Identification and molecular characterisation of Lausanne Institutional Biobank participants with familial hypercholesterolaemia – a proof-of-concept study
DOI: https://doi.org/10.4414/smw.2016.14326
Fabienne
Maurer, Sylvain
Pradervand, Isabelle
Guilleret, David
Nanchen, Ali
Maghraoui, Laurence
Chapatte, Karolina
Bojkowska, Zahurul Alam
Bhuiyan, Nathalie
Jacquemont, Keith
Harshman, Vincent
Mooser
Summary
AIMS: We aimed to identify familial hypercholesterolaemia mutation carriers among participants to the Lausanne Institutional Biobank (BIL). Our experimental workflow was designed as a proof-of-concept demonstration of the resources and services provided by our integrated institutional clinical research support platform.
METHODS: Familial hypercholesterolaemia was used as a model of a relatively common yet often underdiagnosed and inadequately treated Mendelian disease. Clinical and laboratory information was extracted from electronic hospital records. Patients were selected using elevated plasma cholesterol levels (total cholesterol ≥7.5 mM or low-density lipoprotein cholesterol ≥5 mM), premature coronary artery disease status and age (18–60 yr) as main inclusion criteria. LDLR, APOB and PCSK9were analysed by high-throughput DNA sequencing. The most relevant mutations were confirmed by Sanger sequencing.
RESULTS: Of 23 737 patients contacted by the BIL, 17 760 individuals consented to participate and 13 094 wished to be recontacted if there were findings requiring clinical action. Plasma cholesterol records were available for 5111 participants, of whom 94 were selected for genetic screening. Twenty-five of the tested patients presented with premature coronary artery disease while 69 had no such diagnosis. Seven heterozygous carriers of eight rare coding missense variants were identified. Three mutations were pathogenic (APOB p.R3527Q) or likely pathogenic (LDLR p.C27W, LDLR p.P526S) for hypercholesterolaemia, while the others were either benign or of unknown significance. One patient was a double heterozygote for variants APOB p.R3527Q and LDLR p.P526S.
CONCLUSION: This work illustrates how clinical and translational research can benefit from a dedicated platform integrating both a hospital-based biobank and a data support team.
Introduction and background
The PSRC, a centralised support hub for translational research and genomic medicine
In 2013, the Centre Hospitalier Universitaire Vaudois (CHUV) and the University of Lausanne (UNIL) launched the Lausanne Institutional Biobank (BIL), a repository of biological samples and hospital records collected from CHUV patients. This facility is dedicated to research and, ultimately, genomic medicine [1]. In 2015, in partnership with competence centres based in Lausanne, in particular VITAL-IT, the high performance computing centre of the Swiss Institute of Bioinformatics (SIB), the Genomic Technologies Facility (GTF) of UNIL and the information security ISC-GE laboratory of EPFL, the Institutions further established an integrated in-house clinical research support platform, the PSRC. The platform combines expertise covering medical, scientific, regulatory, bioinformatic and technical specialties. Its mission is to provide in-house support to clinical research, with a primary focus on clinical monitoring, clinical trials, regulatory affairs, pre-analytics, information technology (IT) and bioinformatics. It is further responsible for collecting general consent forms signed by CHUV patients, providing assistance in the recruitment of clinical trial and research study participants, informing about clinical research activities conducted within the Institution, supervising the collection, storage and management of biological samples for the BIL, providing support to other institutional biobanks, and providing local researchers with secured access to coded medical data and information.
Familial hypercholesterolaemia as a model disease
Familial hypercholsterolaemia (FH) was selected as a generic model of genetic disease that matched our needs for a proof-of-concept study. FH is relatively common, autosomal dominant and of limited genetic complexity [2]. Depending on ethnicity, its frequency ranges from 1:500 to 1:200 heterozygous and around 1:1000000 homozygous affected carriers in the general population [3, 4]. In most cases, a single pathogenic variant is involved, primarily affecting one of three genes: LDLR (60–80% of the cases), APOB(1–5%), and PCSK9(up to 3%). De novo mutations and mutations in other genes have been described, yet remain rare. Oligogenic or polygenic forms have been observed too. Although the number of implicated genes is limited, the diversity of the underlying variants is broad, with over 1500 variants documented for LDLR [5]. Disease penetrance is high (90–100%), but expressivity varies depending on the nature of the mutation [6].
The clinical significance of FH is well established. Male carriers are at ≈50% risk of experiencing a major cardiovascular insult before the age of 55 and females at ≈30% before that of 60 [3]. These percentages are up to twenty-fold higher than in subjects not carrying a mutation and add on to classical risk factors for cardiovascular disease, typically obesity, hypertension, diabetes, smoking and sedentariness [3, 7]. FH is clinically actionable: well-tolerated statins and other lipid-lowering compounds allow for first-line preventive and therapeutic management, altogether contributing to an up to two-fold reduction in cardiovascular risk [8, 9]. However, although effective, these regimens are often insufficient to achieve recommended low-density lipoprotein cholesterol (LDL-C) goals in affected subjects. Owing to their marked potency at further reducing plasma LDL-C in patients receiving maximally tolerated statin therapy, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors recently emerged as a promising novel class of biological agents capable of minimising residual cardiovascular risk [9–12].
Whereas genetic testing is essential to confirm the clinical diagnosis of FH, it is neither routinely performed nor reimbursed by the Swiss healthcare system. In the absence of any dedicated screening programme, the percentage of undiagnosed FH carriers likely exceeds 80% in this country [8]. In practice, diagnosis essentially rests on the calculation of clinical risk scores pertaining to criteria of personal or familial history of premature cardiovascular disease, elevated levels of blood cholesterol – in particular LDL-C – and lipid (cholesterol) deposits around tendons [6, 8]. Assessing those scores in a multicentre, Switzerland-based cohort of 4478 participants, Nanchen et al. recently estimated that up to 50% of those hospitalised for premature acute coronary syndrome (ACS), defined as ACS affecting males before the age of 55 and females before the age of 60, presented with a possible, probable or definite risk for FH [13]. While those results still need genetic confirmation, unpublished analyses of longitudinal CoLaus [14] data further indicate that >60% of adults with a clinical diagnosis of possible FH are inappropriately treated over 5 years of follow-up (Nanchen et al. personal communication 2015). Taken together, these observations highlight the patent need for improved clinical management and guidelines for FH in Switzerland.
LDLR or APOB mutations are detected in approximately 60% of patients with a definite risk for FH and in 20 to 30% of those with a possible risk [15]. Accordingly, the likelihood of carrying pathogenic mutations in FH genes strongly correlates with LDL-C levels. Circulating concentrations of total and LDL cholesterol in heterozygous FH patients typically range from 9–14 mM and 5–10 mM, respectively, reaching up to 17–26 mM and >15.5 mM in homozygous patients [6]. Interestingly, using extreme values of total cholesterol as sole stratification criterion for genetic testing is sufficient to enrich for LDLRmutation carrier detection [16]. Since blood values of (mainly total) cholesterol are routinely monitored at the CHUV, we reasoned that the number of BIL participants with documented cholesterol measurements would be high, and that these data would be easily retrievable from the corresponding hospital records.
For the reasons listed above, FH was an optimal model to test the procedures and support provided by the PSRC.
Materials and methods
Clinical and laboratory data extraction
All data and biological samples relate to BIL participants who signed the general consent for clinical research (for additional details, see [1]). Data extraction and crossing through clinical and/or personal information were performed by authorised personnel of the Institution, following secured procedures validated by the Institution. The following categories of information were extracted and the results were provided to the researchers in the form of independent lists of coded or anonymised data, depending on the nature of each item:
‒ general information: date of birth, gender, body weight, height, date(s) of hospital stay(s);
‒ laboratory values spanning years 2004 to 2014, each with the corresponding date of measurement: LDL-C and TC for 5111 participants; triglycerides (TG), creatinine, glycaemia, thyroid stimulating hormone (TSH), triiodothyronine (T3) and thyroxine (T4) for the set of 94 selected patients;
‒ prescriptions of statin(s) and/or other lipid-lowering therapies at time of hospitalisation and/or during hospital stay: name, type, dose, date of prescription;
‒ International Classification of Diseases (ICD10) diagnostic codes for any of the following: CAD or ACS, diabetes, stroke, peripheral arterial disease, acquired immunodeficiency syndrome (AIDS), nephrotic syndrome, renal failure, dialysis, alcohol dependence, hyperglycaemia, hypothyroidism.
Patients positive for human immunodeficiency virus (HIV), nephrotic syndrome, renal failure or a prescription for dialysis, or for whom the quality of genomic DNA did not comply with high throughput DNA sequencing requirements were excluded from further analyses.
Purification of genomic DNA
Genomic DNA was extracted by LGC Genomics, Hoddesdon (UK), starting from frozen buffy coat collected from EDTA-blood samples. Procedures complied with good laboratory practice (GLP). DNA quantities and qualities were verified at the Genomic Technologies Facility (GTF) of UNIL, using the Nanodrop (Thermo Scientific) and Fragment Analyzer (Advanced Analytical) systems.
High-throughput DNA sequencing
The ADH MASTR v2 kit (Multiplicom), an amplicon-based gene panel targeting all exons, 5’UTRs and 300 to 500 base-pair promoter regions of LDLR,PCSK9 and APOE, together with exon 26 of APOB was used for high throughput DNA sequencing (supplementary table 2). An additional 12 common single nucleotide polymorphisms (SNPs) are included in the assay, allowing for polygenic risk score calculation for FH [34]. Library preparation and 250 nucleotide paired-end sequencing on the Illumina MiSeq system were performed at the GTF, closely following the recommendations of the supplier. Ten samples were tested in a pilot phase and 84 were multiplexed in a second, larger assay.
Approximately 2×20 mio sequencing reads were produced in each run. Illumina software was used for base-calling and demultiplexing on the MiSeq instrument. Given its link with Alzheimer’s disease, APOEwas masked from downstream analyses, while in absence of control patients, polygenic risk scores were not assessed.
The variant calling workflow consisted of a set of standard open source tools [35]. The main steps were: (1) trimming of low-quality bases and sequencing adapters at read ends using FastqMcf; (2) sequencing read mapping onto the human reference genome (hg19) using BWA-MEM [36]; (3) local realignment around small insertions/deletions using GATK; (4) base quality score recalibration using GATK; (5) removal of polymerase chain reaction (PCR) primers using a custom Perl script; (6) variant calling using the GATK unified genotyper tool; and (7) removal of low-quality variants. At the end of the process, variant call format (VCF) files were imported into Annovar (wannovar.usc.edu) to annotate variants. The most relevant annotations for variant assessment include those of ClinVar ( http://www.ncbi.nlm.nih.gov/clinvar ), OMIM ( http://www.omim.org /), LOVD ( http://www.lovd.nl ), HGMD public ( http://www.hgmd.cf.ac.uk ), ExAC (exac.broadinstitute.org), dbSNP ( http://www.ncbi.nlm.nih.gov/SNP ), 1000 genomes ( http://www.1000genomes.org ) and EVS (evs.gs.washington.edu) databases and servers. The full list of annotation databases is available at annovar.openbioinformatics.org/en/latest/user-guide/download/.
Confirmatory Sanger sequencing
Genomic fragments covering LDLR and APOB variants were amplified by touch-down PCR using specific primers (Microsynth). For APOB p.R3527Q: 5’-GAGTCATCTACCAAAGGAGATG-3’ and 5’-TGCTCCCAGAG-GGAATATATGC-3’; for LDLR p.C27W: 5’-GACTGTTCCTGATCGGATGAC-3’ and 5’-TCTCCTGGGACTCATCAGAGC-3’; for LDLR p.P526S: 5’-CAGAGGCTGAGGCTGCAGTGG-3’ and 5’-GTGAGACATTGTCACTATCTCC-3’; and for LDLR p.V736I: 5’-CTGTGGACTGGATCCACAGC-3’ and 5’-CACTAACCAGTTCCTGAAGC-3’. PCR conditions consisted of 3 min denaturation at 94°C, 46 cycles of 1 min denaturation at 94°C, 1 min annealing at progressively lower temperatures (i.e. starting from 1 cycle at 68°C, followed by 2 cycles at 66°C, 3 cycles at 64°C, 4 cycles at 62°C, 5 cycles at 60°C, 6 cycles at 58°C and 25 cycles at 56°C), and 1 min elongation at 72°C, followed by a final extension of 10 min at 72°C. Bi-directional sequencing of the PCR-amplified genomic fragments was performed on an ABI 3500 sequencer (PE Biosystems) using standard methods.
Sample identity coding
Data storage management and confidentiality were provided by the PSRC information technology unit in close collaboration with the CHUV information systems department. In agreement with US Food and Drug Administration (FDA) [37], European Union (EU) [38] and Organization for Economic Collaboration and Development (OECD) [39] recommendations for privacy and security of genomic data, a double encoding procedure was used, where a coded identifier, different from the permanent patient identifier of the hospital, was assigned to each BIL participant, unique 2D-barcodes were assigned to each biological sample, and distinct unique identifiers were used for genetic data.
Ethics
The protocol of the present study was submitted to the Research Ethics Committee of the Canton of Vaud (CER-VD, Lausanne, Switzerland) in June 2015 and granted authorisation on 21 August 2015. Protocol number: 273/15.

Figure 1
Selection scheme of 94 BIL participants for FH genetic testing and analysis workflow.
*As of April 26, 2015.
BIL = Lausanne Institutional Biobank; CAD = coronary artery disease; LDL-C = low-density lipoprotein cholesterol; PSRC = an integrated in-house clinical research support platform; TC = total cholesterol
Results
Data extraction
At time of data extraction (26 April 2015), 23 737 CHUV patients had been contacted by the BIL and PSRC. Of the 17 760 (74.8%) consenting participants, 13 094 (73.7%) wished to be recontacted if there were findings requiring clinical action. For 5111 of the latter, a minimum of one plasma total cholesterol (TC) value could be extracted from electronic hospital records, spanning the years 2004 to 2014 (fig. 1). Among the 5111 patients, 2291 had also been tested at least once for LDL-C.
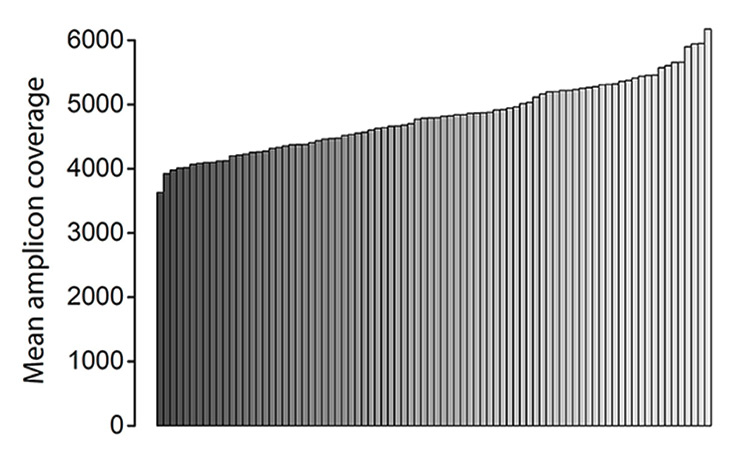
Figure 2
Mean amplicon coverage. Each bar corresponds to one of the 84 samples tested in the second sequencing run. Samples are ranked from the lowest to the highest fold-coverage value. Fold-coverage values of the first 10 samples ranged from 44 978 to 53 336 (data not shown).
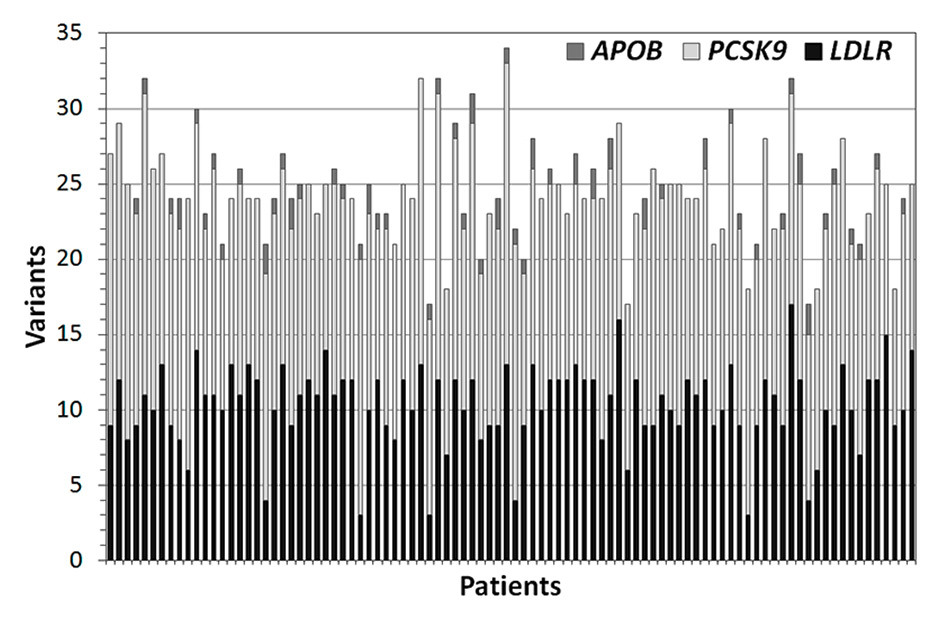
Figure 3
Number of variants identified in each of the 94 patients for APOB, LDLR, and PCSK9. Each bar corresponds to a single sample.
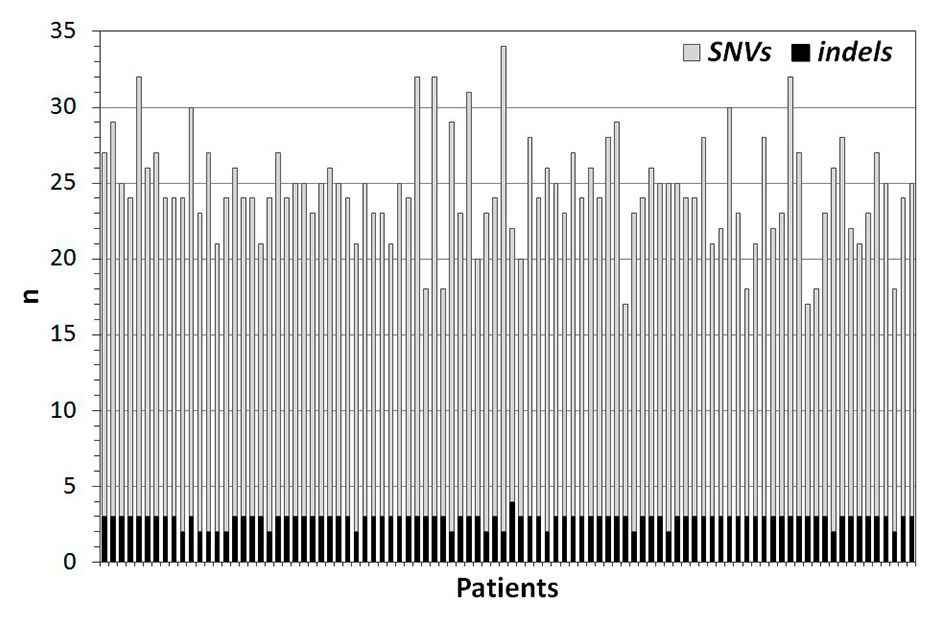
Figure 4
Number of SNVs and indels identified in each of the 94 patients for APOB, LDLR, and PCSK9. Each bar corresponds to a single sample.
SNV = single nucleotide variant; indel = short insertion and/or deletion
Selection of a priority set of 94 patients
Given the 1:500 to 1:200 prevalence of FH in Switzerland, a sample of 5111 unselected patients was expected to include 10 to 25 FH mutation carriers. To enrich for those individuals in our selection procedure, we relied on criteria of elevated TC and/or LDL-C as well as young age (18–55 yr in males, 18–60 yr in females) and presence of premature coronary artery disease (CAD) at the time of cholesterol testing. Our initial intention was to select subjects presenting at least once with TC >8.5 mM, a cut-off suggested as clinically useful for the detection of FH by genetic testing [16]. Since only 41 such patients were retrieved, while up to 94 samples could be sequenced in parallel, we relaxed our inclusion criteria, ultimately prioritising subjects presenting either with (1) early CAD, and TC ≥7.5 mM or LDL-C ≥5 mM at least once, or (2) no CAD, but TC >7.7 mM or LDL-C ≥5 mM at least once, irrespective of treatment (fig. 1). A concentration of LDL-C ≥5 mM matches the minimum criterion for possible FH, as defined by the Dutch Lipid Clinic network [6]. Here, all patients presenting with at least one such value were selected. While treatment status was not considered for selection, retrospective inspection indicated that all subjects with at least one value of plasma LDL-C ≥4 mM when under statin(s) or lipid-lowering therapy were included.
Genetic testing
Genetic testing was performed by high throughput DNA sequencing of LDLR, APOB, and PCSK9 (see Methods). Resulting mean amplicon coverage was far above the usual requirements for germline sequencing, exceeding 4000-fold in almost all samples (fig. 2). Moreover, coverage uniformity was close to complete, with 100% of the targeted bases covered 30-fold or higher in 87 samples and >99.9% in the others. Overall, these results demonstrate excellent sequencing quality.
On average, 25 ± 3 (mean ± standard deviation) variants were detected in each patient (minimum 17, maximum 34; fig. 3). The vast majority were single nucleotide variants (SNVs) and the number of short insertions or deletions (indels) did not exceed three in any sample (fig. 4). Of the 84 distinct variants identified in total, 43 affected LDLR, 5 APOB and 36 PCSK9(see appendix, supplementary table 1). According to the ExAC browser (http://exac.broadinstitute.org, version of July 2015), 23 SNVs were rare [i.e. occurring at global minor allele frequencies (MAF) <1% in the general population], while the others were common polymorphisms. Among the rare SNVs, eight coded for missense amino acids and five for synonymous variants, one was in a 3’UTR and nine were intronic (table 1).
None of the intronic variants was predicted to affect messenger RNA (mRNA) splicing (data not shown). Therefore, to identify likely causative mutations for FH, we restricted our investigations to low-frequency (MAF <1% in ExAC) missense coding variants. All of these heterozygous variants were documented in ExAC, indicating that none was novel.
We excluded two known SNVs from the list of candidates because of lack of evidence for a causal link with FH in the literature and/or databases relevant to FH (namely ClinVar [17], UCL [18], LOVD [5] and HGMD public [19]). The first one, PCSK9 p.A443T (rs28362263; genomic coordinates: chr1:55523855 G>A), is associated with reduced plasma LDL-C [20]. The second one, APOB p.Q3432E (rs1042023; genomic coordinates: chr2:21229446 G>C), was detected in four carriers and considered of low interest because of a lack of cosegregation with hypercholesterolaemia [21–24] and a classification as “likely benign for FH” in ClinVar.
Assessments of the remaining six missense coding variants were based on allele frequencies, predicted and/or experimentally demonstrated functional impact (focusing on predictive CADD scores of nonfunctionality [25] and on an extensive functional study of LDLR mutations [26]), and clinical relevance. Three variants were considered as pathogenic (APOB p.R3527Q) or likely pathogenic (LDLR p.C27W and LDLR p.P526S) for hypercholesterolaemia, whereas the others were of either uncertain or unknown significance (table 2). The most significant variants were verified by Sanger sequencing (fig. 5). Although the number of pathogenic mutation carriers was small, thus preventing further statistical demonstration, we note that LDL-C and TC plasma levels of patients 65 and 82 were among the highest values documented for the set of 94 high-cholesterol subjects, a trend expected to FH (fig. 6). In contrast, mean cholesterol levels of patients 39, 50 and 73, who carried potentially less damaging variants, were lower than the clinically relevant thresholds of 4.9 mM for LDL-C and 7.5 mM for TC.
|
Table 1:Types of genetic variants identified in the set of 94 selected patients. |
|
|
APOB
|
LDLR
|
PCSK9
|
TOTAL
|
|
Variants, all
|
5 |
45 |
36 |
86 |
| 1. MAF(ExAC) ≤1% |
3 |
12 |
8 |
23 |
| - SNVs |
3 |
12 |
8 |
23 |
| - Coding |
3 |
6 |
4 |
13 |
| - Non-synonymous |
2 |
5 |
1 |
8 |
| - Nonsense |
|
0 |
0 |
0 |
| - Frameshift |
|
0 |
0 |
0 |
| - Upstream |
|
0 |
0 |
0 |
| - 3’UTR |
|
0 |
1 |
1 |
| - Intronic |
|
6 |
3 |
9 |
| - Indels |
|
0 |
0 |
0 |
| 2. MAF(ExAC) = 1–2% |
|
2 |
0 |
2 |
| - Intronic |
|
1 |
0 |
1 |
|
- ncRNA |
|
1 |
0 |
1 |
| MAF(ExAC): minor allele frequency as reported in the ExAC database; SNV = single nucleotide variant; UTR = untranslated region |
|
Table 2:Details of the primary rare, missense, coding variants. Data are presented for five variant carriers, two females, three males. All of these patients were aged 18-60 yr at time of top cholesterol testing and presented with plasma triglyceride levels ≤2.2 mM. |
|
|
Patient 39
|
Patient 50
|
Patient 65
|
Patient 73
|
Patient 82
|
| Variant(s) |
LDLR p.R595Q |
LDLR p.G269D |
LDLR p.C27W |
LDLR p.V736I |
(1) APOB p.R3527Q
(2) LDLR p.P526S |
| Genomic coordinates |
19:11227613 G>A |
19:11217352 G>A |
19:11210912 C>T |
19:11233915 G>A |
(1) 2:21229160 C>T
(2) 19:11224428 C>T |
| dbSNP ID |
rs201102492 |
rs143992984 |
NA |
rs547268730 |
(1) rs5742904;
(2) NA |
| MAF (ExAC) |
4.942×10–5
|
0.0002 |
1.65×10–5
|
2.475×10–5
|
(1) 0.0002
(2) 1.66×10-5
|
| CADD score [25] |
22.8 (high); see also [26] |
11.28 (moderate) |
16.97 (high) |
6.505 (low) |
(1) 19.81 (high)
(2) 20.1 (high) |
| LOVD/HGMD annotation and assertion |
FH, probably pathogenic |
FH, Rome allele |
FH, San Francisco
allele |
NA |
(1) ApoB deficiency; cosegregates with ADH; is the most frequent mutation for FH/ADH
(2) FH, Cincinatti allele, reduced LDLR activity |
| CLINVAR assertion |
Not asserted |
Uncertain significance for FH |
Not referenced |
Not referenced |
(1) Pathogenic, likely pathogenic for FH (see OMIM #107730 [40]; to be reported (ACMG, [31])
(2) Not asserted |
| Functional testing and clinical assertion by Thormaehlen et al. [26] |
Not disruptive; found in a patient with MI and in an unaffected control; pathogenic for MI |
Not disruptive (see also [41]); found in a patient with MI; pathogenic for MI |
Not tested |
Not tested |
(1) NA
(2) Disruptive; found in a patient with high LDL-C; pathogenic for high LDL-C |
| Confirmation by Sanger (fig. 5) |
NA |
NA |
YES |
YES |
(1) YES
(2) YES |
| Diagnostic |
No CAD |
No CAD |
Premature CAD |
Premature CAD |
Premature CAD |
| Statins/LLT |
No |
No |
Yes |
Yes |
Yes |
| ACMG: American college of medical genetics; HGMD: human gene mutation database; LLT: lipid-lowering therapy; NA: not applicable; LOVD: Leiden open (source) variation database. |
Discussion
The major finding of this work consists of the identification of seven carriers of rare, missense heterozygous coding variants in FH genes among 94 high-cholesterol participants in the BIL. Given the proof-of-concept nature of our study, these results are remarkable as they were obtained by applying a relatively crude analytical workflow, where laboratory and clinical data extracted from electronic hospital records of 5111 consenting participants were used to enrich for mutation carriers within a small test set of 94 selected individuals, and high-throughput DNA sequencing allowed for targeted genetic screening of the major FH genes.
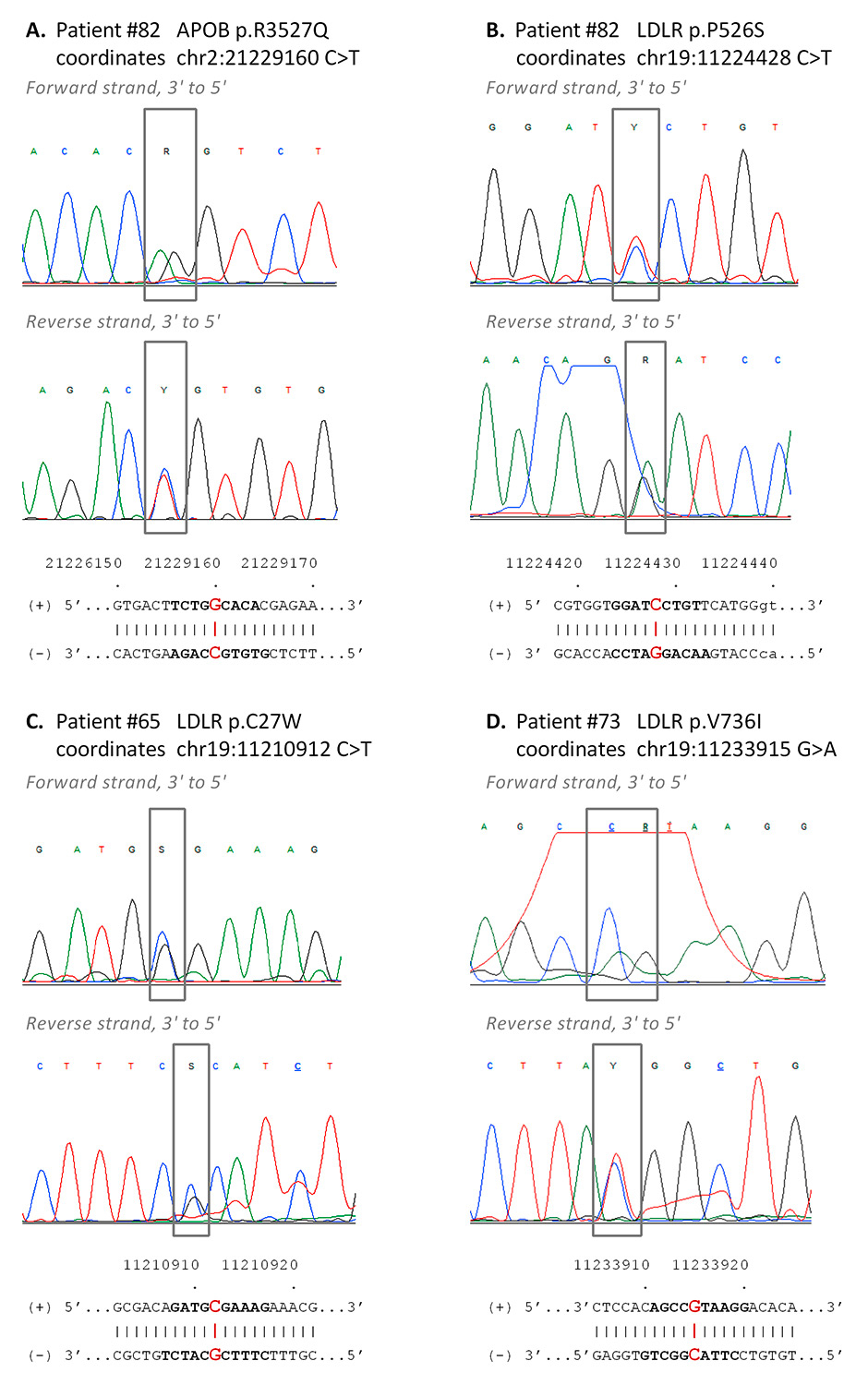
Figure 5
Confirmatory Sanger sequencing. The most clinically relevant variants of patients 82 (panels A, B), 65 (panel C) and 73 (panel D) were confirmed by bidirectional Sanger sequencing. Each panel shows chromatograms of the forward (top) and reverse (bottom) sequencing reactions, together with the corresponding reference sequences on the (+) and (–) strands (hg19). The variants are boxed in grey on the chromatograms; the corresponding positions on the reference sequence are highlighted in red; the stretch of reference sequence shown in the respective chromatograms is in bold letters. APOB is coded by the (-) strand, LDLR is coded by the (+) strand.
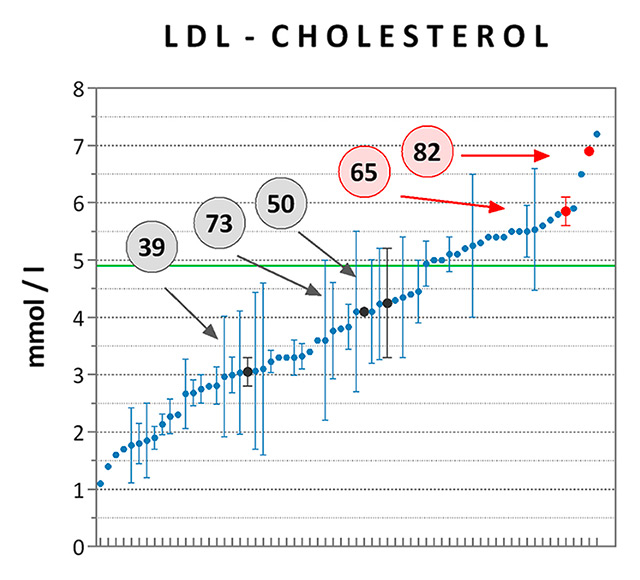
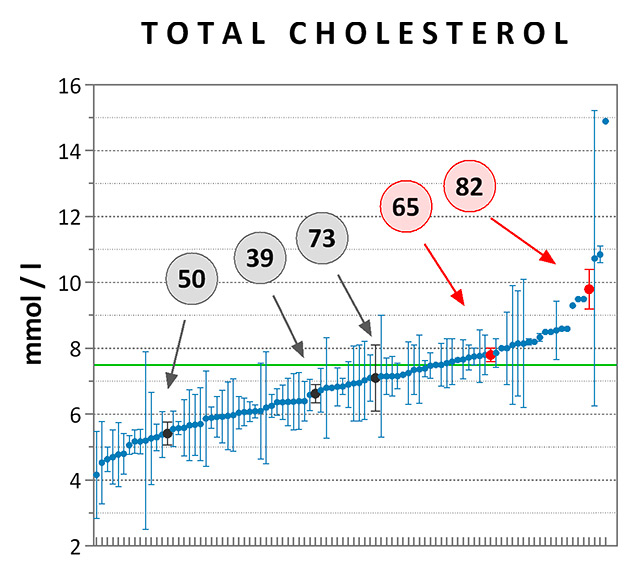
Figure 6
Cholesterol levels measured in carriers vs non carriers of rare, missense APOB and LDLR variants. Average LDL-C (panel A) and TC (panel B) levels were computed for each of the high-cholesterol patients tested by sequencing, irrespective of treatment. At least one value of LDL-C was available for 65 of the 94 subjects [n = 2 ± 4 data points per patient (mean ± SD)], while TC was measured at least once in all of them [n = 5 ± 6 data points per patient (mean ± SD)]. Data are presented as means ± standard error of the mean and are ranked from the lowest to the highest mean value. Data points referring to the carriers of pathogenic (patient #82) or likely pathogenic (patient #65) variants for FH are highlighted in red, while those referring to carriers of rare variants of unknown significance are highlighted in dark grey (patients #39, 73, and 50). The clinical thresholds of high LDL-C (4.9 mM) and TC (7.5 mM) are indicated by the green lines.
FH = familial hypercholesterolaemia; LDL-C = low-density lipoprotein cholesterol; TC = total cholesterol
Given the prevalence of FH, an estimated 10 to 20 carriers of pathogenic mutations were expected. In reality, eight rare LDLR and/or APOB missense variants were identified, a number slightly below our predictions. Although presumably not reaching top performance in terms of sensitivity, our results compare well with the approximately 2% pathogenic mutation carriers identified by Norsworthy et al. upon testing a population of 193 hypercholesterolaemic primary care patients and applying selection criteria similar to ours [27]. Moreover, Futema et al. clearly demonstrated that using elevated cholesterol as the sole inclusion criterion for genetic testing was particularly efficient when considering extreme laboratory values (i.e. above 9 to 10 mM for TC), while specificity dropped quickly at lower levels, typically reaching less than 5% when applying relaxed thresholds of 7.5 to 7.7 mM TC, as was done here [16]. More recently, these observations were confirmed in the large study of Khera et al. who identified <2% FH mutation carriers in a cohort of 20 485 multiethnic participants presenting plasma LDL-C >4.9 mM [7]. Interestingly, this study also showed that, for a given LDL-C level in plasma, carriers of FH-causing mutations were at a much higher risk than noncarriers, providing strong support for the concept of adding genetic information when predicting CAD risk in suspected FH patients.
There are several explanations for suboptimal detection sensitivity. For the sake of simplicity, patient stratification and selection were performed regardless of treatment type and status, and no correction for age, gender or treatment was applied. Therefore, a number of mutation carriers may have escaped prioritisation, in particular those for whom cholesterol was monitored only in the presence of lipid-lowering treatment, reaching nominal values below 7.5 mM for TC and 4 mM for LDL-C. To improve detection efficiency, these data could be corrected for the type and intensity of therapy used [7, 28], but such a task was beyond our basic aims. A second limitation relates to the identification of (large) structural variants (SVs), which are not detected by amplicon-based sequencing. Also, while preliminary investigations did not point at copy number variants (CNVs) in our dataset (data not shown), we lacked proper positive controls to verify the reliability of those results. Further, one should keep in mind that a few genes other than LDLR, APOB, and PCSK9 are known to contribute to rare forms of FH [29, 30], but were not tested. And last, a few of the hypercholesterolaemic patients may be affected by oligo- or polygenic forms of FH, which we did not investigate.
Three of the rare APOBand LDLRvariants comply with “pathogenic” or “likely pathogenic” clinical classifications [31]. Essentially based on existing variant annotations and published data, these assertions warrant further assessment and clinical validation by a panel of expert reviewers. Tentative scoring of the rare variant carriers using the data in table 2 and the criteria of the Dutch Lipid Clinic network [6] tend to suggest that patients 65 and 82, who carry the most damaging mutations, suffered stronger clinical manifestations than subjects 39, 50 and 73 (data not shown). In the absence of more comprehensive information, complementary clinical investigations will be necessary to confirm risk scoring and diagnosis. Nonetheless, our results provide important insights into information feedback to research study participants, a process formally supported by the general consent of the BIL [1]. Of note, the design of an appropriate feedback procedure will be the task of a dedicated working group, whose conclusions and recommendations will be the topic of a separate report.
Linking data derived from electronic hospital records to biological samples stored in biobanks such as the BIL enables cost- and time-effective research. According to comparisons with more traditional approaches, the total time and study cost necessary to recruit a cohort of a few hundred participants, phenotype each subject, collect biological specimens and proceed to genotyping can be reduced by up to 8- and 15-fold, respectively [32]. In a large biobank like the BIL, storage of biological specimens is often restricted to a small array of samples, typically DNA and plasma. However, the principle of the general consent [1] makes it possible to recontact consenting participants, thereby enabling subsequent enrichment with additional specimens and/or clinical data. These benefits may be amplified further by pooling analyses across collaborative networks, leading to an increase in sample sizes and minimisation of biases [33]. Networks will be instrumental in investigating rare diseases and/or rare variants, screening for specific variant carriers in larger pools of samples, crossing or replicating results with information gained in independent cohorts, or accessing complementary information or samples from other repositories. As recognised earlier, strong support structures will be necessary to streamline efficient and effective communication and management among the research teams and networks involved [32].
At the local level, the present study illustrates the capabilities of the PSRC and its partners to provide efficient assistance and support to research centered on genomic medicine, in particular for the recruitment of consenting patients, the secured management of high-quality biological samples and medical data, and the generation and analysis of genetic profiles. To address similar needs, and also to ascertain research quality and interconnectedness across biobanks at the national level, the Swiss National Science Foundation, the Swiss Academy of Medical Sciences (SAMS) and a dedicated Project Group recently launched the Swiss Biobanking Platform (SBP; http://www.swissbiobanking.ch/). The mission of the SBP will be to develop a reliable customer-oriented network across Swiss biobanks, with harmonised processes for general information and consent, pre-analytical sample processing and storage, laboratory informatics management systems, data storage and confidentiality, annotation of samples and data, traceability, access and distribution of samples and data to researchers, support to investigators and biobank sustainability.
Appendix
Supplementary tables http://www.smw.ch/fileadmin/smw/pdf/SMW-14326-Appendix.pdf
Acknowledgements: We thank Drs Grégoire Würzner, Cindy Roth and Christine Currat for helpful discussions and insights during the preparation and realization of the project, and Fady Fares, Didier Foretay, Farid Ahmad and Sébastien Rocher for technical assistance.
References
1 Mooser V, Currat C. The Lausanne Institutional Biobank: a new resource to catalyse research in personalised medicine and pharmaceutical sciences. Swiss Med Wkly. 2014;144:w14033.
2 Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161–72.
3 Youngblom E, Knowles JW, Familial Hypercholesterolemia, in GeneReviews(R), R.A. Pagon, et al., Editors. 1993: Seattle (WA).
4 Brun N, Rodondi N. How to deal with familial dyslipidemia in clinical practice? Rev Med Suisse. 2012;8(331):494–6, 498–500. French.
5 Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76(5):387–401.
6 Hovingh GK, Davidson MH, Kastelein JJ, and O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34(13):962–71.
7 Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, et al. Diagnostic Yield of Sequencing Familial Hypercholesterolemia Genes in Patients with Severe Hypercholesterolemia. J Am Coll Cardiol. 2016;67(22):2578–89.
8 Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–90a.
9 Reiner Z. Management of patients with familial hypercholesterolaemia. Nat Rev Cardiol. 2015;12(10):565–75.
10 Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–9.
11 Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–99.
12 Ballantyne CM, Neutel J, Cropp A, Duggan W, Wang EQ, Plowchalk D, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo-controlled, dose-ranging study in statin-treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115(9):1212–21.
13 Nanchen D, Gencer B, Auer R, Raber L, Stefanini GG, Klingenberg R, et al. Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Eur Heart J. 2015;36(36):2438–45.
14 Firmann M, Mayor V, Vidal PM, Bochud M, Pecoud A, Hayoz D, et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc Disord. 2008;8:6.
15 Damgaard D, Larsen ML, Nissen PH, Jensen JM, Jensen HK, Soerensen VR, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis. 2005;180(1):155–60.
16 Futema M, Kumari M, Boustred C, Kivimaki M, Humphries SE. Would raising the total cholesterol diagnostic cut-off from 7.5 mmol/L to 9.3 mmol/L improve detection rate of patients with monogenic familial hypercholesterolaemia? Atherosclerosis. 2015;239(2):295–8.
17 Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–5.
18 Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72(Pt 4):485–98.
19 Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9.
20 Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78(3):410–22.
21 Gaffney D, Hoffs MS, Cameron IM, Stewart G, O’Reilly DS, Packard CJ. Influence of polymorphism Q3405E and mutation A3371V in the apolipoprotein B gene on LDL receptor binding. Atherosclerosis. 1998;137(1):167–74.
22 Pullinger CR, Love JA, Liu W, Hennessy LK, Ghassemzadeh M, Newcomb KC, et al. The apolipoprotein B Q3405E polymorphism has no effect on its low-density-lipoprotein receptor binding affinity. Hum Genet. 1996;98(6):678–80.
23 Ludwig EH, Hopkins PN, Allen A, Wu LL, Williams RR, Anderson JL, et al. Association of genetic variations in apolipoprotein B with hypercholesterolemia, coronary artery disease, and receptor binding of low density lipoproteins. J Lipid Res. 1997;38(7):1361–73.
24 Leren TP, Bakken KS, Hoel V, Hjermann I, Berg K. Screening for mutations of the apolipoprotein B gene causing hypocholesterolemia. Hum Genet. 1998;102(1):44–9.
25 Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
26 Thormaehlen AS, Schuberth C, Won HH, Blattmann P, Joggerst-Thomalla B, Theiss S, et al. Systematic cell-based phenotyping of missense alleles empowers rare variant association studies: a case for LDLR and myocardial infarction. PLoS Genet. 2015;11(2):e1004855.
27 Norsworthy PJ, Vandrovcova J, Thomas ER, Campbell A, Kerr SM, Biggs J, et al. Targeted genetic testing for familial hypercholesterolaemia using next generation sequencing: a population-based study. BMC Med Genet. 2014;15:70.
28 Fouchier SW, Hutten BA, and Defesche JC. Current novel-gene-finding strategy for autosomal-dominant hypercholesterolaemia needs refinement. J Med Genet. 2015;52(2):80–4.
29 Hegele RA, Ban MR, Cao H, McIntyre AD, Robinson JF, Wang J. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol. 2015;26(2):103–13.
30 Stitziel NO, Peloso GM, Abifadel M, Cefalu AB, Fouchier S, Motazacker MM, et al. Exome sequencing in suspected monogenic dyslipidemias. Circ Cardiovasc Genet. 2015;8(2):343–50.
31 Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
32 Bowton E, Field JR, Wang S, Schildcrout JS, Van Driest SL, Delaney JT, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci Transl Med. 2014;6(234):234cm3.
33 Ioannidis JP, Trikalinos TA, Khoury MJ. Implications of small effect sizes of individual genetic variants on the design and interpretation of genetic association studies of complex diseases. Am J Epidemiol. 2006;164(7):609–14.
34 Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet. 2013;381(9874):1293–301.
35 DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
36 Li H and Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
37 FDA. E15 definitions for genomic biomarkers, pharmacogenomics, pharmacogenetics, genomic data and sample coding categories. 2008.
38 EU. Proposal for a regulation of the European Parliament and of the Council on the protection of individuals with regard to the processing of personal data and on the free movement of such data; General data protection regulation. 2015.
39 OECD. OECD guidelines on human biobanks and genetic research databases recommendations for data privacy and security. 2009.
40 Online Mendelian Inheritance in Man, OMIM®. 2011, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University: Baltimore, MD.
41 Etxebarria A, Palacios L, Stef M, Tejedor D, Uribe KB, Oleaga A, et al. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum Mutat. 2012;33(1):232–43.