Regulatory T cells: balancing protection versus pathology
DOI: https://doi.org/10.4414/smw.2016.14343
Nikolas
Rakebrandt, Katharina
Littringer, Nicole
Joller
Summary
Foxp3+ regulatory T cells (Tregs) maintain immune tolerance, prevent autoimmunity and modulate immune responses during infection and cancer. Recent studies have revealed considerable heterogeneity and plasticity within the Treg compartment, depending on the immunological context, which may result in Tregs losing their suppressive function in inflammatory environments. We review how dysfunctional Tregs contribute to disease pathogenesis in inflammatory conditions and how inappropriate regulatory responses may hamper protective immunity in the context of infection and cancer. We also discuss how Tregs might be targeted therapeutically to re-establish a proper balance between regulatory and effector responses in autoimmunity, infections, and cancer.
Introduction
Regulatory T cells (Tregs) are a subset of CD4+ T cells that are crucial for immune homeostasis. Tregs are defined by their expression of the transcription factor forkhead-box protein P3 (Foxp3), which is essential for their development and suppressive function [1]. Loss of Foxp3 function leads to severe lymphoproliferative disease and autoimmunity, and highlights the essential role of Tregs in maintaining immune tolerance [2, 3]. In addition to preventing autoimmunity and inflammatory diseases, Tregs ensure a controlled immune response upon pathogen encounter and thereby prevent immune pathology. Conversely, excessive suppression by Tregs can hamper pathogen clearance and promote chronic infection [4, 5]. In addition, Tregs can also restrain anti-tumour immune responses and thus promote tumour progression [6]. Properly balanced Treg function and activation is therefore essential to prevent immune pathology but allow for protective immune responses against tumours and pathogens.
To accomplish these tasks, Tregs must adapt to their immune environment and specialise into subsets with distinct functional properties that work together to ensure an adequate immune response while maintaining immune tolerance [7]. However, the plasticity required for this specialisation also bears the risk of instability, and recent studies have revealed the ability of Tregs to loose suppressive and acquire effector function [8, 9]. These observations reveal a potential causative role of unstable or dysfunctional Tregs in inflammatory settings. Therefore, understanding the factors that control Treg stability, plasticity and function is an important step towards improving safety and efficacy of therapeutic applications directed against Tregs. In this review, we discuss the role of Tregs in autoimmunity, infectious diseases, and cancer by addressing: the importance of a fine-tuned regulatory response for maintaining a healthy balance between effector and regulatory T cell responses; Treg specialisation and heterogeneity and its impact on Treg function and stability in inflammatory settings; therapeutic approaches used to manipulate Treg numbers and function.
Regulatory T cells in autoimmunity
The importance of Tregs in preventing autoimmunity becomes evident in patients with IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome), who suffer from multi-organ autoimmunity because of a mutation in the FOXP3 gene and hence impaired Treg function [2, 3]. In addition, perturbations in the pathways of the Treg suppressive network, including the interleukin (IL)-2 / IL-2 receptor α-chain (IL-2Rα, CD25) axis or the co-inhibitory receptor CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), predispose to autoimmune disorders [10, 11]. Whereas it is generally accepted that Tregs are essential for maintaining self-tolerance, studies addressing whether altered Treg frequencies and/or function affect the disease course or severity of e.g. type 1 diabetes [12], systemic lupus erythematosus [13], or rheumatoid arthritis [14] have led to largely confounding findings. However, differences in the markers used to define the Treg population and hence the purity and composition of the analysed Treg population might in part account for the divergent findings, and highlight the importance of a standardised set of markers that allows for cross-comparison between independent studies (table 1). Nowadays a general consensus exists that in humans FoxP3-expressing Tregs are most accurately defined as CD4+CD25+CD127- cells [15, 16].
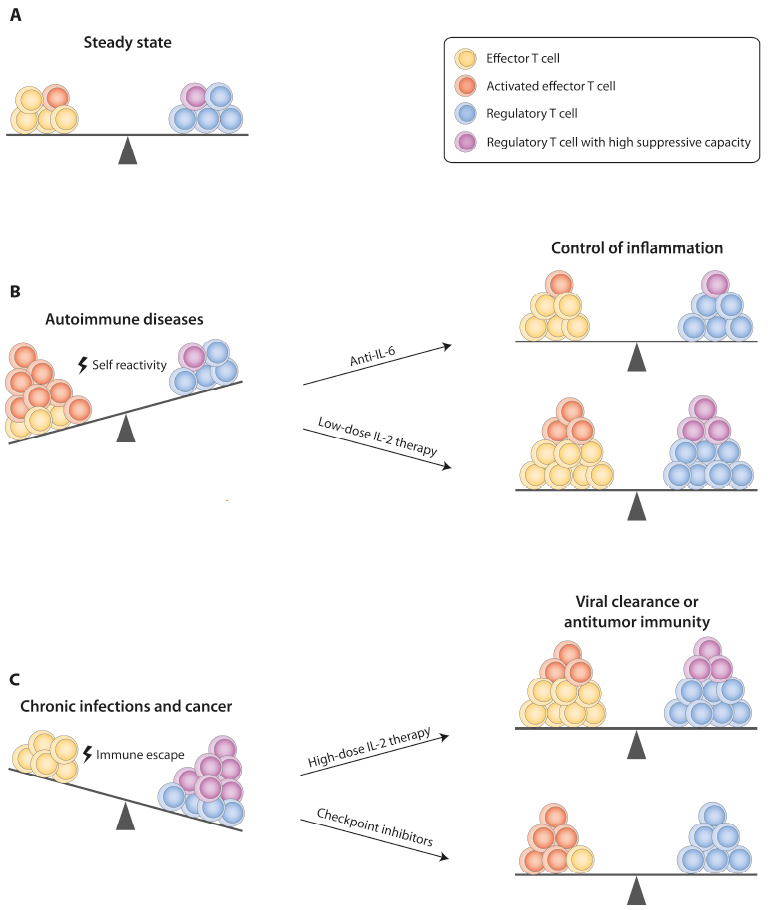
Figure 1
Therapeutic targeting of the effector T cell–Treg balance.Depicted are approaches that can restore a dysfunctional ratio of effector T cells and Tregs in different diseases to the healthy state (A). (B) In autoimmune diseases, self-tolerance is lost and autoreactive effector T cells expand. In Th17-driven autoimmune diseases, blocking of IL-6 signalling favours Treg and inhibits Th17 differentiation. Low-dose IL-2 therapy, which leads to expansion of Tregs and thus restores a healthy effector T cell–Treg balance represents a more general approach for treatment of autoimmunity. (C) Application of high-dose IL-2 results in expansion of effector T cells and induction of antitumor immune responses in cancer patients. Checkpoint inhibitors block signalling through inhibitory receptors such as PD-1 or CTLA-4 and can thus reverse T cell exhaustion, which favours immune evasion of tumour cells or pathogens. Blockade of these pathways can additionally support the function of effector T cells by reducing the suppressive capacity of Tregs.
CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; IL = interleukin; PD-1 = programmed death 1; Th = T helper cell; Treg = regulatory T cell
The role of Tregs defined on the basis of these markers has been studied most extensively in multiple sclerosis. Multiple sclerosis patients with relapsing-remitting disease have reduced frequencies of Tregs with impaired suppressive capacity [17, 18]. Decreased expression levels of FoxP3 [17] as well as of CTLA-4 [19] detected in multiple sclerosis patients were attributed to Treg dysfunction. Importantly, treatment of multiple sclerosis with interferon-beta (IFN-β) [17] and glatiramer acetate [20] restores Treg numbers and FoxP3 expression levels, supporting a protective role of Tregs in multiple sclerosis.
Within their reduced Treg pool, patients with multiple sclerosis show an accumulation ofTh1-like Tregs that produce interferon-gamma (IFN-γ) [21] (Th1: type 1 T helper cell; table 2). The production of IFN-γ in these Tregs was accompanied by upregulation of the Th1 master transcription factor T-bet and loss of suppressive function. Blockade of IFN-γ could re-establish the suppressive capacity of the Tregs while the initial induction of the Th1-like phenotype was dependent on IL-12 signalling. Importantly, IFN-β treatment reduced frequencies of IFN-γ-producing Th1-Tregs in multiple sclerosis patients to normal levels [21]. Similar findings were reported in patients with type 1 diabetes [22], supporting a protective role of Tregs in the disease.
Mouse models in which Tregs lack the ability to co-express Foxp3 together with effector Th lineage-specific transcription factors revealed the ability of Tregs to specialise during the course of inflammatory responses. Driven by the corresponding cytokine environment, co-expression of T-bet or Stat3 was shown to be indispensable for control of Th1 or Th17 effector T cell responses, respectively [23, 24]. In the mouse model, delayed induction of the IL-12Rβ2 chain prevented Th1-specialised Tregs from fully differentiating into potentially pathogenic IFN-γ-producing Tregs [25]. The appearance of IFN-γ-producing Tregs in patients with multiple sclerosis suggests that this pathway is defective in these patients. A recent study reported the accumulation of Tregs expressing T-bet in the central nervous system of mice during experimental autoimmune encephalomyelitis, the mouse model of multiple sclerosis. However, in contrast to the initial reports, T-bet deficiency in Tregs had no impact on the control of the proinflammatory T cell response in this study [26]. These findings indicate that specialisation might not be universally required for Treg function and homing. It will thus be important to evaluate whether Treg specialisation is beneficial or potentially harmful in different inflammatory diseases.
In patients with rheumatoid arthritis, Tregs are counterintuitively enriched in the synovial fluid, the site of inflammation [27, 28], raising the question of whether they are dysfunctional and thus unable to prevent disease. When examined for their functional properties, Tregs isolated from the synovial fluid of patients with rheumatoid arthritis retained their ability to suppress effector T cell proliferation in vitro [27, 28], but displayed defects in suppression of effector cytokine production (table 2) [14]. Furthermore, the inflammatory environment in rheumatoid arthritis, particularly the presence of tumour necrosis factor-alpha (TNF-α), led to a loss in the suppressive function of Tregs [14, 29]. Whether this is also the case in other autoimmune settings is still unclear [30]. Parallel to the IFN-γ-producing Th1-Tregs observed in multiple sclerosis, Tregs from rheumatoid arthritis patients have the potential to secrete the proinflammatory cytokine IL-17 when re-stimulated ex vivo [31]. However, in contrast to IFN-γ-producing Th1-Tregs, they retain their ability to suppress effector cell proliferation in vitro[32, 33]. IL-17-secreting FoxP3+ Tregs were first described under steady-state conditions, and co-express FoxP3 and the Th17 master transcription factor RORγt (retinoic acid receptor-related orphan receptor gamma-t) [32–34]. Despite the fact that they retain their suppressive capacity, FoxP3+RORγt+ Tregs secrete IL-17 in response to IL-1β or IL-6 and thereby potentially contribute to inflammation [32, 33]. The relevance of these Th17-like Treg cells in autoimmune inflammation still remains unclear, but again highlights the importance of considering Treg heterogeneity and subset distribution in disease settings.
The pathological importance of Treg plasticity and the ability of Tregs to adapt effector function under inflammatory conditions were investigated in the mouse model of collagen-induced arthritis. Exposure of CD25lowFoxp3+ T cells to high levels of IL-6, as encountered during arthritis, resulted in a loss of Foxp3 expression in this still plastic population and thus converted them into IL-17-secreting T cells driving pathology [35]. This highlights the importance of Treg stability when addressing the imbalance between effector T cells and Tregs in inflammatory diseases. A number of studies have focused on the question of whether Tregs are stable or if they can convert into other potentially pathogenic T cell phenotypes. In addition to sustained expression of Foxp3 and CD25, the unique epigenetic signature of Tregs, marked by hypomethylation of CpG motifs in key genes such as the Foxp3 conserved noncoding sequence 2 (CNS2), Il2ra(CD25), Ikzf4(Eos) andCtla4, ensures Treg stability and maintenance of a functional Treg phenotype [36–38]. The epigenetic programming of Tregs may even ensure their long-term stability when Foxp3 expression is transiently lost [39, 40]. However, this topic is still somewhat controversial and some studies suggest that Tregs can permanently lose Foxp3 expression under inflammatory conditions and convert to “ex-Tregs” exhibiting a pathogenic phenotype [41, 42]. Whether these “ex-Tregs” actually underwent epigenetic reprogramming or rather represent precursors that only transiently upregulated Foxp3 is also not entirely clear. Regardless of this debate, a better understanding of Treg stability is a prerequisite for the development of Treg cell therapy, especially for treatment in inflammatory settings.
On the basis of these observations it is clear that in both Th1- and Th17-dominated autoimmune settings, Treg numbers as well as their function are critical determinants for disease progression. Therapeutic approaches should thus aim at restoring a healthy balance between effector cells and functional Tregs (fig. 1B). Th17 and Treg cells share the dependence on transforming growth factor-beta (TGF-β) for their development, but the presence of IL-6 drives their differentiation towards the Th17 lineage [43–45]. Interference with the IL-6-derived signal, for example with a blocking antibody to the IL-6 receptor (tocilizumab), thus inhibits Th17 but supports Treg differentiation. This therapeutic approach has successfully been used in rheumatoid arthritis, where treatment resulted in an increase in Treg but not Th17 numbers, restoring the Treg/Th17 balance [46].
Ongoing studies on IL-2 treatment, as key cytokine in Treg development and maintenance, have demonstrated a specific activation and expansion of Tregs, but not effector T cells, when low doses of IL-2 are administered, suggesting a more general approach for modulation of the T effector / Treg balance to treat autoimmune diseases [47]. Similarly, adoptive immunotherapy with ex-vivo expanded CD4+CD25+CD127- polyclonal Tregs is another interesting approach to restoring the T effector / Treg balance in multiple autoimmune diseases [48]. However, many open issues regarding the stability and functionality of Tregs expanded in vitro still need to be addressed before this approach could become broadly applicable.
In summary, Tregs are a very heterogeneous population harbouring subsets with a high level of plasticity and possibly variable thresholds for stability under inflammatory conditions. The assessment of Tregs defined by their stability and suppressive properties in the relevant inflammatory setting of each autoimmune disease will be key in ensuring the efficacy of re-establishing the T effector / Treg balance through therapeutic approaches.
|
Table 1: Marker combinations to identify regulatory T cells (Tregs). |
|
Marker combination
|
Represented population
|
Organism
|
Comments
|
References
|
| CD4+CD25+
|
Tregs and activated effector T cells |
Mouse/human |
Activated effector T cells upregulate CD25. |
|
| CD4+CD25highFoxP3+
|
Tregs |
Mouse |
Exclusive marker combination to identify Tregs. |
|
| Tregs and activated effector T cells |
Human |
Activated effector T cells transiently upregulate FoxP3. |
|
| CD4+CD25highCD127–
|
Primarily Tregs |
Human |
Most accurate marker combination to identify FoxP3+ Treg cells. Combination widely-used for Treg isolation when cell permeabilisation for FoxP3 detection is not possible (e.g., for functional assays). |
[15, 16, 48] |
|
Table 2: Subsets of human regulatory T cells (Tregs) and their potential role in autoimmune disease. |
|
Markers
|
Function
|
References
|
|
FoxP3
|
CD25
|
Other markers
|
Suppression
|
Effector cytokines
|
Comments
|
| + |
++ |
CD45RA+CTLA-4low
|
+++ |
– |
Resting or naive Tregs |
|
| +++ |
+++ |
CD45RA–CTLA-4high
|
+++ |
– |
Effector or activated Tregs |
|
| + |
++ |
CD45RA–CTLA-4int
|
– |
IL-2, IFN-γ |
Treg precursor, may lose FoxP3 expression |
|
| +++ |
+++ |
CD45RA–CCR6+CD45RA–HLA-DR–CCR6+CD45RA–CD27+CCR6+
|
+++ |
IL-17 (express RORγt) |
|
[32–34] |
| +++ |
+++ |
CD45RA–CD127–
|
+ |
IFN-γ (express T-bet, CXCR3) |
Subset increased in MS and T1D patients. |
[21, 22] |
| CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; CXCR3 = C-X-C motif chemokine receptor type 3; FoxP3 = forkhead-box protein P3; IFN = interferon; IL = interleukin; MS = multiple sclerosis; RORγt = retinoic acid receptor-related orphan receptor gamma-t; T1D = type 1 diabetes |
Regulatory T cells in infectious diseases
Pathogens and nascent transformed cells that can progress to cancer pose a constant threat to the host, which must be kept in check through appropriate immune responses. However, excessive responses resulting from failure to adequately control the magnitude and extent of the response can result in collateral damage of affected tissues and organs, also known as immunopathology [49]. A number of studies have shown that Tregs can help to minimise such immune-mediated tissue damage. These focused on infectious settings that can result in immunopathology under certain conditions. As of now, only a limited number of studies have addressed the tissue protective role of Tregs in infections in humans. However, observations made in infectious mouse models provide valuable insight on the mechanisms contributing to immune pathology and outline potential therapeutic approaches targeting Tregs in humans.
Persistent herpes simplex virus infection can result in a form of keratitis in the human eye, in which excessive T cell responses cause lesions in the cornea ultimately leading to blindness. In the mouse model, Tregs were able to reduce the severity of viral inflammatory lesions by dampening the Th1 response and influx of CD4+ effector T cells into the cornea, demonstrating the tissue protective role of Tregs in this setting [50]. Similarly, in Helicobacter hepaticus infection, a model of microbe-associated gut inflammation, adoptively transferred Tregs were also able to inhibit T cell-mediated and innate intestinal inflammation [51]. In line with these results, Helicobacter pylori-colonised patients who developed peptic ulcer disease displayed reduced Treg function compared with patients without peptic ulcer disease, suggesting a tissue protective role for Tregs also in human settings [52].
While insufficient suppression by Tregs can favour tissue damage, an imbalance in effector versus regulatory response in favour of inhibitory Treg function can promote pathogen persistence. Consequently, enhanced Treg function or numbers have been linked to poor effector T cell function and pathogen clearance in a number of chronic infections in mice and humans. During retroviral infection of mice inhibition of Treg function by anti-glucocorticoid-induced tumour necrosis factor receptor (anti-GITR) treatment can improve CD8+ T cell function and reduce viral load [53]. This indicates that suppression by Tregs hampers CD8+ T cell responses and thereby promotes viral persistence. In patients with human immunodeficiency virus (HIV) infection, a correlation between viral load and Foxp3 expression in T cells has also been suggested [54]. In extreme cases, inhibition of effector responses by Tregs can lead to host death, as demonstrated in mice infected with the malaria parasite Plasmodium yoelii [55]. Expansion of Tregs and TGF-β signalling also correlates with higher rates of parasite growth in malaria patients, pointing towards a similar role of Tregs in promoting parasite persistence in humans [56].
Tregs have repeatedly been linked to T cell exhaustion, which is commonly observed in chronic infections and characterised by reduced effector function and high expression of inhibitory receptors [57]. This is, for instance, the case in mice chronically infected with lymphocytic choriomeningitis virus (LCMV), where Treg depletion and blockade of the programmed death 1 / programmed death ligand 1 (PD-1/PD-L1) pathway results in recovery of CD8+ T cell effector functions and the reduction of viral titres [58]. Treg-mediated immune suppression may represent a general mechanism for immune evasion during establishment of chronic viral infection [59]. How this can be achieved in an immunological sense is still largely unknown [57]. One possible antagonist of exhaustion is the proinflammatory cytokine IL-21, as absence of IL-21 signalling leads to expansion of Tregs and promotes CD8+ T cell exhaustion during chronic LCMV infection [60]. In the latter study it was also demonstrated that depletion of Tregs during chronic viral infection can lead to lethal immunopathology, highlighting again the importance of an equilibrium between inhibitory and effector immune mechanisms.
Many studies focused on Tregs during chronic infections, but their role in acute infections seemed to be minor and has so far been subject of only a few studies [5]. However, recent reports show that the involvement of Tregs in immune responses during acute infections might indeed be more complex than initially thought. Although depletion of Tregs has no significant effect on the course of primary influenza infection in mice [61], another study observed the emergence of antigen-specific Tregs that persisted after acute infection and were tissue protective in a homologous challenge [62]. Furthermore, through their secretion of amphiregulin in response to the alarmins IL-33 and IL-18, Tregs play a direct role in tissue protection and repair [63–65]. These observations showcase the diverse functions of Tregs during acute infections, and illustrate that their role in this setting is still only poorly understood and probably depends on various factors, including the pathogen itself as well as the site of infection.
Regulatory T cells in cancer
Immune surveillance is essential for eradication of aberrant cells and prevention of their progression to cancer. Based on their regulatory function, an involvement of Tregs in tumour progression was long suspected [66]. In some human cancers, high prevalence of Tregs and low numbers of CD8+ effector T cells are associated with poor prognosis (reviewed in [6]). To date there is still relatively little known about the function and plasticity of tumour-associated Tregs. Nevertheless, Tregs are thought to inhibit potent anti-tumour responses under certain conditions and many approaches to the development of cancer immunotherapies focused on the induction of anti-tumour immunity by reducing the suppressive capacity of Tregs.
A now widely utilised cancer immunotherapy is high-dose IL-2. This has been successfully applied for treatment of metastatic melanoma and renal cell carcinoma [67, 68]. Since IL-2 can promote effector T cell differentiation and stimulate natural killer (NK) cells, the prolonged survival in treated patients may be attributed to increased anti-tumour immune responses [69]. A drawback of this treatment lies in the high doses of IL-2 required, which can lead to serious side effects [69]. However, high doses seem to be necessary for the induction of anti-tumour immune responses, as low doses preferentially expand Tregs owing to their expression of the high-affinity IL-2 receptor, which includes the IL-2 receptor α-chain (CD25) [47]. In contrast, effector T cells are mostly restricted to expressing the low-affinity IL-2 receptor and thus require higher concentrations of IL-2 for activation (fig. 1C). The differential expression of high versus low affinity IL-2 receptors in effector T cells and Tregs might, under certain circumstances, even interfere with the responsiveness of patients to high-dose IL-2 treatment. For instance, one study reported the expansion of an activated Treg subset in melanoma patients treated with high-dose IL-2 [70]. This study also came to the conclusion that more prominent expansion of this Treg subset predicted poorer responsiveness towards high-dose IL-2 therapy. To counteract these unwanted effects, it may be beneficial to combine IL-2 treatment with depletion or inhibition of Tregs. This could potentially increase the efficacy of IL-2 in triggering an anti-tumour response and hence might even allow for administration of less toxic IL-2 dosages. The principle of Treg depletion as a possible cancer treatment was already explored over a decade ago, and was shown to slow tumour growth and enhance anti-tumour immunity [71, 72]. However, when considering Treg depletion via the CD25-blocking antibody daclizumab, it is important to note that CD25 is also expressed on activated effector T cells and, depending on the timing and dose, daclizumab might also deplete effector T cells. Furthermore, Treg depletion generally harbours the risk of causing autoimmunity. Selective stimulation of specific lymphocyte subsets by use of IL-2-antibody complexes has also been suggested as an alternative to classic IL-2 immunotherapy in order to minimise its shortcomings [69]. Depending on the antibody clone, IL-2-antibody complexes direct activity of IL-2 towards CD25+ cells such as Tregs or selectively expand CD122high effector cells in mice [73, 74]. Therefore, the use of IL-2-antibody complexes might facilitate a more tailored approach to balancing the Treg / effector T cell ratio therapeutically.
Targeting of immune checkpoint inhibitors represents another promising approach to activate anti-tumour immune responses (fig. 1C). Current immunotherapies are directed against the co-inhibitory receptors CTLA-4 and PD-1, and exhibit unprecedented efficacy in several cancer indications [75]. Combination therapy against CTLA-4 (ipilimumab) and PD-1 (nivolumab) was shown to have synergistic effects in melanoma patients and significantly prolonged progression-free survival [76]. In the mouse model, this combined treatment resulted in increased tumour infiltration of cytotoxic T cells, reduced frequencies of Tregs and, ultimately, tumour regression in up to 75% of mice [77]. These results highlight the similarity between the T cell responses in cancer and chronic infection, where the same approach results in superior viral clearance [58]. Importantly, these co-inhibitory molecules are also highly expressed on Tregs, where they contribute to Treg suppressive function. Immune checkpoint blockade is therefore likely to both interfere with Treg function and enhance effector responses, and thereby promote the effector response through two synergistic pathways. This dual action of checkpoint inhibitors was investigated in detail in a recent set of studies on the novel co-inhibitory receptor TIGIT (T cell immunoglobulin and immunoreceptor tyrosine-based inhibition motif [ITIM] domain), which recently gained attention as another potential checkpoint inhibitor for cancer therapy. Like CTLA-4 and PD-1, TIGIT is expressed on effector as well as regulatory T cells, and mechanistic studies in mice revealed that anti-TIGIT therapy affects both CD8+ T cell responses as well as Tregs, supporting an impact of checkpoint inhibitors on both effector as well as regulatory T cell function [78–80].
As illustrated by both IL-2 treatment and the checkpoint inhibitor approaches, a key aspect in effective cancer immunotherapies lies in skewing the balance of Tregs and effector T cells towards an increased effector response. A better understanding of the dynamics of marker expression on both effector and regulatory T cells in the different disease settings, as well as a detailed assessment of their expression patterns in patients will be an important step to improve therapeutic efficacy and minimise side effects.
Conclusions
The studies discussed here emphasise the broad impact Tregs have on the development of tolerance versus immunity. Consequently their manipulation harbours enormous therapeutic potential for the treatment of a wide spectrum of diseases ranging from autoimmunity to chronic infection and cancer. However, in order to realise this potential, a better understanding of the underlying Treg biology is essential. In particular, deeper insight into the mechanisms that govern Treg stability, plasticity, and specialisation will be necessary to allow tailored manipulation of Treg numbers and function aimed at re-establishing a healthy balance between tolerance and protective immunity in this broad range of diseases.
References
1 Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6.
2 Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanov, JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20.
3 Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1.
4 Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7(11):875–88.
5 Veiga-Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev. 2013;255(1):182–96.
6 Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127(4):759–67.
7 Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11(2):119–30.
8 Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. 2014;259(1):159–72.
9 Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. 2014;259(1):173–91.
10 Maier LM, Lowe CE, Cooper J, Downes K, Anderson DE, Severson C, et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009;5(1):e1000322.
11 Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423(6939):506–11.
12 Battaglia M, Roncarolo MG. Immune intervention with T regulatory cells: past lessons and future perspectives for type 1 diabetes. Semin Immunol. 2011;23(3):182–94.
13 Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45(2):344–55.
14 Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200(3):277–85.
15 Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7):1701–11.
16 Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–700.
17 Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens JL, et al. Compromised CD4+ CD25(high) regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. 2008;123(1):79–89.
18 Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–9.
19 Sellebjerg F, Krakauer M, Khademi M, Olsson T, Sorensen PS. FOXP3, CBLB and ITCH gene expression and cytotoxic T lymphocyte antigen 4 expression on CD4(+) CD25(high) T cells in multiple sclerosis. Clin Exp Immunol. 2012;170(2):149–55.
20 Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci U S A. 2005;102(18):6449–54.
21 Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. 2011;17(6):673–5.
22 McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol. 2011;186(7):3918–26.
23 Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell, DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10(6):595–602.
24 Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326(5955):986–91.
25 Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37(3):501–10.
26 McPherson RC, Turner DG, Mair I, O’Connor RA, Anderton SM. T-bet Expression by Foxp3(+) T Regulatory Cells is Not Essential for Their Suppressive Function in CNS Autoimmune Disease or Colitis. Front Immunol. 2015;6:69.
27 Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6(4):R335–46.
28 Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, et al. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201(11):1793–803.
29 Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108(1):253–61.
30 Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest. 2010;120(12):4558–68.
31 Afzali B, Mitchell PJ, Edozie FC, Povoleri GA, Dowson SE, Demandt L, et al. CD161 expression characterizes a subpopulation of human regulatory T cells that produces IL-17 in a STAT3-dependent manner. Eur J Immunol. 2013;43(8):2043–54.
32 Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood. 2009;113(18):4240–9.
33 Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112(6):2340–52.
34 Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci U S A. 2009;106(21):8635–40.
35 Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–8.
36 Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37(5):785–99.
37 Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science. 2010;329(5999):1667–71.
38 Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463(7282):808–12.
39 Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-Driven Activation Induces Instability of Regulatory T Cells during an Inflammatory Autoimmune Response. Immunity. 2013;39(5):949–62.
40 Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36(2):262–75.
41 Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. Pathogenic conversion of Foxp3(+) T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–8.
42 Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10(9):1000–7.
43 Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–8.
44 Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4.
45 Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89.
46 Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(8):2499–503.
47 Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. 2015;15(5):283–94.
48 Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315):315ra189.
49 Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335(6071):936–41.
50 Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172(7):4123–32.
51 Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197(1):111–9.
52 Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57(10):1375–85.
53 Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, et al. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20(3):293–303.
54 Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, et al. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J Immunol. 2005;174(6):3143–7.
55 Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, et al. Escape of malaria parasites from host immunity requires CD4+ CD25+ regulatory T cells. Nat Med. 2004;10(1):29–30.
56 Walther M, Tongren JE, Andrews L, Korbel D, King E, Fletcher H, et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity. 2005;23(3):287–96.
57 Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
58 Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. 2014;211(9):1905–18.
59 Zelinskyy G, Dietze K, Sparwasser T, Dittmer U. Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog. 2009;5(8):e1000406.
60 Schmitz, I, Schneider, C, Frohlich, A, Frebel, H, Christ, D, Leonard, WJ, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog, 2013. 9(5): p. e1003362.
61 Betts RJ, Ho AW, Kemeny DM. Partial depletion of natural CD4(+)CD25(+) regulatory T cells with anti-CD25 antibody does not alter the course of acute influenza A virus infection. PLoS One. 2011;6(11):e27849.
62 Brincks EL, Roberts AD, Cookenham T, Sell S, Kohlmeier JE, Blackman MA, et al. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J Immunol. 2013;190(7):3438–46.
63 Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162(5):1078–89.
64 Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–95.
65 Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, et al. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44(2):355–67.
66 Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol. 2013;171(1):36–45.
67 Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, et al. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113(2):293–301.
68 Smith FO, Downey SG, Klapper JA, Yang JC, Sherry RM, Royal RE, et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin Cancer Res. 2008;14(17):5610–8.
69 Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–90.
70 Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014;124(1):99–110.
71 Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59(13):3128–33.
72 Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ, Jr., et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134ra62.
73 Boyman O, Surh CD, Sprent J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther. 2006;6(12):1323–31.
74 Letourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, et al. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc Natl Acad Sci U S A. 2010;107(5):2171–6.
75 Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561–84.
76 Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373(1):23–34.
77 Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73(12):3591–603.
78 Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–58.
79 Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell. 2014;26(6):923–37.
80 Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053–62.