Prolonged administration of β-lactam antibiotics – a comprehensive review and critical appraisal
DOI: https://doi.org/10.4414/smw.2016.14368
Michael
Osthoff, Martin
Siegemund, Gianmarco
Balestra, Mohd Hafiz
Abdul-Aziz, Jason A
Roberts
Summary
Prolonged infusion of β-lactam antibiotics as either extended (over at least 2 hours) or continuous infusion is increasingly applied in intensive care units around the world in an attempt to optimise treatment with this most commonly used class of antibiotics, whose effectiveness is challenged by increasing resistance rates.
The pharmacokinetics of β-lactam antibiotics in critically ill patients is profoundly altered secondary to an increased volume of distribution and the presence of altered renal function, including augmented renal clearance. This may lead to a significant decrease in plasma concentrations of β-lactam antibiotics. As a consequence, low pharmacokinetic/pharmacodynamic (PK/PD) target attainment, which is described as the percentage of time that the free drug concentration is maintained above the minimal inhibitory concentration (MIC) of the causative organism (fT>MIC), has been documented for β-lactam treatment in these patients when using standard intermittent bolus dosing, even for the most conservative target (50% fT>MIC).
Prolonged infusion of β-lactams has consistently been shown to improve PK/PD target attainment, particularly in patients with severe infections. However, evidence regarding relevant patient outcomes is still limited. Whereas previous observational studies have suggested a clinical benefit of prolonged infusion, results from two recent randomised controlled trials of continuous infusion versus intermittent bolus administration of β-lactams are conflicting. In particular, the larger, double-blind placebo-controlled randomised controlled trial including 443 patients did not demonstrate any difference in clinical outcomes.
We believe that a personalised approach is required to truly optimise β-lactam treatment in critically ill patients. This may include therapeutic drug monitoring with real-time adaptive feedback, rapid MIC determination and the use of antibiotic dosing software tools that incorporate patient parameters, dosing history, drug concentration and site of infection.
Universal administration of β-lactam antibiotics as prolonged infusion, even if supported by therapeutic drug monitoring, is not yet ready for “prime time”, as evidence for its clinical benefit is modest. There is a need for prospective randomised controlled trials that assess patient-centred outcomes (e.g. mortality) of a personalised approach in selected critically ill patients including prolonged infusion of β-lactams compared with the current standard of care.
Introduction
Beta-lactam antibiotics have been the cornerstone of antibiotic treatment since the early 1940s [1]. Owing to their wide spectrum of antibiotic activity and favourable safety profile, they remain the primary choice for treatment of severe infections worldwide. However, increasing resistance rates have challenged their widespread application in clinical practice. Rapid spread of β-lactamases in Gram-negative bacteria represents a genuine threat to successful treatment of both uncomplicated and serious infections [2, 3]. To make matters worse, the research and development pipeline for new antibiotics has declined over recent decades, and novel treatment strategies have mostly yielded disappointing results in sepsis trials [4, 5].
For decades, development of doses for new antibiotics for clinical registration was based on in vitro studies using historical models of pharmacokinetics (PK) and pharmacodynamics (PD) in healthy volunteers or non-critically ill individuals. Traditionally, individualising antibiotic therapy was more focused on the choice of antibiotic rather than the optimal dosage and mode of administration. With the exception of renal impairment, patient characteristics were largely neglected when choosing the dose of β-lactam antibiotics – a one-size-fits-all approach. However, recent evidence highlights that one size cannot fit all [6–10]. Importantly, optimal antibiotic exposure may not be achieved with traditional dosing strategies in a significant number of patients (e.g. critically ill or infected by resistant organisms), which may lead to microbiological and clinical failure, and may promote the emergence of antibiotic resistance [8–10].
Given increasing resistance rates and the limited availability of new treatment options, clinical researchers have concentrated their efforts on optimising treatment with β-lactam antibiotics. This includes identifying patient populations at risk for underdosing and applying PK/PD principles to define optimal dosing strategies. As a result, prolonged infusion of β-lactam antibiotics has been suggested as one of the dosing strategies to improve achievement of PK/PD targets and may improve patient outcomes, particularly in the intensive care unit (ICU). Although evidence supporting its efficacy is currently scarce, prolonged infusion dosing of β-lactam antibiotics is being increasingly adopted in many ICUs around the world.
In this article, we review the arguments and theory underlying the use of prolonged infusion dosing of β-lactam antibiotics, current evidence and caveats, and identify areas for future research.
Pharmacokinetic/pharmacodynamic targets for β-lactam antibiotics
The two main areas of pharmacology are PK and PD. PK refers to the time-course of drug concentration in tissue and body fluids, whereas PD, in the case of antibiotics, describes their antibiotic activity, clinical effects and toxicology. Antibiotic PD helps to define which dosing strategies should be used for different antibiotic classes (table 1), and is mainly dependent on the minimal inhibitory concentration (MIC) of the pathogen and the presence of a post-antibiotic effect [11].
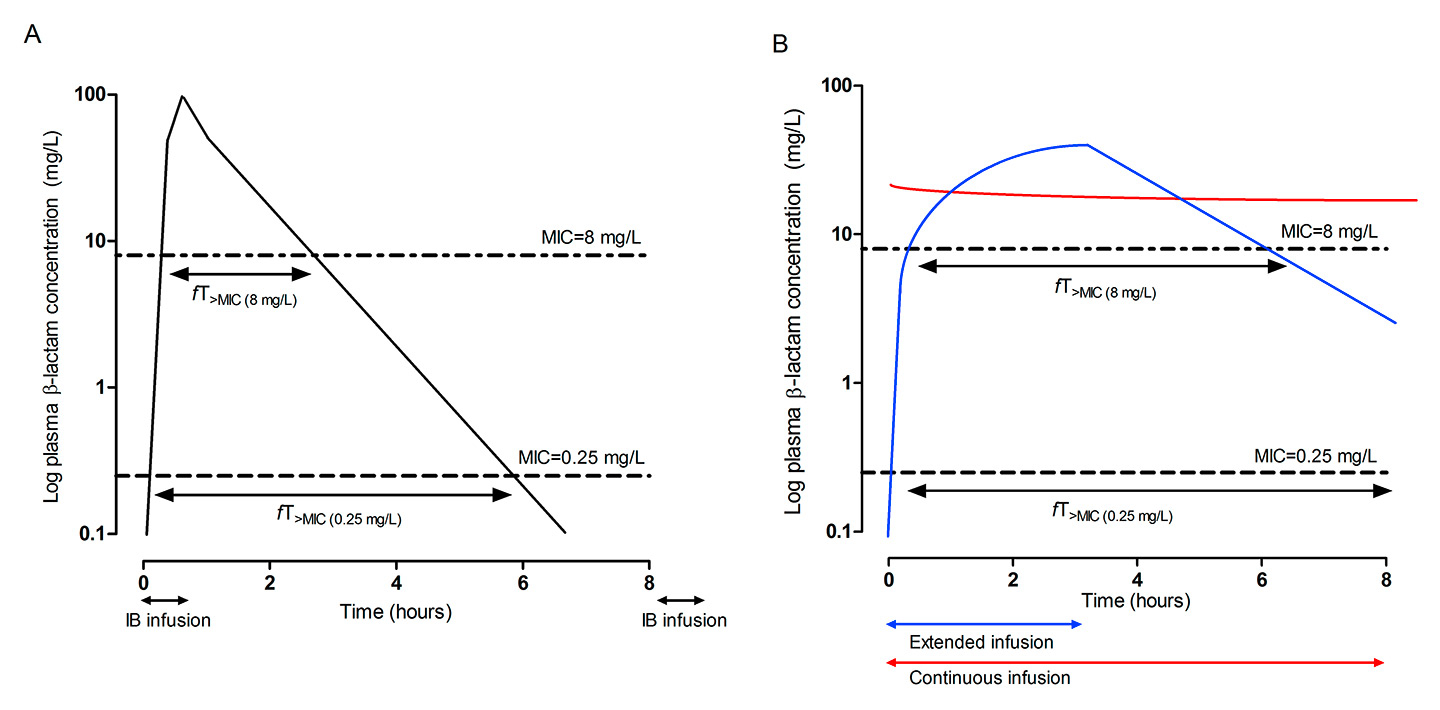
Figure 1
Differences in the time that β-lactam concentrations exceed the MIC (fT>MIC) of two different pathogens (MIC of 0.125 mg/l and 8 mg/l, respectively) according to the mode of β-lactam administration. (A) Intermittent bolus administration. (B) Extended infusion (blue line) and continuous infusion (red line).
IB = intermittent bolus administration; MIC = minimal inhibitory concentration; fT>MIC = time that the free drug concentration is above the MIC
For β-lactams, the time that the free (unbound) drug concentration remains above the MIC (fT>MIC) has been described as the PK/PD index that best correlates with bactericidal activity [12, 13] (fig. 1). Maximal killing rates for β-lactams are attained at low multiples of the MIC (2–4 x MIC), which is related to the fact that low drug concentrations are sufficient to saturate all possible binding sites and consequently inhibit peptidoglycan synthesis [14]. Conversely, drug concentrations below the MIC may permit regrowth of many organisms within a short period of time secondary to a lack of a relevant post-antibiotic effect [15]. Previous animal and clinical studies have found that the time interval in which the free drug concentration is above the MIC is the central parameter for optimal bacterial killing and clinical efficacy (45–100% for cephalosporins, 40–50% for penicillins and 40–75% for carbapenems [16]). However, these targets were mainly derived from experiments involving neutropenic animal models [11] and relatively susceptible bacterial strains, and do not account for the variable penetration of β-lactams into various tissues. For example, the patient’s immune system may be able to clear a minor pulmonary infection even if antibiotic treatment only achieves bacteriostasis. Conversely, higher drug exposures may be required to clear serious pulmonary infections in an immunocompromised host, such as a critically ill patient, or for a β-lactam with limited penetration into the lung.
In addition, although rarely considered clinically at this time, different targets may be used to suppress emergence of resistance, to attenuate selective pressure or to successfully kill pathogens which have already acquired certain resistance mechanisms (mutant population). Clinical cure was higher when concentrations of β-lactams were maintained above the MIC for extended periods (fT>MIC ≥75–100%) [17, 18], and several studies have identified the time above four times the MIC (fT>4xMIC) as a target for achieving maximal bactericidal activity and microbiological success [15, 19–21], taking into account of reduced antibiotic penetration in infected tissues. Although the optimal PK/PD target is still a matter of debate [22], recent clinical studies have shown that extending β-lactam exposure to more than 50% of the dosing interval is associated with improved outcome in critically ill patients with severe infections [7, 18, 23–25]. Limitations of these studies include the lack of data on actual MICs of the causative pathogen (a “worst-case scenario” was used in many instances), on drug concentration at the site of infection, on free (non-protein-bound) drug concentrations (only free drug is microbiological active [14, 26], and free drug concentrations are often derived from published protein binding values [24]), and the inclusion of patients with concomitant (active) antibiotic treatment.
In summary, the magnitude of PK/PD indices required for clinical efficacy is still controversial and may vary according to the severity and site of infection. Conservative targets such as 50% fT>MIC are probably sufficient when treating less severe infections with a removable focus (e.g. catheter-related or urinary tract infection), whereas increased drug exposure (100% fT>MIC or 100% fT>4xMIC) may be needed for treatment of serious infections, which often involve resistant organisms with a high bacterial load (e.g. hospital-acquired pulmonary infections) and/or limited penetration of β-lactams into the site of infection [19, 20, 27]. When interpreting studies on pharmacology of β-lactam antibiotics, it is important to realise that PK/PD endpoints vary considerably between studies, including significant differences in the definition of target/theoretical MICs (as exact MICs are rarely measured and reported). As a consequence, these studies often report “worst-case” scenarios from a pharmacological perspective, and correlation with clinical outcomes is clearly desirable.
|
Table 1:Pharmacokinetic/pharmacodynamic properties of selected antibiotics that correlate with efficacy. |
|
|
Pharmacodynamic kill characteristics
|
|
Time dependent
|
Concentration dependent
|
Concentration dependent with time dependence
|
| Antibiotic |
Penicillins
Cephalosporins
Carbapenems
Linezolid
Clarithromycin
Clindamycin |
Aminoglycosides
Metronidazole
Daptomycin
Fluoroquinolones |
Fluoroquinolones
Azithromycin
Glycopeptides
Tetracyclines
Tigecycline
Linezolid
Aminoglycosides |
| Optimal PK/PD index (and target examples for selected drugs) |
T>MIC
e.g. 40–100% T>MIC for β-lactams |
Cmax:MIC
e.g. Cmax:MIC 8–10 for aminoglycosides |
AUC0–24:MIC
e.g. AUC0–24:MIC ≥400 for vancomycin |
| Objective |
Maximise duration of exposure |
Maximise concentration |
Maximise amount of drug exposure |
| Measures |
Frequent administration or prolonged infusion dosing |
Infrequent (once daily) administration of high doses |
Administration of a high total daily dose |
| MIC = minimal inhibitory concentration; PK/PD = pharmacokinetics/pharmacodynamics; AUC0-24:MIC = the ratio of the area under the concentration time curve during a 24-hour period to MIC; Cmax:MIC = the ratio of the maximum plasma concentration to MIC; T>MIC = time that the drug concentration is above the MIC;
Note: For some antibiotics therapeutic efficacy may be correlated with more than one pharmacokinetic/pharmacodynamic parameter (e.g. aminoglycosides or fluoroquinolones). |
Pathophysiological alterations that may influence β-lactam pharmacokinetics
Interpatient variability in drug exposure is considerable when administering β-lactams at a fixed dose and time interval. The PK of antibiotics is complex in hospitalised patients, particularly in critically ill and obese patients, and is inadequately explained by traditional patient factors such as age, gender, disease severity or glomerular filtration rate. Two parameters markedly influence β-lactam exposure in critically ill patients (fig. 2): altered renal function and an increased volume of distribution [28].
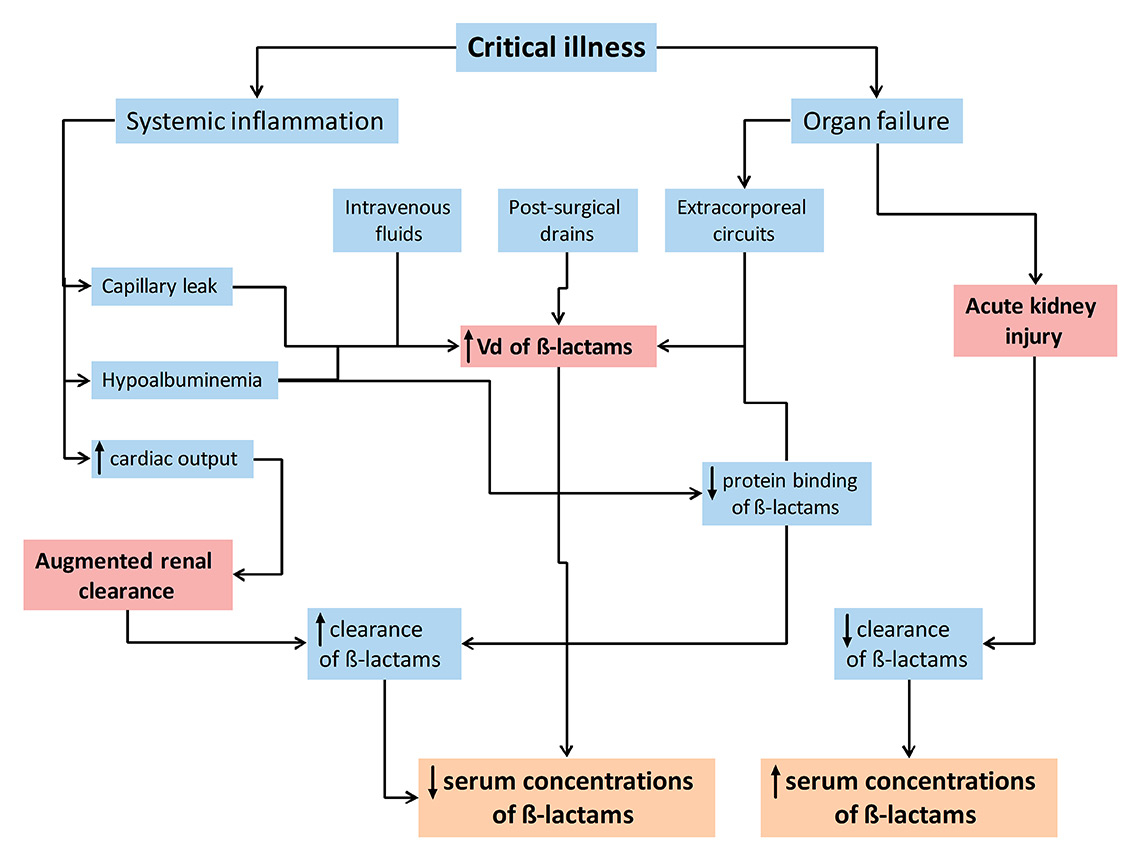
Figure 2
Pathophysiological alterations in critically-ill patients and their predicted influence on β-lactam pharmacokinetics.
Vd = volume of distribution.
Changes of volume of distribution
Target site distribution
Endothelial dysfunction with increased vascular permeability secondary to a systemic inflammatory response and/or direct endothelial damage is a hallmark of critically ill patients, particularly in patients with severe inflammatory conditions (e.g. pancreatitis, burn injuries) and septic shock [29]. Capillary leakage results in fluid extravasation into the interstitial space and systemic hypotension [30]. In response, large amounts of intravenous fluids are administered. As a consequence, the volume of distribution of some drugs may increase substantially within a short period of time (within hours to a few days) [31]. The presence of mechanical ventilation, extracorporeal circuits, surgical drains and hypoalbuminaemia may further expand the volume of distribution in critically ill patients [28]. For hydrophilic drugs such as β-lactam antibiotics, volume of distribution is heavily influenced by extracellular water volume and hence may increase several-fold in critically ill patients [27, 32]. In addition, observed interpatient variability is substantial compared with healthy individuals [27]. Drug concentrations may be considerably lower in the early period of critical illness before stable serum concentrations are reached.
Hypoalbuminaemia
Hypoalbuminaemia, defined as a serum albumin concentration <25 g/l, is present in 40–50% of critically ill patients [33], and has two prominent effects on the PK of β-lactam antibiotics [34]. Firstly, it increases the concentration of unbound antibiotic, which in turn is available for distribution and renal clearance. Secondly, it increases the volume of distribution of β-lactam antibiotics by augmenting fluid shifts into the interstitial space. This is particularly relevant for highly protein bound β-lactam antibiotics such as flucloxacillin, ertapenem and ceftriaxone [34, 35]. While hypoalbuminaemia may temporarily result in higher concentrations of highly protein bound β-lactam antibiotics, a reduced fT>MIC will eventually result as a consequence of an increased dilution and drug clearance [34].
Changes of drug clearance
Augmented renal clearance
In contrast to the commonly perceived risk of overdosing, the presence of altered renal function actually exposes ICU patients to significant underdosing, particularly in two settings. Firstly, systemic inflammation, increased cardiac output, fluid resuscitation and administration of vasopressors may result in increased renal perfusion and subsequently increased renal clearance. Augmented renal clearance is defined as enhanced elimination of solutes (including β-lactam antibiotics) or, more specifically, as a creatinine clearance of ≥130 ml/min [36]. Risk factors for the phenomenon include younger age, sepsis, trauma, febrile neutropenia, burn injury and cystic fibrosis [36]. Recent studies reported the presence of augmented renal clearance on at least one day in up to 50–60% of critically ill patients during their ICU stay [37, 38]. Consequently, β-lactam exposure is markedly reduced in these patients and aggressive PK/PD targets such as 100% fT>MIC or 100% fT>4xMIC may not be achieved in a considerable proportion of critically ill patients [6, 39, 40].
Similarly, moderate to severe renal failure may put ICU patients at risk for underdosing, in particular in the early treatment period. Clinicians may choose inappropriately low β-lactam doses secondary to the inappropriate use of formulas for estimating renal function [41], fear of overdosing and side effects, and limited acknowledgement of rapid changes in volume of distribution (e.g. during fluid resuscitation).
Renal replacement therapy
Renal replacement therapy prescribed for acute kidney injury increases the complexity of antibiotic dosing owing to variability in the mode of renal replacement therapy and in its differential effect on β-lactam antibiotics. Consequently, both inadequate and (infrequently) excessive β-lactam drug exposures have been documented in this setting with adjusted and standard doses, highlighting the current lack of knowledge of how to dose during renal replacement therapy in this situation.
Pharmacokinetic/pharmacodynamic target attainment in ICU patients
The prevalence of subtherapeutic β-lactam concentrations and inadequate drug exposure was explored in a recent large, multicentre study [7]. Plasma concentrations at 50 and 100% of the dosing interval were determined once for eight different β-lactam antibiotics in 361 patients from 68 hospitals. The key results of this analysis included the presence of an extreme variability in free β-lactam concentrations (up to 500-fold) and nonachievement of the most conservative (50% fT>MIC) and the most aggressive (100% fT>4xMIC) PK/PD targets in 21% and 75% of patients, respectively. In addition, a significant association of a positive clinical outcome with increasing antibiotic concentrations at 50 and 100% of the dosing interval was observed. Of note, increasing creatinine clearance and use of intermittent bolus dosing emerged as significant risk factors for target nonattainment in this study [42].
To make the situation even more complex, intraindividual serum concentrations vary considerably over time [43] and, furthermore, plasma concentrations may not necessarily correspond to concentrations measured at the infection site. This is particularly true for many β-lactams in the case of pulmonary infections [44, 45], the most common site of severe infection [46]. Penetration into pulmonary epithelial lining fluid ranges from 20–25% in the case of ceftazidime [47] and ceftobiprole [48] to 50% for piperacillin [44, 49] and 100% for cefepime [50]. Interpretation of these values is hampered by the fact that significant variability in lung penetration of the same antibiotic has been documented (up to 100%) [44, 51] and because the effect of the local immune system is not considered. In addition, PK/PD targets of different β-lactam antibiotics may vary, which has not been assessed in detail in clinical studies.
Prolonged infusion of β-lactam antibiotics – PK/PD target attainment
Modulation of β-lactam dosing is required to address subtherapeutic drug exposures in critically ill patients. This may include increasing the dose, shortening the dosing interval, prolonging the infusion time either for the entire dosing interval (continuous infusion) or for 40–50% of the dosing interval (3–4 hours; extended infusion), or a combination of these (fig. 1). In theory, current PK/PD targets may be attained with all the above dosing strategies, depending on the known/likely MIC. However, disadvantages of dose escalation include unnecessarily high peak concentrations (which may increase the risk of side effects, including seizures) and higher costs, the latter also being the case for more frequent administration. Limited drug stability, drug-drug incompatibilities or the need for constant availability of vascular access may present a challenge for implementing continuous infusion dosing. Nevertheless, continuous infusion dosing has gained widespread popularity as a promising solution for enhancing the activity of current β-lactam antibiotics against increasingly resistant (Gram-negative) bacteria.
The majority of studies have shown that prolonged infusion dosing of β-lactam antibiotics improves PK/PD target attainment, albeit often using Monte Carlo simulations based on drug concentration measurements in a limited number of patients [52, 53]. A unique feature of these simulations is prediction of target attainment for a chosen dosing strategy against the distribution of MICs and renal clearance, which may be used to select the target population that will most likely benefit from prolonged infusion. For example, Asin-Prieto et al. demonstrated that standard intermittent bolus administration of piperacillin/tazobactam (4.5 g eight hourly) may be sufficient to achieve 100% fT>MIC in patients with moderate renal impairment up to an MIC of 4 mg/l (which is the case for the majority of Enterobacteriaceaein Europe), whereas extended or even continuous infusions are required for treating organisms with higher MICs or in patients with augmented renal clearance [54]. This was illustrated by Udy et al., who found target nonattainment (100% fT>MIC) for intermittent bolus dosing in the majority of patients with a creatinine clearance >90 ml/min when targeting an MIC of at least 8 mg/l [40].
However, desirable PK/PD targets may not be achieved even with the use of extended infusion dosing in patients with augmented renal clearance [55] or resistant organisms [56], as highlighted by Carlier et al. [57], who reported that 55% of patients on extended infusion did not achieve 100% fT>MIC, when targeting the MIC susceptibility breakpoints for piperacillin/tazobactam and meropenem. Furthermore, 100% fT>MIC was achieved in only approximately 70% of febrile neutropenic patients on extended infusion dosing of piperacillin/tazobactam [58]. In this setting – augmented renal clearance and high MICs – continuous infusion is more likely than extended infusion dosing to achieve PK/PD targets of β-lactam antibiotics [54, 59].
There is a paucity of data regarding the effect of prolonged infusion dosing on resistance development. In theory, altered dosing schemes may result in drug concentrations that lie in the mutant selection window (the concentration range between the MIC and the mutant prevention concentration) for a longer period of time than with intermittent bolus dosing. Clinical studies have suggested a neutral effect of optimised piperacillin dosing with respect to resistance development [60], and a recent in vitro hollow-fibre infection model with Pseudomonas aeruginosa suggested similar rates of resistance emergence when comparing intermittent bolus with extended infusion dosing. However, achievement of higher trough concentrations seems to be required in the case of extended infusion versus intermittent bolus dosing for suppression of resistance (trough concentration [Cmin] / MIC of 10.4 vs 3.4, respectively) [61].
Prolonged infusion of β-lactam antibiotics – clinical outcome data
A number of observational and randomised controlled trials have compared prolonged infusion with intermittent bolus dosing of β-lactams in different patient populations. Overall, two recent meta-analyses have documented a mortality benefit favouring prolonged infusion over intermittent bolus dosing [60, 62], but with conflicting results in terms of clinical cure and with a lack of mortality benefit when the analysis was restricted to meropenem treatment only [60, 62]. The observed reduction in all-cause mortality was mainly driven by results from observational trials, whereas a mortality benefit was lacking if only data from randomised controlled trials were included [62, 63]. Adverse events were similar. Inclusion of a homogeneous patient population that would most likely benefit from optimised administration (Gram-negative infections, higher severity of illness, multi-drug resistant pathogens) in observational studies may explain the differences observed.
Two major trials of continuous infusion versus intermittent bolus administration in patients with severe sepsis have recently been published. In a multicentre, double-blind, double-dummy placebo-controlled trial, Dulhunty et al. randomised 443 patients with severe sepsis to continuous infusion or intermittent bolus dosing of β-lactam antibiotics, of whom 432 were analysed [64]. This trial failed to demonstrate any benefit of continuous infusion over intermittent bolus administration with regards to all endpoints analysed, including 90-day all-cause mortality and clinical cure after 14 days after antibiotic cessation. Several limitations of this study need to be acknowledged. Firstly, 26% of the patients were on renal replacement therapy, which is associated with a reduced likelihood of subtherapeutic β-lactam concentrations in patients on intermittent bolus dosing compared with patients not on renal replacement therapy [65]. Secondly, patients were receiving continuous infusion treatment on average for only 3.2 days, a duration that may have been too short to test for a significant difference between the treatment groups. Thirdly, causative organisms were identified in less than 20% (only bloodstream isolates were reported) without exact MIC determination, and therapeutic drug monitoring was not performed. Hence, achievement of therapeutic concentrations could not be verified. This is of importance, as even some patients on continuous infusion therapy may not achieve sufficient drug levels [24], and as therapeutic drug monitoring results may have provided explanations for the observed lack of benefit. For example, attainment of therapeutic concentrations may have been the same in both groups or only slightly different (without clinical relevance), given that most cases of severe sepsis in the study region are caused by susceptible pathogens with low MICs [66]. Lastly, combination treatment was utilised in a substantial number of participants (continuous infusion vs intermittent bolus dosing: aminoglycoside use in 11 and 15%, quinolone use in 9 and 14%, glycopeptide use in 36 and 31%, respectively), which might have obscured any treatment effect.
The second study by Abdul-Aziz et al. [67] was an open-label, randomised controlled trial of continuous infusion versus intermittent bolus dosing of β-lactam antibiotics in 140 patients with severe sepsis in two ICUs in Malaysia. Clinical cure at 14 days after cessation of antibiotic treatment was higher in the continuous infusion group (56% vs 34%, p = 0.011), particularly in patients receiving piperacillin/tazobactam, without concomitant antibiotic treatment and with pulmonary infection. Survival and ICU-free days were similar. Importantly, this study also demonstrated that PK/PD target attainment (albeit using surrogate MICs) was higher for continuous infusion patients, particularly when the more aggressive target (100% fT>MIC) was analysed. Limitations of this study include the open-label design, a larger antibiotic dose on day 1 in the continuous infusion arm (due to administration of a loading dose in this group only), concomitant antibiotic therapy in 47% of patients and a lack of exact MIC determination. Major differences from the first study are a longer treatment duration (median 7, interquartile range [IQR] 5–9 days vs 3, IQR 2–6 days), exclusion of patients on renal replacement therapy, infrequent use of Gram-negative combination therapy (6% vs >15%) and more frequent isolation of causative pathogens (74% vs 20%) with a higher incidence of difficult to treat Gram-negative organisms (41% vs <10% of isolates were Acinetobacter baumannii or P. aeruginosa).
A more recently published meta-analysis of individual patient data (n = 632 patients with severe sepsis) [68] included both randomised controlled trials mentioned above plus a previous pilot study of continuous infusion versus intermittent bolus dosing of β-lactam antibiotics in patients with severe sepsis [24]. In this analysis, continuous infusion was superior to intermittent bolus dosing with respect to 30-day in-hospital mortality (odds ratio 0.62, p = 0.03), but not with respect to clinical cure, ICU-free days at Day 28 and ICU mortality. The impact of continuous infusion was more evident in patients with higher APACHE II scores, not on renal replacement therapy and treated with piperacillin/tazobactam (APACHE: acute physiology and chronic health evaluation). Similar high-quality randomised controlled trials of extended infusion dosing of β-lactams and comparing continuous infusion with extended infusion dosing in severe sepsis patients are lacking.
The future: personalised medicine including improved dosing strategies, therapeutic drug monitoring and rapid MIC determination
What are the lessons learned from these real-world studies? Firstly, a one-size-fits-all approach (which failed so many times in sepsis trials in the last three decades [5]) to dosing of β-lactam antibiotics in a heterogeneous population irrespective of disease severity, causative organism, infection site and requirement for renal replacement therapy is not necessarily successful in improving patient outcomes. Secondly, a definitive randomised controlled trial is clearly desirable to quantify any effects of prolonged infusion dosing of β-lactams on patient-centred outcomes. Thirdly, to move the field forward, a more holistic and personalised approach should ideally be assessed, in analogy to the successfully established bundled approaches in infection prevention [69].
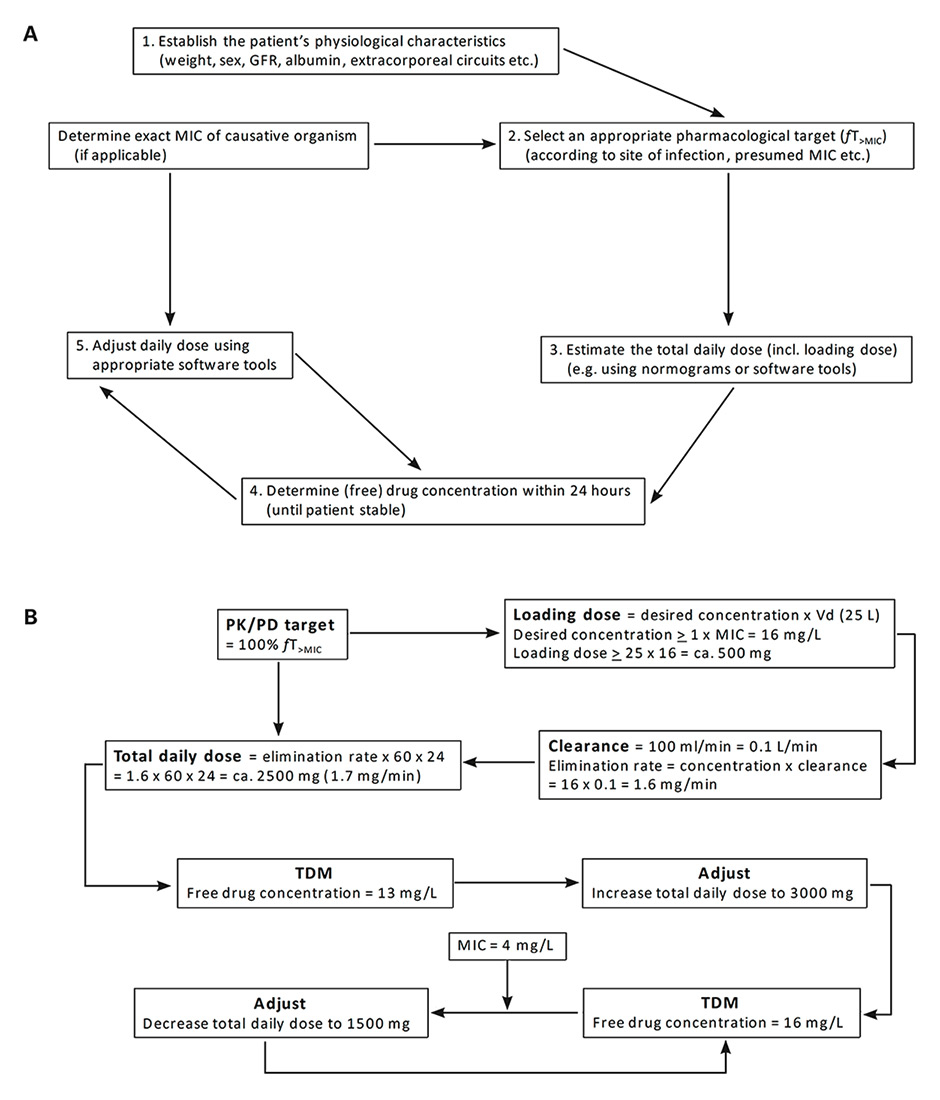
Figure 3
(A) Algorithm for personalised β-lactam dose optimisation in critically-ill patients. (B) Calculation of meropenem dose for empirical and definitive treatment of severe pulmonary sepsis caused by P. aeruginosa in a neutropenic fever patient (38 years old, 70 kg). The following parameters are assumed for this hypothetical calculation: presumed MIC for P. aeruginosa ≤16 mg/L (local surveillance data), calculated creatinine clearance of 100 ml/min, treatment with vasoactive agents. Based on concept from Choi et al. [85].
GFR = glomerular filtration rate; MIC = minimal inhibitory concentration; PK/PD = pharmacokinetics/pharmacodynamics; fT>MIC = time that the free drug concentration is above the MIC; TDM = therapeutic drug monitoring; Vd = volume of distribution
A bundled approach for dose personalisation of β-lactams might include the following (fig. 3A). After diagnosis of an infection and selection of the most appropriate antibiotic, the first loading dose and subsequent 24-hour total dose for continuous infusion of the chosen empirical β-lactam regimen should be estimated. The use of a loading dose is advocated to rapidly achieve therapeutic concentrations at the site of infection [70]. The main determinants for estimating the dosing on the first day are the volume of distribution, serum protein concentration and the presumed MIC of the causative pathogen (derived from local MIC surveillance data) [71]. Consequently, normograms derived from similar critically ill patients and taking into account parameters such as age, sex, weight, renal function, presence of extracorporeal support, volume of distribution and albumin concentration are paramount in order to select an appropriate loading dose and initial 24-hour total dose [59, 72]. This may include different targets for different patient groups and/or sites of infection, as MIC distributions and need for more aggressive targets may vary accordingly (e.g. neutropenic fever vs community-acquired sepsis vs ventilator-associated pneumonia) [73, 74]. Essentially, this corresponds to the development of standardised clinical pathways for selection of empirical doses for patients at risk for underdosing of β-lactams [75].
During the first dosing interval of the continuous infusion, one or more blood samples for therapeutic drug monitoring should be drawn. Ideally, turn-around should be quick with the unbound (microbiologically active [76]) drug concentration results being available within the dosing interval for the drug, so that the can be dose adjusted at the time of the next dose. Therapeutic drug monitoring is an integral part of this approach, as classical patient variables such as renal function or age poorly predict serum β-lactam concentrations and hence much of the variance in concentrations observed remains unexplained [77].
Subsequently, a personalised dosing recommendation for the next dosing interval may be generated using antibiotic dosing software (fig. 3B). These software tools use either Bayesian forecasting with embedded population PK models or nonlinear regression. Both approaches can incorporate patient parameters, dosing history, drug concentration and even site of infection (assuming differential penetration of different β-lactams) [28, 78]. Importantly, the first dose adaptation should ideally occur within 24 hours after start of empirical therapy to ensure rapid achievement of therapeutic antibiotic exposure. Although the therapeutic window for β-lactams is broad, dosing recommendations should also consider dose reductions, at least for certain antibiotics and clinical scenarios (e.g. cefepime in patients at risk for neurotoxicity [79, 80]). As intraindividual β-lactam concentrations may vary over time in critically ill patients [43], daily therapeutic drug monitoring with adaptive feedback may be required until the patient is stable, in particular after new interventions including major surgery and introduction of extracorporeal circuits [71]. However, at this point, additional parameters, such as susceptibility data of the causative pathogen (if isolated) and the need for extracorporeal support, should be taken into account. In this regard, rapid and exact (as opposed to imputed) determination of the MIC is important to enable verification of PK/PD target attainment. Alternatively, a “worst-case” scenario may be applied assuming that the MIC for a particular organism is equal to either the MIC90 (concentration that inhibits 90% of pathogens) or the susceptibility breakpoint of the antibiotic used [58], which may lead to a higher target dose than necessary for more susceptible pathogens.
Using this holistic approach, an evidence-based and personalised dosing regimen may be generated for each individual patient. In our opinion, evaluation of a bundled approach in future prospective clinical studies (in a selected target population) is more promising than investigating single interventions (such as investigating only therapeutic drug monitoring with or without additional population PK models or only optimised β-lactam administration). Furthermore, comparing a bundled approach to intermittent bolus dosing without therapeutic drug monitoring in a clinical trial is ethically justifiable, as this standard approach is still used in the majority of hospitals worldwide [7, 81, 82], although some hospitals that already use therapeutic drug monitoring and/or prolonged infusion dosing may have ethical concerns with regards to clinical equipoise of the two approaches. In fact, in a recent survey of 328 hospitals in 53 countries, therapeutic drug monitoring and extended infusion dosing of β-lactams was utilised in less than 5 and 30% of all ICUs, respectively [83].
Results from several clinical studies have proven that therapeutic drug monitoring with real-time feedback and dose adjustment is feasible and successful with regards to optimising target attainment [58, 65, 84]. In a pilot-study, therapeutic drug monitoring with subsequent dose adjustment was applied in 236 ICU patients [65], of whom 50% required a dose increase after the first measurement. In a second study, Sime et al. [58] randomised 32 febrile neutropenia patients to therapeutic drug monitoring with immediate dose adjustment during the first 3 days or to standard care. This study is notable, as many elements of the outlined holistic approach were implemented, including prolonged infusion dosing, therapeutic drug monitoring with real-time dose adaptation and, if possible, incorporation of the exact MIC of the pathogen. Patients initially received intermittent bolus dosing of piperacillin/tazobactam at standard doses (4.5 g eight hourly), which yielded a low target attainment (target was 100% fT>MIC) of 19% (intervention) and 25% (control) after 24 hours. After dose optimisation (including extended infusion dosing and more frequent administration), day 2 trough levels and target attainment were significantly increased in the intervention group (69% vs 19%, p = 0.012) with similar effects on day 3. Clinical outcomes were not different in this feasibility study.
There is a need for prospective randomised controlled trials that compare the benefit of a personalised approach in selected critically ill patients with standard of care. In the meantime, hospitals may apply selected interventions of the outlined bundled approach in patient populations with unpredictable PK or with difficult-to-treat infections. Implementation of extended infusion dosing of β-lactam antibiotics may be feasible in many settings without much effort and additional costs. Introducing continuous infusion dosing is certainly more challenging as several important issues and practicalities need to be considered. Regarding therapeutic drug monitoring, costs, including infrastructure, staff and the assay itself, may certainly be an issue for many hospitals.
Conclusion
Universal administration of β-lactams via prolonged infusion dosing is not yet ready for “prime time”, as evidence for its potential benefit is modest and indirect. Importantly, reasonable PK/PD targets may be achieved with standard intermittent bolus dosing in many patients with less severe disease and infections caused by susceptible organisms. Today, few strategies are left to successfully treat resistant organisms and hence we argue that prolonged infusion administration should be considered in the sickest patients at risk for infections with less susceptible organisms and a high bacterial load – ideally in combination with therapeutic drug monitoring and real-time dose adaptation. Future studies should clarify the role of such a combined approach for the treatment of severe infections.
Disclosure statement: J.A.R. is funded by a Career Development Fellowship from the National Health and Medical Research Council of Australia (APP1048652). The authors would like to acknowledge other funding to the Burns Trauma Critical Care Research Centre from the National Health and Medical Research Council of Australia for a Project Grant (APP1044941) and Centre for Research Excellence (APP1099452). J.A.R. has served as a consultant for Astellas and Infectopharm and has given lectures for Merck. MO has received research grants from the University of Basel (Career Development Grant) and the Fondation Machaon. The authors declare that they have no conflict of interest related to this article.
References
1 Keefer CS, Blake FG, Marshall EK, Jr, Lockwood JS, Wood BW. Penicillin and treatment of infections: a report of 500 cases. Statement by the Committee on Chemotherapeutic and Other Agents, Division of Medical Sciences, National Research Council. JAMA. 1943;122(18):1217–24.
2 Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13(9):785–96.
3 Johnson AP, Woodford N. Global spread of antibiotic resistance: the example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J Med Microbiol. 2013;62(Pt 4):499–513.
4 Boucher HW, Talbot GH, Benjamin DK, Jr, Bradley J, Guidos RJ, Jones RN, et al.; Infectious Diseases Society of America. 10 x ’20 Progress – development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(12):1685–94.
5 Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20(4):195–203.
6 Huttner A, Von Dach E, Renzoni A, Huttner BD, Affaticati M, Pagani L, et al. Augmented renal clearance, low β-lactam concentrations and clinical outcomes in the critically ill: an observational prospective cohort study. Int J Antimicrob Agents. 2015;45(4):385–92.
7 Roberts JA, Paul SK, Akova M, Bassetti M, De Waele JJ, Dimopoulos G, et al.; DALI Study. DALI: defining antibiotic levels in intensive care unit patients: are current β-lactam antibiotic doses sufficient for critically ill patients? Clin Infect Dis. 2014;58(8):1072–83.
8 Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37(3):311–20.
9 Olofsson SK, Cars O. Optimizing drug exposure to minimize selection of antibiotic resistance. Clin Infect Dis. 2007;45(Suppl 2):S129–36.
10 Roberts JA, Kruger P, Paterson DL, Lipman J. Antibiotic resistance – what’s dosing got to do with it? Crit Care Med. 2008;36(8):2433–40.
11 Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26(1):1–10, quiz 11–2.
12 Eagle H, Fleischman R, Levy M. “Continuous” vs. “discontinuous” therapy with penicillin; the effect of the interval between injections on therapeutic efficacy. N Engl J Med. 1953;248(12):481–8.
13 Flückiger U, Segessenmann C, Gerber AU. Integration of pharmacokinetics and pharmacodynamics of imipenem in a human-adapted mouse model. Antimicrob Agents Chemother. 1991;35(9):1905–10.
14 Drusano GL. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat Rev Microbiol. 2004;2(4):289–300.
15 Craig WA, Ebert SC. Killing and regrowth of bacteria in vitro: a review. Scand J Infect Dis Suppl. 1990;74:63–70.
16 Abdul-Aziz MH, Lipman J, Mouton JW, Hope WW, Roberts JA. Applying pharmacokinetic/pharmacodynamic principles in critically ill patients: optimizing efficacy and reducing resistance development. Semin Respir Crit Care Med. 2015;36(1):136–53.
17 Ariano RE, Nyhlén A, Donnelly JP, Sitar DS, Harding GK, Zelenitsky SA. Pharmacokinetics and pharmacodynamics of meropenem in febrile neutropenic patients with bacteremia. Ann Pharmacother. 2005;39(1):32–8.
18 McKinnon PS, Paladino JA, Schentag JJ. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents. 2008;31(4):345–51.
19 Tam VH, McKinnon PS, Akins RL, Rybak MJ, Drusano GL. Pharmacodynamics of cefepime in patients with Gram-negative infections. J Antimicrob Chemother. 2002;50(3):425–8.
20 Manduru M, Mihm LB, White RL, Friedrich LV, Flume PA, Bosso JA. In vitro pharmacodynamics of ceftazidime against Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob Agents Chemother. 1997;41(9):2053–6.
21 Li C, Du X, Kuti JL, Nicolau DP. Clinical pharmacodynamics of meropenem in patients with lower respiratory tract infections. Antimicrob Agents Chemother. 2007;51(5):1725–30.
22 Wong G, Brinkman A, Benefield RJ, Carlier M, De Waele JJ, El Helali N, et al. An international, multicentre survey of β-lactam antibiotic therapeutic drug monitoring practice in intensive care units. J Antimicrob Chemother. 2014;69(5):1416–23.
23 Lodise TP, Jr, Lomaestro B, Drusano GL. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: clinical implications of an extended-infusion dosing strategy. Clin Infect Dis. 2007;44(3):357–63.
24 Dulhunty JM, Roberts JA, Davis JS, Webb SA, Bellomo R, Gomersall C, et al. Continuous infusion of beta-lactam antibiotics in severe sepsis: a multicenter double-blind, randomized controlled trial. Clin Infect Dis. 2013;56(2):236–44.
25 Muller AE, Punt N, Mouton JW. Exposure to ceftobiprole is associated with microbiological eradication and clinical cure in patients with nosocomial pneumonia. Antimicrob Agents Chemother. 2014;58(5):2512–9.
26 Odenholt I, Holm SE, Cars O. Effect of antibiotic protein binding on the killing rate of Staphylococcus aureus and on the paradoxical phenomenon. Chemotherapy. 1987;33(5):331–9.
27 Gonçalves-Pereira J, Póvoa P. Antibiotics in critically ill patients: a systematic review of the pharmacokinetics of β-lactams. Crit Care. 2011;15(5):R206.
28 Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, et al.; International Society of Anti-Infective Pharmacology and the Pharmacokinetics and Pharmacodynamics Study Group of the European Society of Clinical Microbiology and Infectious Diseases. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis. 2014;14(6):498–509.
29 van der Poll T. Immunotherapy of sepsis. Lancet Infect Dis. 2001;1(3):165–74.
30 Hinshaw LB. Sepsis/septic shock: participation of the microcirculation: an abbreviated review. Crit Care Med. 1996;24(6):1072–8.
31 Blot SI, Pea F, Lipman J. The effect of pathophysiology on pharmacokinetics in the critically ill patient – concepts appraised by the example of antimicrobial agents. Adv Drug Deliv Rev. 2014;77:3–11.
32 Georges B, Conil JM, Seguin T, Ruiz S, Minville V, Cougot P, et al. Population pharmacokinetics of ceftazidime in intensive care unit patients: influence of glomerular filtration rate, mechanical ventilation, and reason for admission. Antimicrob Agents Chemother. 2009;53(10):4483–9.
33 Finfer S, Bellomo R, McEvoy S, Lo SK, Myburgh J, Neal B, et al.; SAFE Study Investigators. Effect of baseline serum albumin concentration on outcome of resuscitation with albumin or saline in patients in intensive care units: analysis of data from the saline versus albumin fluid evaluation (SAFE) study. BMJ. 2006;333(7577):1044.
34 Ulldemolins M, Roberts JA, Rello J, Paterson DL, Lipman J. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin Pharmacokinet. 2011;50(2):99–110.
35 Joynt GM, Lipman J, Gomersall CD, Young RJ, Wong EL, Gin T. The pharmacokinetics of once-daily dosing of ceftriaxone in critically ill patients. J Antimicrob Chemother. 2001;47(4):421–9.
36 Udy AA, Roberts JA, Lipman J. Implications of augmented renal clearance in critically ill patients. Nat Rev Nephrol. 2011;7(9):539–43.
37 Udy AA, Baptista JP, Lim NL, Joynt GM, Jarrett P, Wockner L, et al. Augmented renal clearance in the ICU: results of a multicenter observational study of renal function in critically ill patients with normal plasma creatinine concentrations*. Crit Care Med. 2014;42(3):520–7.
38 Claus BO, Hoste EA, Colpaert K, Robays H, Decruyenaere J, De Waele JJ. Augmented renal clearance is a common finding with worse clinical outcome in critically ill patients receiving antimicrobial therapy. J Crit Care. 2013;28(5):695–700.
39 Udy AA, Varghese JM, Altukroni M, Briscoe S, McWhinney BC, Ungerer JP, et al. Subtherapeutic initial β-lactam concentrations in select critically ill patients: association between augmented renal clearance and low trough drug concentrations. Chest. 2012;142(1):30–9.
40 Udy AA, Lipman J, Jarrett P, Klein K, Wallis SC, Patel K, et al. Are standard doses of piperacillin sufficient for critically ill patients with augmented creatinine clearance? Crit Care. 2015;19:28.
41 Sunder S, Jayaraman R, Mahapatra HS, Sathi S, Ramanan V, Kanchi P, et al. Estimation of renal function in the intensive care unit: the covert concepts brought to light. J Intensive Care. 2014;2(1):31.
42 De Waele JJ, Lipman J, Akova M, Bassetti M, Dimopoulos G, Kaukonen M, et al. Risk factors for target non-attainment during empirical treatment with β-lactam antibiotics in critically ill patients. Intensive Care Med. 2014;40(9):1340–51. Corrected in: Intensive Care Med. 2015 May;41(5):969. Dosage error in article test.
43 Carlier M, Carrette S, Stove V, Verstraete AG, De Waele JJ. Does consistent piperacillin dosing result in consistent therapeutic concentrations in critically ill patients? A longitudinal study over an entire antibiotic course. Int J Antimicrob Agents. 2014;43(5):470–3.
44 Felton TW, McCalman K, Malagon I, Isalska B, Whalley S, Goodwin J, et al. Pulmonary penetration of piperacillin and tazobactam in critically ill patients. Clin Pharmacol Ther. 2014;96(4):438–48.
45 Rodvold KA, George JM, Yoo L. Penetration of anti-infective agents into pulmonary epithelial lining fluid: focus on antibacterial agents. Clin Pharmacokinet. 2011;50(10):637–64.
46 Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al.; ACCESS Study Group. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154–62.
47 Boselli E, Breilh D, Rimmelé T, Poupelin JC, Saux MC, Chassard D, et al. Plasma and lung concentrations of ceftazidime administered in continuous infusion to critically ill patients with severe nosocomial pneumonia. Intensive Care Med. 2004;30(5):989–91.
48 Rodvold KA, Nicolau DP, Lodise TP, Khashab M, Noel GJ, Kahn JB, et al. Identifying exposure targets for treatment of staphylococcal pneumonia with ceftobiprole. Antimicrob Agents Chemother. 2009;53(8):3294–301.
49 Boselli E, Breilh D, Rimmelé T, Guillaume C, Xuereb F, Saux MC, et al. Alveolar concentrations of piperacillin/tazobactam administered in continuous infusion to patients with ventilator-associated pneumonia. Crit Care Med. 2008;36(5):1500–6.
50 Boselli E, Breilh D, Duflo F, Saux MC, Debon R, Chassard D, et al. Steady-state plasma and intrapulmonary concentrations of cefepime administered in continuous infusion in critically ill patients with severe nosocomial pneumonia. Crit Care Med. 2003;31(8):2102–6.
51 Lodise TP, Sorgel F, Melnick D, Mason B, Kinzig M, Drusano GL. Penetration of meropenem into epithelial lining fluid of patients with ventilator-associated pneumonia. Antimicrob Agents Chemother. 2011;55(4):1606–10.
52 Kuti JL, Dandekar PK, Nightingale CH, Nicolau DP. Use of Monte Carlo simulation to design an optimized pharmacodynamic dosing strategy for meropenem. J Clin Pharmacol. 2003;43(10):1116–23.
53 Roberts JA, Kirkpatrick CM, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Meropenem dosing in critically ill patients with sepsis and without renal dysfunction: intermittent bolus versus continuous administration? Monte Carlo dosing simulations and subcutaneous tissue distribution. J Antimicrob Chemother. 2009;64(1):142–50.
54 Asín-Prieto E, Rodríguez-Gascón A, Trocóniz IF, Soraluce A, Maynar J, Sánchez-Izquierdo JA, et al. Population pharmacokinetics of piperacillin and tazobactam in critically ill patients undergoing continuous renal replacement therapy: application to pharmacokinetic/pharmacodynamic analysis. J Antimicrob Chemother. 2014;69(1):180–9.
55 Cotta MO, Gowen B, Truloff N, Bursle E, McWhinney B, Ungerer JP, et al. Even high-dose extended infusions may not yield desired concentrations of β-lactams: the value of therapeutic drug monitoring. Infect Dis (Lond). 2015;47(10):739–42.
56 Reese AM, Frei CR, Burgess DS. Pharmacodynamics of intermittent and continuous infusion piperacillin/tazobactam and cefepime against extended-spectrum beta-lactamase-producing organisms. Int J Antimicrob Agents. 2005;26(2):114–9.
57 Carlier M, Carrette S, Roberts JA, Stove V, Verstraete A, Hoste E, et al. Meropenem and piperacillin/tazobactam prescribing in critically ill patients: does augmented renal clearance affect pharmacokinetic/pharmacodynamic target attainment when extended infusions are used? Crit Care. 2013;17(3):R84.
58 Sime FB, Roberts MS, Tiong IS, Gardner JH, Lehman S, Peake SL, et al. Can therapeutic drug monitoring optimize exposure to piperacillin in febrile neutropenic patients with haematological malignancies? A randomized controlled trial. J Antimicrob Chemother. 2015;70(8):2369–75.
59 Roberts JA, Kirkpatrick CM, Roberts MS, Dalley AJ, Lipman J. First-dose and steady-state population pharmacokinetics and pharmacodynamics of piperacillin by continuous or intermittent dosing in critically ill patients with sepsis. Int J Antimicrob Agents. 2010;35(2):156–63.
60 Falagas ME, Tansarli GS, Ikawa K, Vardakas KZ. Clinical outcomes with extended or continuous versus short-term intravenous infusion of carbapenems and piperacillin/tazobactam: a systematic review and meta-analysis. Clin Infect Dis. 2013;56(2):272–82.
61 Felton TW, Goodwin J, O’Connor L, Sharp A, Gregson L, Livermore J, et al. Impact of Bolus dosing versus continuous infusion of Piperacillin and Tazobactam on the development of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2013;57(12):5811–9.
62 Teo J, Liew Y, Lee W, Kwa AL. Prolonged infusion versus intermittent boluses of β-lactam antibiotics for treatment of acute infections: a meta-analysis. Int J Antimicrob Agents. 2014;43(5):403–11.
63 Shiu J, Wang E, Tejani AM, Wasdell M. Continuous versus intermittent infusions of antibiotics for the treatment of severe acute infections. Cochrane Database Syst Rev. 2013;3(3):CD008481.
64 Dulhunty JM, Roberts JA, Davis JS, Webb SA, Bellomo R, Gomersall C, et al.; BLING II Investigators for the ANZICS Clinical Trials Group *. A Multicenter Randomized Trial of Continuous versus Intermittent β-Lactam Infusion in Severe Sepsis. Am J Respir Crit Care Med. 2015;192(11):1298–305.
65 Roberts JA, Ulldemolins M, Roberts MS, McWhinney B, Ungerer J, Paterson DL, et al. Therapeutic drug monitoring of beta-lactams in critically ill patients: proof of concept. Int J Antimicrob Agents. 2010;36(4):332–9.
66 Harris P, Paterson D, Rogers B. Facing the challenge of multidrug-resistant gram-negative bacilli in Australia. Med J Aust. 2015;202(5):243–7.
67 Abdul-Aziz MH, Sulaiman H, Mat-Nor MB, Rai V, Wong KK, Hasan MS, et al. Beta-Lactam Infusion in Severe Sepsis (BLISS): a prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med. 2016 Jan 11. [Epub ahead of print].
68 Roberts JA, Abdul-Aziz MH, Davis JS, Dulhunty JM, Cotta MO, Myburgh J, et al. Continuous versus Intermittent β-Lactam Infusion in Severe Sepsis. A Meta-analysis of Individual Patient Data from Randomized Trials. Am J Respir Crit Care Med. 2016;194(6):681–91.
69 Blot K, Bergs J, Vogelaers D, Blot S, Vandijck D. Prevention of central line-associated bloodstream infections through quality improvement interventions: a systematic review and meta-analysis. Clin Infect Dis. 2014;59(1):96–105.
70 Roberts JA, Paul SK, Akova M, Bassetti M, De Waele JJ, Dimopoulos G, et al. Reply to Rhodes et al. Clin Infect Dis. 2014;59(6):907–8.
71 Ulldemolins M, Vaquer S, Llauradó-Serra M, Pontes C, Calvo G, Soy D, et al. Beta-lactam dosing in critically ill patients with septic shock and continuous renal replacement therapy. Crit Care. 2014;18(3):227.
72 Ulldemolins M, Rello J. The relevance of drug volume of distribution in antibiotic dosing. Curr Pharm Biotechnol. 2011;12(12):1996–2001.
73 Nicasio AM, Eagye KJ, Nicolau DP, Shore E, Palter M, Pepe J, et al. Pharmacodynamic-based clinical pathway for empiric antibiotic choice in patients with ventilator-associated pneumonia. J Crit Care. 2010;25(1):69–77.
74 Zelenitsky SA, Ariano RE, Zhanel GG. Pharmacodynamics of empirical antibiotic monotherapies for an intensive care unit (ICU) population based on Canadian surveillance data. J Antimicrob Chemother. 2011;66(2):343–9.
75 Roberts JA, Roberts DM. Antibiotic dosing in critically ill patients with septic shock and on continuous renal replacement therapy: can we resolve this problem with pharmacokinetic studies and dosing guidelines? Crit Care. 2014;18(3):156.
76 Wong G, Briscoe S, Adnan S, McWhinney B, Ungerer J, Lipman J, et al. Protein binding of β-lactam antibiotics in critically ill patients: can we successfully predict unbound concentrations? Antimicrob Agents Chemother. 2013;57(12):6165–70.
77 Casu GS, Hites M, Jacobs F, Cotton F, Wolff F, Beumier M, et al. Can changes in renal function predict variations in β-lactam concentrations in septic patients? Int J Antimicrob Agents. 2013;42(5):422–8.
78 Felton TW, Roberts JA, Lodise TP, Van Guilder M, Boselli E, Neely MN, et al. Individualization of piperacillin dosing for critically ill patients: dosing software to optimize antimicrobial therapy. Antimicrob Agents Chemother. 2014;58(7):4094–102.
79 Chapuis TM, Giannoni E, Majcherczyk PA, Chioléro R, Schaller MD, Berger MM, et al. Prospective monitoring of cefepime in intensive care unit adult patients. Crit Care. 2010;14(2):R51.
80 Fugate JE, Kalimullah EA, Hocker SE, Clark SL, Wijdicks EF, Rabinstein AA. Cefepime neurotoxicity in the intensive care unit: a cause of severe, underappreciated encephalopathy. Crit Care. 2013;17(6):R264.
81 George JM, Colton BJ, Rodvold KA. National survey on continuous and extended infusions of antibiotics. Am J Health Syst Pharm. 2012;69(21):1895–904.
82 Buyle FM, Decruyenaere J, De Waele J, Tulkens PM, Van Audenrode T, Depuydt P, et al. A survey of beta-lactam antibiotics and vancomycin dosing strategies in intensive care units and general wards in Belgian hospitals. Eur J Clin Microbiol Infect Dis. 2013;32(6):763–8.
83 Tabah A, De Waele J, Lipman J, Zahar JR, Cotta MO, Barton G, et al.; Working Group for Antimicrobial Use in the ICU within the Infection Section of the European Society of Intensive Care Medicine (ESICM). The ADMIN-ICU survey: a survey on antimicrobial dosing and monitoring in ICUs. J Antimicrob Chemother. 2015;70(9):2671–7.
84 Hayashi Y, Lipman J, Udy AA, Ng M, McWhinney B, Ungerer J, et al. β-Lactam therapeutic drug monitoring in the critically ill: optimising drug exposure in patients with fluctuating renal function and hypoalbuminaemia. Int J Antimicrob Agents. 2013;41(2):162–6.
85 Choi G, Gomersall CD, Tian Q, Joynt GM, Freebairn R, Lipman J. Principles of antibacterial dosing in continuous renal replacement therapy. Crit Care Med. 2009;37(7):2268–82.