Pulmonary hypertension associated with chronic lung diseases
DOI: https://doi.org/10.4414/smw.2016.14363
Manuela
Funke, Thomas
Geiser, Otto D.
Schoch
Summary
In 2015, the international guidelines for diagnosis and treatment of pulmonary hypertension were updated. Group 3 of the current classification includes pulmonary hypertension associated with chronic obstructive pulmonary disease (COPD), interstitial lung disease, other pulmonary diseases with restrictive and obstructive pattern, sleep-disordered breathing, alveolar hypoventilation disorders, chronic exposure to high altitude, and developmental lung diseases. Pulmonary hypertension associated with COPD and interstitial lung disease is common but difficult to manage, as no long-term randomised controlled trial has been conducted with specific pulmonary arterial hypertension drugs in pulmonary hypertension associated with COPD, and the one recent RCT in pulmonary hypertension associated with interstitial lung disease had to be stopped early due to a possible increased risk of death and side effects in the treatment group. Specific treatment may thus be harmful and may worsen gas exchange as a result of possible recruitment of less oxygenised areas, i.e. by increasing ventilation-perfusion mismatch. Management of pulmonary hypertension patients with chronic lung diseases requires careful evaluation and understanding of pathophysiological mechanisms. Interestingly, mediators in PAH and in fibrotic lung disease show some overlaps. Several drugs effective in pulmonary arterial hypertensio have been evaluated for anti-fibrotic treatment in interstitial lung disease, but until today without consistent success. While new drugs with anti-fibrotic effects are now accessible, a specific evidence-based treatment for pulmonary hypertension in interstitial lung disease or COPD with or without emphysema is not yet available.
Abbreviations
BMPR2 bone morphogenetic protein receptor, type 2
COPD chronic obstructive pulmonary disease
CPET cardiopulmonary exercise testing
CPFE combined pulmonary fibrosis and emphysema
GOLD global initiative for chronic obstructive pulmonary disease
HRCT high resolution CT scan
HIF-1α hypoxia inducible factor 1-alpha
HPV hypoxic pulmonary vasoconstriction (Euler-Liljestrand mechanism)
ILD interstitial lung disease
IPF idiopathic pulmonary fibrosis
IIP idiopathic interstitial pneumonia
mPAP mean pulmonary arterial pressure
NYHA New York Heart Association Functional Classification
PAH pulmonary arterial hypertension (= group 1)
PH pulmonary hypertension
PH-COPD PH associated with COPD
PH-ILD PH associated with ILD
PH-CPFE PH associated with CPFE
PDE5i phosphodiesterase-5 inhibitor
PPARγ peroxisome proliferator-activated receptor γ
PVR pulmonary vascular resistance
RCT randomised controlled trial
TGF-β transforming growth factor-beta
Introduction
Pulmonary hypertension (PH) is defined by a mean pulmonary artery pressure (mPAP) ≥25 mm Hg at rest measured during right heart catheterisation [1]. According to its aetiology, PH is classified into five diagnostic groups [1]. Pulmonary arterial hypertension (PAH, group 1) describes patients with precapillary PH, documented by a pulmonary artery wedge pressure (PAWP) ≤15 mm Hg, a pulmonary vascular resistance (PVR) >3 Wood units (WU) and the absence of other causes of pre-capillary PH, e.g. hypoxemia or chronic parenchymal lung diseases (group 3) or chronic thromboembolic disease (group 4) [1]. Chronic lung diseases induce PH and, because they are very prevalent, may also occur occasionally concomitant with PAH [1].
Group 3 PH includes chronic obstructive pulmonary disease (COPD) (group 3.1), interstitial lung disease (ILD, group 3.2) (e.g. idiopathic pulmonary fibrosis [IPF]), other lung diseases with mixed restrictive and obstructive pattern (group 3.3, e.g. combined pulmonary fibrosis with emphysema [CPFE]), sleep-disordered breathing (group 3.4), alveolar hypoventilation syndromes, where hypercapnic pulmonary vasoconstriction might have a role (group 3.5), chronic exposure to high altitude (group 3.6), and developmental abnormalities (group 3.7) [1]. Rare diseases involving the lung, like sarcoidosis or pulmonary Langerhans cell histiocytosis or lymphangioleiomyomatosis, are included in group 5.2 with unclear or multifactorial mechanisms [1]. In the following review we will focus on PH associated with the lung diseases COPD with or without emphysema, interstitial lung diseases, ILD (as an example IPF) and CPFE: PH group 3.1 to 3.3.
Pathophysiology of pulmonary hypertension in parenchymal lung diseases
Depending on the underlying lung disease, rarefication of vessels (for example in emphysematous areas), parenchymal or vascular remodelling (for example in IPF), cytokine release, hypoxaemia, and to some extent pulmonary vascular abnormalities and thrombosis may contribute to the development of PH. In contrast, in PH due to sleep-disordered breathing (3.4), alveolar hypoventilation disorders (3.5) and chronic exposure to high altitude (3.6) hypoxemia is present although lung parenchymal structure is preserved.
Physiologically, the entire cardiac output flows through the pulmonary circulation and for a given cardiac output, pulmonary resistance decreases by pulmonary arterial dilatation or recruitment of under-recruited pulmonary vessels [2]. While pulmonary arteries regulate their diameters and are orchestrated by vasoconstrictive signalling, including hypoxaemia-induced, resistance of alveolar capillaries is mainly dependent on alveolar pressure and volume [2]. The hypoxic regulation (HPV or Euler-Liljestrand mechanism) is relevant for pulmonary vasoconstriction in acute or chronic pulmonary diseases such as pneumonia, COPD or pulmonary embolism, which may all consecutively develop into acute or chronic PH. This pathophysiological consideration is particularly important to look at if vasodilating therapy targeting pulmonary arteries is considered in patients with PH due to heterogeneous lung diseases. Vasodilating therapy may recruit areas with worse oxygenation and may lead to worsening of hypoxia.
Hypoxaemia
Hypoxaemia, and on a cellular level hypoxia, is found in advanced stages of all diseases of group 3 PH, although in parenchymal diseases, destruction of lung tissue and reduction of total number of alveoli and vessels might additionally increase pulmonary arterial pressure. On one hand, destroyed lung parenchyma has reduced blood flow and to some extend increased pulmonary resistance; on the other hand, hypoxia induces release of cytokines and mediators which itself may lead to pulmonary vasoconstriction. The physiological response to alveolar hypoxia helps to regulate blood flow to alveolar areas with high oxygen partial pressure and is referred to as hypoxaemia-induced vasoconstriction (HPV or Euler-Liljestrand mechanism). Although the precise mechanisms of the regulation of vascular tone by hypoxaemia is still unknown, hypoxic effects are thought to be mediated through reactive oxygen species (ROS), redox couples and adenosine monophosphate-activated kinases [3]. L-type calcium channels, nonspecific cation channels and voltage-dependent potassium channels are effectors of the hypoxia induced pulmonary vasoconstriction [3].
Another mediator of hypoxia induced vasoconstriction, the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α), contributes to hypoxia-mediated pulmonary vascular remodelling and hypertension [4] and has been linked to pulmonary hypertension in chronic fibrotic lung disease [5]. Interestingly, vasonconstricting effects of hypoxaemia are not linear: if hypoxaemia is very severe, hypoxaemic pulmonary vasodilation will be observed [6].
Proliferative mediators in PH overlapping with fibrotic pathways
In addition to hypoxia-induced signalling, increased cytokines potentially promote vascular remodelling and pulmonary (arterial) hypertension [7]. Similar to pulmonary hypertension, fibrotic lung disease typically is characterised by increased cell proliferation and impaired apoptosis. Lung fibroblasts differentiate into myofibroblasts and destroy normal lung tissue by accumulation and extracellular matrix production. In pulmonary hypertension typically smooth muscle cells of pulmonary arterial vessels show increased proliferation and accumulation. Cytokines and other signalling mediators orchestrate cell migration, proliferation and survival, and can overlap between PH and fibrotic lung disease. One important cytokine is endothelin, a potent vasoconstrictor, which contributes to vascular remodelling in PH but also fibroblast accumulation in pulmonary fibrosis [8].
Endothelin is a vasoactive peptide, with three subtypes, ET-1, ET-2 and ET-3. It is expressed in several cell types such as endothelial cells, epithelial cells, smooth muscle cells and many more [8]. Endothelin binds to two G-protein coupled receptors, endothelin receptor A and B [8]. Endothelin receptor B (specifically B1) is found on endothelial cells, which are known to stimulate release of nitric oxide and prostacyclin and thus vasodilation. Endothelin receptor B (B2) mediates also vasoconstrictive effects. Endothelin A and B receptors are found on fibroblasts and smooth muscle cells [8]. Endothelin receptor A is mainly responsible for vasoconstriction [8]. Unselective or selective endothelin receptor antagonism is a well-established treatment option for patients with PAH [9]. For fibrotic diseases, the role of endothelin has been extensively studied in different models, and increased levels of endothelin have been found in patients with IPF [8]. This double signalling role of endothelin contributing to pulmonary hypertension and fibrosis has led to several clinical trials investigating endothelin antagonism in IPF or other idiopathic interstitial pneumonia (IIP) patients, although none of them found positive results so far [10–14].
Platelet derived growth factor (PDGF) has been associated with pulmonary hypertension and lung fibrosis. PDGF is found in several cell types including epithelial cells and smooth muscle cells and acts via its two receptors PDGFα and PDGFβ, with an intracellular tyrosine kinase domain which can be inhibited by specific tyrosine kinase inhibitors [7, 15]. PDGF activation promotes proliferation, migration and survival of smooth muscle cells in PAH and targeting PDGF has been considered a target in PH [7, 16]. In lung fibrosis, PDGF is a potent mitogen for fibroblasts [17] and recognized pro-fibrotic mediator in different organs [18].
Peroxisome proliferator-activated receptors (PPARγ) are transcription factors involved in several physiological processes such as adipogenesis, lipid metabolism and inflammation. They regulate amongst others cell proliferation and migration [7]. PPARγ agonists are beneficial to reduce PH in different models [7]. In addition, PPARγ agonists oppose the pro-fibrotic effects of transforming growth factor-beta in fibrotic disease [19].
Transforming growth factor-beta(TGF-β) signalling regulates various biological processes. Amongst others it has potent effects on cell proliferation, apoptosis and differentiation. PH can present with impaired TGF-β signalling due to possible BMPR2 mutations [7]. In pulmonary fibrosis, TGF-β signalling is considered the main pro-fibrotic mediator, with effects on epithelial cells and fibroblasts [20]. It is a potential inducer of differentiation of fibroblasts into myofibroblasts, the key effector cells in fibrosis.
Epidemiology and clinical presentation
Pulmonary hypertension associated with chronic obstructive pulmonary disease
COPD, as defined by the presence of pulmonary symptoms including chronic cough, sputum production and the presence of an airway obstruction in spirometry, is a very common and mainly smoking-associated lung disease with an estimated 350 000 cases living in Switzerland [21]. COPD is a syndrome affecting several organ systems, including peripheral muscles and the cardiovascular system. The clinical presentation of COPD comprises a broad spectrum of different phenotypes, characterised by various degrees of lung emphysema and bronchiectasis, nonspecific symptoms and chronic disease progression over several years.
The prevalence of PH in COPD populations is clearly related to the severity of the underlying disease. If patients are grouped according to the global initiative on obstructive lung diseases (GOLD), the prevalence of PH is around 5% for GOLD stage 1 or 2 (previously defined by 1-second forced expiratory volume [FEV1] values >50% predicted), 25% in stage 3 (previously defined by FEV1 values 30–49% predicted) and almost 40% for stage 4 disease (previously defined by FEV1 values <30% predicted) [22, 23]. The annual rate of increase in pulmonary pressure for PH-COPD is thought to be relatively low at 0.39–0.65 mm Hg/year [24]. In a cohort of 409 COPD GOLD 4 patients, 36% showed an mPAP of >25 mm Hg, but severe PH with values >35 mm Hg was found in only 3.9% [25], and PH-COPD with mPAP >40 mm Hg is rare [26].
During acute COPD exacerbations PAP has been shown to increase about 16 mm Hg [27, 28]. However, PAP increases only in a subgroup of COPD patients [29]. PH itself might influence occurrence and severity of COPD exacerbation, as pulmonary artery enlargement has been associated with more severe COPD exacerbations [30].
Pulmonary hypertension associated with interstitial lung disease
In comparison to COPD and emphysema, chronic fibrosing interstitial lung diseases (ILD) are less common. Idiopathic pulmonary fibrosis (IPF) is the most prevalent idiopathic form of ILD, with prevalence estimates below 5000 cases for Switzerland [31]. PH in IPF is also associated with advanced disease stages, (i.e. the severity of the underlying IPF) and ranges from 8–15% [32, 33] at the time of diagnosis to 35–45% [34, 35] for patients before lung transplantation [23]. In IPF patients with mild-to-moderate restriction, who were recruited for a study of ambrisentan in IPF patients (ARTEMIS-IPF), PH could be found in 14% [36]. In patients with advanced or end-stage IPF, pulmonary hypertension occurs in 30-50% or >60% respectively [23]. However, in a cohort of patients awaiting lung transplantation, only 9% had an mPAP of >40 mm Hg [37]. Lung functional or radiological impairment (high resolution computed tomography; HRCT) do not correlate well with severity of PH [23, 25, 37]. The evolution of PH-ILD is characterised by a progressive increase in mPAP of 0.17–3.8 mm Hg/month over time [34, 36]. PH represents an independent predictor of survival for IPF [33].
Pulmonary hypertension associated with combined pulmonary fibrosis and emphysema (CPFE) is a characteristic combination of basal fibrotic and apical emphysematous lung lesions affecting elderly male smokers, but the prevalence of the disease is rather low. CPFE has a typical appearance on chest computed tomography and is characterised by a severely impaired diffusion capacity but preserved lung volumes in functional testing. PH-CPFE is found in up to 50% of patients, and 68% of PH-CPFE patients had mPAP >35 mm Hg [23, 38]. Presence of PH in these patients is associated with worse outcome, with a median survival of less than 1 year in the subgroup of patients with a PVR of >485 dyn·s·cm-5 [39].
Assessment and diagnosis
Patients with COPD (with or without emphysema), ILD or CPFE and suspected PH require careful staging of the parenchymal lung disease. Lung disease assessment includes lung functional testing by body plethysmography, diffusion capacity measurements, arterial blood gas analysis, HRCT of the chest and eventually bronchoscopy with bronchoalveolar lavage and lung biopsy (if possible) to evaluate lung parenchymal structure. Functional exercise tests including cardiopulmonary exercise testing (CPET) and the 6 minutes walking test or sit-to-stand test [40] are part of the assessment to diagnose and define disease stages accurately. Non-invasive screening for PH-COPD, PH-ILD and PH-CPFE is performed with echocardiography [1]. The combination of the clinical and functional assessment with brain natriuretic peptide measurements has been shown to be useful to predict the presence of PH [41]. Nevertheless, echocardiographic measurements might be inaccurate, especially in COPD patients [42]. However, while echocardiography and referral to an expert centre is recommended if echocardiographic signs of severe PH are present (class I, level C) in the recently updated European guidelines [1], right heart catheterisation is discouraged for suspected PH in patients with lung diseases (class III, level C), unless therapeutic consequences are to be expected (e.g. lung transplantation, alternative diagnosis such as PAH or chronic thromboembolic pulmonary hypertension). Contributing factors with an impact on treatment decisions are to be searched for, including full night polysomnography in the search for obstructive sleep apnoea or nocturnal hypoventilation, left heart catheterisation and coronary angiography in suspected coronary or left heart disease and ventilation-perfusion scintigraphy to evaluate for venous thromboembolism [1].
Sometimes it is difficult to differentiate whether lung disease and PH are coincidental or related. In suspected PH-COPD or PH-ILD, previously proposed criteria [23] favouring PAH, with FEV1 >60% or forced vital capacity (FVC) >70%, the absence of parenchymal abnormalities on chest computed tomography and preserved breathing reserve with reduced oxygen pulse and a low cardiac output / oxygen consumption ratio (CO/VO2 slope) are uncommon. Most of the time patients show additional signs of parenchymal disease which complicates differentiation between PAH or group 3 PH [23]. If PH is disproportionate to parenchymal lung disease the term “out of proportion” has been previously used [43], but it has been suggested that this term be abandoned because only a loss >80% of normal lung structure will provoke PH [23]. However, a recent study showed that differentiation into mild and severe lung disease as previously suggested [23] and illustrated in table 1 has no impact on survival, but only severe PH-ILD showed poor survival similar to the overall reduced survival of the ILD cohort (fig. 1 and fig. 2) [44]. The current guidelines now categorise patients with parenchymal lung diseases into three groups: lung disease without PH (mPAP <25 mm Hg), lung disease with PH (mPAP ≥35 mm Hg) and lung disease with severe PH (mPAP ≥ 35 mm Hg or ≥25 mm Hg in the presence of low cardiac output (<2.5 l/min/m2), not explained by other causes (table 2) [1, 23]. PH-ILD patients have an overall worse survival when compared with other PH groups as observed in our national Swiss registry for pulmonary hypertension [45].
|
Table 1: Typical findings for lung function, radiology and exercise testing in pulmonary hypertension due to chronic lung diseases and in pulmonary arterial hypertension. |
| |
PH due to parenchymal lung diseases (Group 3) |
PAH (Group 1) |
| Lung function
FEV1
FVC or TLC |
(% predicted)
<60% in PH – COPD
<70% in PH – ILD |
(% predicted)
>60%
>70% |
| High resolution CT scan |
Presence of airway and / or parenchymal abnormalities |
No (or mild) abnormalities |
| CPET
Exercise limitation
Breathing reserve
Oxygen pulse
CO/VO2 slope
Oxygen saturation
CO2 during exercise |
Ventilatory
Reduced
Near normal
Near normal
Mild desaturation
May increase |
Cardiocirculatory
Preserved
Reduced
Low
Pronounced desaturation
No change / decrease |
| COPD = chronic obstructive pulmonary disease; CO/VO2 slope = cardiac output / oxygen consumption ratio; FEV1 = forded expiratory volume in 1 second; FVC = forced vital capacity; ILD = interstitial lung disease; PAH = pulmonary arterial hypertension; PH = pulmonary hypertension; TLC = total lung capacity |
|
Table 2: Current haemodynamic classification of pulmonary hypertension due to lung diseases (adapted from [1]). |
| |
Right heart catheterisation
|
| COPD/ILD without PH |
Mean PAP <25 mm Hg |
| COPD/ILD with PH |
Mean PAP ≥25 mm Hg, but <35 mm Hg |
| COPD/ILD with severe PH |
Mean PAP >35 mm Hg, or ≥25 mm Hg with low cardiac output (CI <2.5 l/min/m2) |
| COPD = chronic obstructive pulmonary disease; ILD = interstitial lung disease; PAP = pulmonary arterial pressure; PH = pulmonary hypertension |
Management
For PH-COPD, PH-ILD and PH-CPFE, recommendations aim at optimal treatment of the underlying lung disease for symptomatic improvement, improvement in quality of life and in survival (class I, evidence level C) [1, 23]. The mainstays for treatment recommendations for lung diseases associated with PH have been listed previously [46].
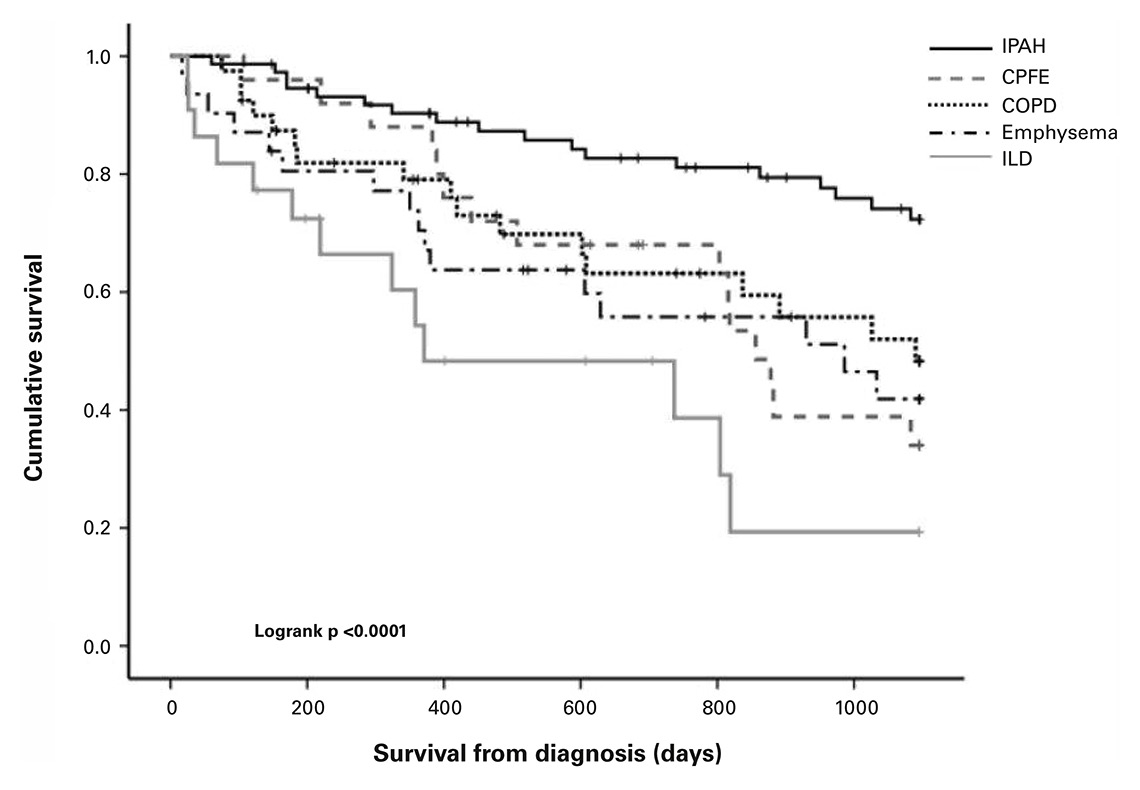
Figure 1
Survival for patients with pulmonary arterial hypertension, pulmonary hypertension associated with CPFE, COPD, emphysema and ILD. Survival for PH-ILD was significantly lower compared with PAH (p <0.0001), PH-COPD (p = 0.0026) and PH-CPFE (p = 0.05), while survival for other lung phenotypes did not differ significantly.
CPFE = combined pulmonary fibrosis and emphysema; COPD = chronic obstructive pulmonary disease; ILD = interstitial lung disease; PAH = pulmonary arterial hypertension; PH = pulmonary hypertension. (Reproduced with permission of the European Respiratory Society © Eur Respir J. 2015;46(5):1378–89; DOI: 10.1183/13993003.02307-2014)
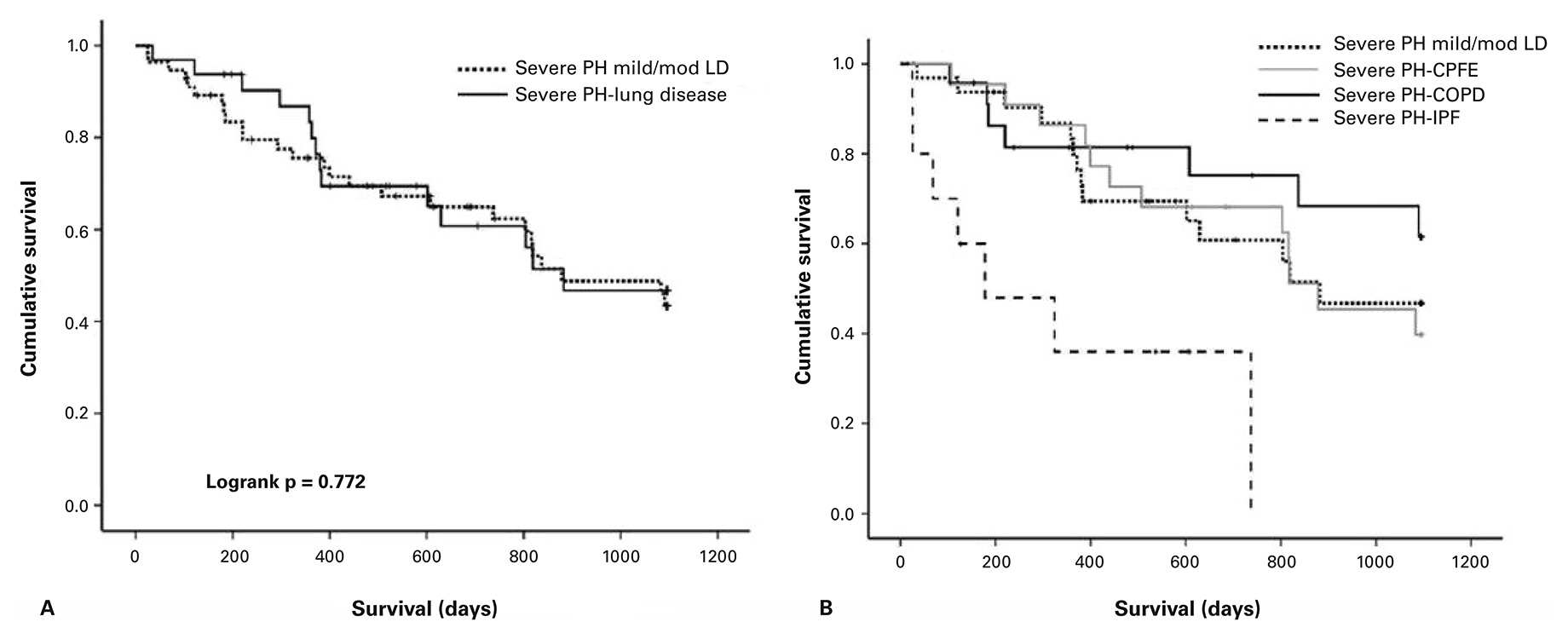
Figure 2
Survival by severity of lung disease.For patients fulfilling the definition of severe PH (mPAP >35 mm Hg or CI <2.0 l/min/m2) and mild/ moderate lung disease (FEV1 >60%), there was no difference in survival observed compared to severe PH/ severe lung disease (Panel A). On the contrary, in patients with severe PH, the underlying lung disease had a significant impact on survival (Panel B). (Reproduced with permission of the European Respiratory Society © Eur Respir J. 2015;46(5):1378–89; DOI: 10.1183/13993003.02307-2014)
CPFE = combined pulmonary fibrosis and emphysema; COPD = chronic obstructive pulmonary disease; ILD = interstitial lung disease; LD = lung disease; PH = pulmonary hypertension
Management of PH-COPD
For PH-COPD, smoking cessation, anti-obstructive treatment and pulmonary rehabilitation are the most important recommendations in all disease stages. As for exercise training, it has recently been shown in a prospective randomised controlled trial in patients with severe PH related to inoperable chronic thromboembolic disease that pulmonary resistance, that right ventricular function as well as CPET parameters and walking distance improved with rehabilitation [47]. Bronchodilators, in the form of long-acting muscarinic antagonists (LAMAs) and long-acting beta agonists (LABAs), often together with topical steroids, are indicated in advanced symptomatic COPD, newly defined GOLD class C (few symptoms, GOLD class 3–4 and/or 2 or more exacerbations per year) and D (more symptomatic, GOLD class 3–4 and/or 2 or more exacerbations per year). Interestingly, in COPD patients, it has been observed in a randomised trial that lung deflation is associated with consistent beneficial effects on cardiac function and pulmonary vasculature [48]. For advanced emphysematous COPD with PH, endoscopic lung volume reduction may be beneficial for both COPD and PH and is preferred over surgical interventions because of the associated perioperative risks in PH-COPD. As hypoxia triggers PH, supplemental oxygen at rest and upon exercise is recommended (class I, evidence level C) [1]. Even though long-term randomised controlled trials are not available, it has recently been shown in a double-blind cross-over trial that nocturnal oxygen was able to improve right ventricular function and walking distance after only 1 week of treatment in patients with precapillary PH and sleep-disturbed breathing [49]. Lung transplantation for PH-COPD is an option if other treatments fail, but post-transplant mortality seems to be increased [50].
PAH specific drugs for PH-COPD
Many trials to treat PH in patients with COPD by drugs targeting pulmonary vessel tone have been disappointing. Inhaled iloprost failed to improve exercise capacity in COPD patients with PH [51]. The endothelin receptor antagonist bosentan was not beneficial, but rather worsened arterial oxygen pressure and health-related quality of life in COPD patients without severe PH [52]. In contrast, sildenafil improved pulmonary haemodynamics in COPD patients with associated PH in a short term study, but long term investigations are lacking [53]. In a more recent study, patients with severe PH-COPD failed to improve functional class for dyspnoea or exercise performance [54]. In a pilot study, riociguat, a soluble guanylate cyclase stimulator, improved haemodynamics without impairing gas exchange in mild PH-COPD, but this needs to be confirmed in a larger study [55]. According to current knowledge and recommendations, specific drugs to induce vasodilatation of pulmonary arteries are thus not a successful treatment option for patients with PH-COPD with or without emphysema at this time [1]. Nevertheless, retrospective studies suggest that some COPD patients with relevant PH (mPAP >30 mm Hg) might profit from specific PH therapy, but additional studies need to be performed [56, 57].
Management of PH-ILD
For PH-COPD, smoking cessation, pulmonary rehabilitation and supplemental oxygen to maintain saturations above 90% at rest and upon exercise are recommended. In patients with nonspecific interstitial lung diseases, a trial of immunsuppressive therapy may be indicated. Anti-fibrotic treatment with pirfenidone and nintedanib have recently been approved for the treatment of IPF in Switzerland. These drugs may potentially stabilise PH in fibrotic lung disease by reduction of fibrosis progression. In addition, novel anti-fibrotic treatments, specifically tyrosine kinase inhibitors, target several of the overlapping signalling pathways between IPF and PH. Nintedanib inhibits VEGF and PDGF tyrosine kinases [58]. Unclear are the effects of VEGF inhibition by nintedanib in PH-IPF, as VEGF is beneficial for PH but might have pro-fibrotic effects [15]. Interestingly, tyrosine kinase inhibitors have previously shown to be promising in PAH treatment [59]. However, long-term safety data on treatment of PAH with imatinib showed severe adverse events and imatinib is no longer approved for PAH treatment [60]. Whether novel anti-fibrotic treatment impacts PH-IPF merits future investigation. Although anti-fibrotic treatments are now able to slow down disease progression in IPF, lung transplantation remains the ultimate treatment for respiratory failure in selected PH-IPF patients. A recent large retrospective analysis of a transplant registry showed no difference in post-transplant survival for IPF patients with or without associated PH [61].
PAH specific drugs in IPF and PH-IPF
In fibrotic lung disease, several studies have investigated the effects of specific PH treatment on PH and also on anti-fibrotic effects of the underlying parenchymal lung disease. For both aspects results have been disappointing. Some very interesting preliminary studies with encouraging results led to the hypothesis that inhaled vasodilators for fibrotic pulmonary disease may reach only well-ventilated areas [62, 63], but until today, these observations have not been published in larger studies. Bosentan, a dual endothelin receptor antagonist, was well tolerated in patients with IPF [10, 64], but failed to reduce progression of lung fibrosis in a phase III trial [11, 23]. Ambrisentan, a selective endothelin A antagonist, was investigated in a randomised clinical trial (ARTEMIS-IPF) for patients with IPF. The study was stopped early for lack of benefit and high likelihood of harm [12]. International guidelines recommend against the use of ambrisentan in IPF-associated PH [65].
Recent data from a large prospective registry (COMPERA) suggested short-term functional improvement in idiopathic ILD patients with PH treated with any vasodilating therapy, although impact on survival remains unclear [66]. Many of the included patients were treated with a phosphodiesterase-5 inhibitor (PDE5i), probably because a recent study of IPF patients with low diffusion capacity (STEP-IPF) suggested that ILD patients with PH might benefit from treatment with sildenafil [67]. In a small study with selected lung fibrosis patients responding to NO vasodilating treatment, in the acute setting sildenafil did not worsen gas exchange due to dilatation of areas with increased hypoxic pulmonary vasoconstriction [68]. Retrospective studies found possible survival benefits of patients with chronic lung disease and severe PH under PDE5i therapy [69].
However, these results need to be confirmed in large prospective controlled trials before this treatment may be recommended. In one recent phase II clinical study on the effects of riociguat in patients with PH associated with IIP (RISE-IIP), riociguat was associated with a possible increased risk for death and other serious adverse events as compared with patients in the placebo group, and the study was terminated by the independent drug monitoring committee [70].
Conclusion and current recommendations
In COPD, ILD (IPF) and CPFE, PH is more prevalent in advanced disease stages and is associated with more symptoms and worse prognosis. Current guidelines recommend that patients with suspected PH-COPD, PH-ILD and PH-CPFE are referred to expert centres with experience in assessment and management of both diseases [23]. Echocardiography is recommended for the non-invasive diagnostic assessment of suspected PH in patients with lung disease. For clarification of the differential diagnosis, lung function tests, chest HRCT, CPET and ventilation-perfusion scans may be helpful. The optimal treatment of the underlying lung disease including long-term oxygen therapy in patients with chronic hypoxaemia at rest or upon exertion is recommended in patients with PH due to lung diseases. Right heart catheterisation is not routinely recommended for suspected PH-COPD and PH-ILD, unless therapeutic consequences are to be expected. In summary, the use of drugs approved for PAH is not recommended in patients with PH due to chronic parenchymal lung diseases [1]. Further studies focusing on novel therapeutic approaches are therefore urgently needed.
References
1 Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–75.
2 Ma KC, Berlin DA. Pathophysiology of pulmonary hypertension in chronic parenchymal lung disease. Am J Med. 2016;129(4):366-71.
3 Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, Schulz R, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J. 2008;32(6):1639–51.
4 Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al., Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. 2014;189(3):314–24.
5 Bryant AJ, Carrick RP, McConaha ME, Jones BR, Shay SD, Moore CS, et al. Endothelial Hif Signaling Regulates Pulmonary Fibrosis-Associated Pulmonary Hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310(3):L249-62.
6 Marshall BE, Marshall C, Magno M, Lilagan P, Pietra GG. Influence of bronchial arterial PO2 on pulmonary vascular resistance. J Appl Physiol (1985). 1991;70(1):405–15.
7 Malenfant S, Neyron AS, Paulin R, Potus F, Meloche J, Provencher S, et al. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ. 2013;3(2):278–93.
8 Fonseca C, Abraham D, Renzoni EA. Endothelin in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;44(1):1–10.
9 Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016;71(1):73–83.
10 King TE Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(1):75–81.
11 King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–9.
12 Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–9.
13 Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J, Music Study Group. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622–32.
14 Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014;190(2):208–17.
15 Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al., Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434–45.
16 Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–38.
17 Clark JG, Madtes DK, and Raghu G, Effects of platelet-derived growth factor isoforms on human lung fibroblast proliferation and procollagen gene expression. Exp Lung Res. 1993;19(3):327–44.
18 Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15(4):255–73.
19 Lakatos HF, Thatcher TH, Kottmann RM, Garcia TM, Phipps RP, Sime PJ. The Role of PPARs in Lung Fibrosis. PPAR Res. 2007. 2007: p. 71323.
20 Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132(4):1311–21.
21 Russi EW, Karrer W, Brutsche M, Eich C, Fitting JW, Frey M, et al. Diagnosis and management of chronic obstructive pulmonary disease: the Swiss guidelines. Official guidelines of the Swiss Respiratory Society. Respiration. 2013;85(2):160–74.
22 Andersen KH, Iversen M, Kjaergaard J, Mortensen J, Nielsen-Kudsk JE, Bendstrup E, et al. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J Heart Lung Transplant. 2012;31(4):373–80.
23 Seeger W, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D109–16.
24 Weitzenblum E, Apprill M, Oswald M, Chaouat A, Imbs JL. Long-term course of pulmonary arterial pressure in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1984;130(6):993–8.
25 Zisman DA, Ross DJ, Belperio JA, Saggar R, Lynch JP 3rd, Ardehali A, et al. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2007;101(10):2153–9.
26 Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189–94.
27 Abraham AS, Cole RB, Green ID, Hedworth-Whitty RB, Clarke, SW, Bishop JM. Factors contributing to the reversible pulmonary hypertension of patients with acute respiratory failure studies by serial observations during recovery. Circ Res. 1969;24(1):51–60.
28 Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32(5):1371–85.
29 Weitzenblum E, Apprill M, Oswald M, Chaouat A, Imbs JL. Pulmonary hemodynamics in patients with chronic obstructive pulmonary disease before and during an episode of peripheral edema. Chest. 1994;105(5):1377–82.
30 Wells JM, Washko GR, Han MK, Abbas N, Nath H, Mamary AJ, et al. Pulmonary arterial enlargement and acute exacerbations of COPD. N Engl J Med. 2012;367(10):913–21.
31 Funke M, Geiser T. Idiopathic pulmonary fibrosis: the turning point is now! Swiss Med Wkly. 2015;145: p. w14139.
32 Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131(3):650–6.
33 Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, et al. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85(6):456–63.
34 Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76(3):288–94.
35 Minai OA, Santacruz JF, Alster JM, Budev MM, McCarthy K, et al. Impact of pulmonary hemodynamics on 6-min walk test in idiopathic pulmonary fibrosis. Respir Med. 2012;106(11):1613–21.
36 Raghu G, Nathan SD, Behr J, Brown KK, Egan JJ, Kawut SM, et al. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild-to-moderate restriction. Eur Respir J. 2015;46(5):1370–7.
37 Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30(4):715–21.
38 Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie-Leblond I, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586–93.
39 Cottin V, Le Pavec J, Prevot G, Mal H, Humbert M, Simonneau G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35(1):105–11.
40 Puhan MA, Siebeling L, Zoller M, Muggensturm P, ter Riet G. Simple functional performance tests and mortality in COPD. Eur Respir J. 2013;42(4):956–63.
41 Palazzuoli A, Ruocco G, Cekorja B, Pellegrini M, Del Castillo G, Nuti R. Combined BNP and Echocardiographic assessment in interstitial lung disease for pulmonary hypertension detection. Int J Cardiol. 2015;178:34–6.
42 Arcasoy SM, Christie JD, Ferrari VA, Sutton MS, Zisman DA. Blumenthal NP et al., Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167(5):735–40.
43 Caminati A, Cassandro R, Harari S. Pulmonary hypertension in chronic interstitial lung diseases. Eur Respir Rev. 2013;22(129):292–301.
44 Brewis MJ, Church AC, Johnson MK, Peacock AJ. Severe pulmonary hypertension in lung disease: phenotypes and response to treatment. Eur Respir J. 2015;46(5):1378–89.
45 Mueller-Mottet S, Stricker H, Domenighetti G, Azzola A, Geiser T, Schwerzmann M, et al. Long-term data from the Swiss pulmonary hypertension registry. Respiration. 2015;89(2):127–40.
46 Maeder MT KR, Weilenmann D, Schoch OD. Pulmonary hypertension associated with left heart and lung diseases. Cardiovascular Medicine. 2014;17(11):320–7.
47 Ehlken N, Lichtblau M, Klose H, Weidenhammer J, Fischer C, Nechwatal R, et al. Exercise training improves peak oxygen consumption and haemodynamics in patients with severe pulmonary arterial hypertension and inoperable chronic thrombo-embolic pulmonary hypertension: a prospective, randomized, controlled trial. Eur Heart J. 2016;37(1):35–44.
48 Stone IS, Barnes NC, James WY, Midwinter D, Boubertakh R, Follows R et al. Lung deflation and cardiovascular structure and function in chronic obstructive pulmonary disease. A randomized controlled trial. Am J Respir Crit Care Med. 2016;193(7):717–26.
49 Ulrich S, Keusch S, Hildenbrand FF, Lo Cascio C, Huber LC, Tanner FC, et al. Effect of nocturnal oxygen and acetazolamide on exercise performance in patients with pre-capillary pulmonary hypertension and sleep-disturbed breathing: randomized, double-blind, cross-over trial. Eur Heart J. 2015;36(10):615–23.
50 Singh VK, George MP, Gries CJ. Pulmonary hypertension is associated with increased post-lung transplant mortality risk in patients with chronic obstructive pulmonary disease. J Heart Lung Transplant. 2015;34(3):424–9.
51 Boeck L, Tamm M, Grendelmeier P, Stolz D. Acute effects of aerosolized iloprost in COPD related pulmonary hypertension – a randomized controlled crossover trial. PLoS One. 2012;7(12):e52248.
52 Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. 2008;32(3):619–28.
53 Blanco I, Gimeno E, Munoz PA, Pizarro S, Gistau C, Rodriguez-Roisin R, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. 2010;181(3):270–8.
54 Girard A, Jouneau S, Chabanne C, Khouatra C, Lannes M, Traclet J, et al. Severe pulmonary hypertension associated with COPD: hemodynamic improvement with specific therapy. Respiration. 2015;90(3):220–8.
55 Ghofrani HA, Staehler G, Grunig E, Halank M, Mitrovic V, Unger S, et al. Acute effects of riociguat in borderline or manifest pulmonary hypertension associated with chronic obstructive pulmonary disease. Pulm Circ. 2015;5(2):296–304.
56 Fossati L, Muller-Mottet S, Hasler E, Speich R, Bloch KE, Huber LC, et al. Long-term effect of vasodilator therapy in pulmonary hypertension due to COPD: a retrospective analysis. Lung. 2014;192(6):987–95.
57 Lange TJ, Baron M, Seiler I, Arzt M, Pfeifer M. Outcome of patients with severe PH due to lung disease with and without targeted therapy. Cardiovasc Ther. 2014;32(5):202–8.
58 Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
59 Speich R, Ulrich S, Domenighetti G, Huber LC, Fischler M, Treder U, et al. Efficacy and Safety of Long-Term Imatinib Therapy for Pulmonary Arterial Hypertension. Respiration. 2015;89(6):515–24.
60 Frost AE, Barst RJ, Hoeper MM, Chang HJ, Frantz RP, Fukumoto Y, et al. Long-term safety and efficacy of imatinib in pulmonary arterial hypertension. J Heart Lung Transplant. 2015;34(11):1366–75.
61 Hayes D Jr, Higgins RS, Black SM, Wehr AM, Lehman AM, Kirkby S, et al. Effect of pulmonary hypertension on survival in patients with idiopathic pulmonary fibrosis after lung transplantation: an analysis of the United Network of Organ Sharing registry. J Heart Lung Transplant. 2015;34(3):430–7.
62 Olschewski H, Ghofrani HA, Walmrath D, Schermuly R, Temmesfeld-Wollbruck, B, Grimminger F, et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis. Am J Respir Crit Care Med. 1999;160(2):600–7.
63 Blanco I, Ribas J, Xaubet A, Gomez FP, Roca J, Rodriguez-Roisin R, et al. Effects of inhaled nitric oxide at rest and during exercise in idiopathic pulmonary fibrosis. J Appl Physiol. (1985), 2011;110(3):638–45.
64 Gunther A, Enke B, Markart P, Hammerl P, Morr H, Behr J, et al. Safety and tolerability of bosentan in idiopathic pulmonary fibrosis: an open label study. Eur Respir J. 2007;29(4):713–9.
65 Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19.
66 Hoeper MM, Behr J, Held M, Grunig E, Vizza CD, Vonk-Noordegraaf A, et al. Pulmonary hypertension in patients with chronic fibrosing idiopathic interstitial pneumonias. PLoS One. 2015;10(12):e0141911.
67 Idiopathic Pulmonary Fibrosis Clinical Research Network. Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–8.
68 Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360(9337):895–900.
69 Tanabe N, Taniguchi H, Tsujino I, Sakamaki F, Emoto, N, et al. Multi-institutional retrospective cohort study of patients with severe pulmonary hypertension associated with respiratory diseases. Respirology. 2015;20(5):805–12.
70 Official press release. [updated 2016 May 12; cited 2016 Jul 4]. Accessed 4.6.2016 at: http://www.prnewswire.com/news-releases/bayer-terminates-phase-ii-study-with-riociguat-in-patients-with-pulmonary-hypertension-associated-with-idiopathic-interstitial-pneumonias-300267616.html.