje.2007.03.006.
46 Shanoudy H, Soliman A, Raggi P, Liu JW, Russell DC, Jarmukli NF. Prevalence of patent foramen ovale and its contribution to hypoxemia in patients with obstructive sleep apnea. Chest. 1998;113(1):91–6. doi:http://dx.doi.org/10.1378/chest.113.1.91.
47 Beelke M, Angeli S, Del Sette M, Gandolfo C, Cabano ME, Canovaro P, et al. Prevalence of patent foramen ovale in subjects with obstructive sleep apnea: a transcranial Doppler ultrasound study. Sleep Med. 2003;4(3):219–23. doi:http://dx.doi.org/10.1016/S1389-9457(02)00256-3.
48 Kraiczi H, Caidahl K, Samuelsson A, Peker Y, Hedner J. Impairment of vascular endothelial function and left ventricular filling : association with the severity of apnea-induced hypoxemia during sleep. Chest. 2001;119(4):1085–91. doi:http://dx.doi.org/10.1378/chest.119.4.1085.
49 Rexhaj E, Rimoldi SF, Pratali L, Brenner R, Andries D, Soria R, et al. Sleep-Disordered Breathing and Vascular Function in Patients With Chronic Mountain Sickness and Healthy High-Altitude Dwellers. Chest. 2016;149(4):991–8. doi:http://dx.doi.org/10.1378/chest.15-1450.
50 Rimoldi SF, Ott SR, Rexhaj E, von Arx R, de Marchi SF, Brenner R, et al. Effect of patent foramen ovale closure on obstructive sleep apnea. J Am Coll Cardiol. 2015;65(20):2257–8. doi:http://dx.doi.org/10.1016/j.jacc.2015.01.062.
51 Bailey DM, Rimoldi SF, Rexhaj E, Pratali L, Salinas Salmòn C, Villena M, et al. Oxidative-nitrosative stress and systemic vascular function in highlanders with and without exaggerated hypoxemia. Chest. 2013;143(2):444–51. doi:http://dx.doi.org/10.1378/chest.12-0728.
52 Rimoldi SF, Rexhaj E, Pratali L, Bailey DM, Hutter D, Faita F, et al. Systemic vascular dysfunction in patients with chronic mountain sickness. Chest. 2012;141(1):139–46. doi:http://dx.doi.org/10.1378/chest.11-0342.
53 Elliott JE, Laurie SS, Kern JP, Beasley KM, Goodman RD, Kayser B, et al. AltitudeOmics: impaired pulmonary gas exchange efficiency and blunted ventilatory acclimatization in humans with patent foramen ovale after 16 days at 5,260 m. J Appl Physiol (1985). 2015;118(9):1100–12. http://dx.doi.org/10.1152/japplphysiol.00879.2014.
54 Brenner R, Pratali L, Rimoldi SF, Murillo Jauregui CX, Soria R, Rexhaj E, et al. Exaggerated pulmonary hypertension and right ventricular dysfunction in high-altitude dwellers with patent foramen ovale. Chest. 2015;147(4):1072–9. doi:http://dx.doi.org/10.1378/chest.14-1353.
55 Johansson MC, Eriksson P, Peker Y, Hedner J, Råstam L, Lindblad U. The influence of patent foramen ovale on oxygen desaturation in obstructive sleep apnoea. Eur Respir J. 2007;29(1):149–55. doi:http://dx.doi.org/10.1183/09031936.00035906.
56 Shaikh ZF, Jaye J, Ward N, Malhotra A, de Villa M, Polkey MI, et al. Patent foramen ovale in severe obstructive sleep apnea: clinical features and effects of closure. Chest. 2013;143(1):56–63. doi:http://dx.doi.org/10.1378/chest.12-0334.
57 Konecny T, Khanna AD, Novak J, Jama AA, Zawadowski GM, Orban M, et al. Interatrial pressure gradients during simulated obstructive sleep apnea: a catheter-based study. Catheter Cardiovasc Interv. 2014;84(7):1138–45. doi:http://dx.doi.org/10.1002/ccd.25433.
58 Schäfer H, Hasper E, Ewig S, Koehler U, Latzelsberger J, Tasci S, et al. Pulmonary haemodynamics in obstructive sleep apnoea: time course and associated factors. Eur Respir J. 1998;12(3):679–84. doi:http://dx.doi.org/10.1183/09031936.98.12030679.
59 Soliman A, Shanoudy H, Liu J, Russell DC, Jarmukli NF. Increased prevalence of patent foramen ovale in patients with severe chronic obstructive pulmonary disease. J Am Soc Echocardiogr. 1999;12(2):99–105. doi:http://dx.doi.org/10.1016/S0894-7317(99)70121-5.
60 Xie A, Skatrud JB, Dempsey JA. Effect of hypoxia on the hypopnoeic and apnoeic threshold for CO(2) in sleeping humans. J Physiol. 2001;535(Pt 1):269–78. doi:http://dx.doi.org/10.1111/j.1469-7793.2001.00269.x.
61 Agnoletti G, Iserin L, Lafont A, Sidi D, Desnos M. Obstructive sleep apnoea and patent foramen ovale: successful treatment of symptoms by percutaneous foramen ovale closure. J Interv Cardiol. 2005;18(5):393–5. doi:http://dx.doi.org/10.1111/j.1540-8183.2005.00072.x.
62 Pinet C, Orehek J. CPAP suppression of awake right-to-left shunting through patent foramen ovale in a patient with obstructive sleep apnoea. Thorax. 2005;60(10):880–1. doi:http://dx.doi.org/10.1136/thx.2004.027508.
63 Silver B, Greenbaum A, McCarthy S. Improvement in sleep apnea associated with closure of a patent foramen ovale. J Clin Sleep Med. 2007;3(3):295–6.